

Abstract
Unlike human malaria parasites that induce persistent infection, some rodent malaria parasites, like Plasmodium yoelii strain 17XNL (Py17XNL), induce a transient (self-curing) malaria infection. Cooperation between CD4 T cells and B cells to produce antibodies is thought to be critical for clearance of Py17XNL parasites from the blood, with major histocompatibility complex (MHC) class II molecules being required for activation of CD4 T cells. In order to better understand the correspondence between murine malaria models and human malaria, and in particular the role of MHC (HLA) class II molecules, we studied the ability of humanized mice expressing human HLA class II molecules to clear Py17XNL infection. We showed that humanized mice expressing HLA-DR4 (DR0401) molecules and lacking mouse MHC class II molecules (EA0) have impaired production of specific antibodies to Py17XNL and cannot cure the infection. In contrast, mice expressing HLA-DR4 (DR0402), HLA-DQ6 (DQ0601), HLA-DQ8 (DQ0302), or HLA-DR3 (DR0301) molecules in an EA0 background were able to elicit specific antibodies and self-cure the infection. In a series of experiments, we determined that the inability of humanized DR0401.EA0 mice to elicit specific antibodies was due to expansion and activation of regulatory CD4+ Foxp3+ T cells (Tregs) that suppressed B cells to secrete antibodies through cell-cell interactions. Treg depletion allowed the DR0401.EA0 mice to elicit specific antibodies and self-cure the infection. Our results demonstrated a differential role of MHC (HLA) class II molecules in supporting antibody responses to Py17XNL malaria and revealed a new mechanism by which malaria parasites stimulate B cell-suppressogenic Tregs that prevent clearance of infection.
INTRODUCTION
Malaria is an Anopheles mosquito-borne infectious disease caused in humans by five different members of the protozoan genus Plasmodium (i.e., P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi). P. falciparum is the most virulent and deadly malaria parasite and infects 1 billion to 2 billion people annually. The number of P. falciparum deaths reported by the WHO for 2010 was 665,000, while the Institute of Health Metrics and Evaluation reported 1.24 million deaths for the same year (1).
Infection is initiated upon a bite of an infected Anopheles mosquito and inoculation of sporozoites into skin, which rapidly invade the bloodstream to infect hepatocytes and develop into liver stage parasites. The liver stage of infection is asymptomatic and lasts 5 to 7 days for human malaria species and 2 to 3 days for rodent malaria species (2). Mature liver stage parasites are then released into the bloodstream to invade red blood cells and to initiate the asexual erythrocyte cycles responsible for the pathological manifestations of malaria. During the course of blood infection, parasites may differentiate into female and male gametocytes that are taken up by the mosquito, where they undergo sexual reproduction and meiosis in the gut and generate sporozoites that migrate to the salivary glands to perpetuate the life cycle (3). Morbidity and mortality associated with P. falciparum occur mostly during first-time infection of infants and pregnant women living in areas where malaria is endemic, as these groups are at the highest risk for development of severe malaria syndromes such as lactic acidosis and anemia as a consequence of hyperparasitemia or cerebral and placental malaria due to sequestration of parasites in organs (1). Travelers are also very vulnerable to severe malaria. Naturally acquired immunity to malaria develops slowly upon repetitive episodes of infection and represents a state of semi-immunity where parasitemia is under control and prevents severe malaria (4).
The major histocompatibility complex (MHC) (HLA in humans) proteins are highly polymorphic glycoproteins consisting of two noncovalently associated α and β chains. A peptide binding groove is generated by the assembly of α1-β1 domains of the MHC class II (MHC-II) heterodimer, which allows binding of peptides of 13 to 17 amino acids in length derived from the exogenous pathway of antigen processing (5). MHC molecules are major players in generating immune responses to microorganisms, since their primary role is to present peptides for activation and differentiation of CD4 T cells. Among the various CD4 T cell subsets, CD4 T helper cells (Th1, Th2, and Th17) are required to orchestrate cellular and/or humoral responses needed to clear infections (6), while regulatory CD4+ Foxp3+ T cells (Tregs) downregulate cellular and/or humoral responses through direct cell-cell interactions or by secretion of suppressive cytokines such as interleukin-10 (IL-10) and transforming growth factor β (TGF-β) (7, 8). Recent studies indicated that some pathogens stimulate Tregs as an immune evasion mechanism (9). When it comes to malaria, studies in rodent models (10–20) and humans (21–23) have demonstrated an increase in Treg frequency shortly upon blood stage infection, although Treg frequency during the course of infection did not correlate with disease severity. Furthermore, attempts to deplete Tregs or to increase Treg frequency in mice prior to infection with a variety of parasite strains (Plasmodium yoelii, P. berghei, or P. chabaudi) have led to conflicting results. Depending on the parasite/mouse strain combination, Tregs have been shown to have either beneficial (10, 17, 18) or deleterious (14–16, 19) effects or no significant effect (12, 16). This has been attributed to the variety of severe malaria syndromes associated with each rodent parasite strain. While Tregs may be deleterious by inhibiting immune responses to malaria parasites, leading to hyperparasitemia, anemia, and metabolic derangement, Tregs may also be beneficial by suppressing strong inflammatory responses that lead to immunopathology (i.e., cerebral malaria). Importantly, Tregs in these models have not been characterized for their ability to downregulate cellular versus antibody (Ab) responses. This is important in considering the different role of cellular versus humoral responses in clearance of various rodent malaria strains. For instance, while clearance of P. yoelii parasites is thought to be mediated by antibodies (24), clearance of P. chabaudi is less dependent on antibodies but more dependent on CD4 T cells (25).
The differential ability of mouse MHC-II molecules versus human HLA class II (HLA-II) molecules to present epitopes to CD4 T cells is a major drawback of preclinical rodent models, which may explain in part why the immunogenicity and/or protective efficacy of many malaria vaccine candidates tested in rodent models did not translate into efficacy in clinical trials. The advent of humanized mice expressing HLA class II molecules and lacking mouse MHC class II molecules (EA0) has significantly advanced the understanding of HLA-II molecules in infectious diseases and autoimmunity. We and others demonstrated that HLA class II molecules expressed in mice function similarly to those in humans, as they present immunodominant epitopes of foreign antigens and self-antigens to the same extent as they do in humans (26, 27). Humanized HLA-II mice also express a diverse set of T cell receptor beta (TCR Vβ) chains similar to that observed in humans and develop clinical and histological similarities of human autoimmune syndromes linked to HLA-II (28). Here we have investigated the role of HLA-II molecules in P. yoelii strain 17XNL (Py17XNL) malaria. Our results indicate that mice expressing HLA-DR0401 molecules, unlike C57BL/6, BALB/c, and humanized mice expressing other HLA-II molecules, succumb to Py17XNL malaria infection due to expansion and activation of CD4+ Foxp3+ T cells that suppress antibody responses by direct cell-cell contact with B cells.
MATERIALS AND METHODS
Mice.
All animal procedures were conducted under IACUC protocols approved by the WRAIR/NMRC and the USUHS in compliance with the Animal Welfare Act and in accordance with the principles set forth in the Guide for the Care and Use of Laboratory Animals (29). DR0401.EA0 (C57BL/6) mice, expressing HLA-DR0401α1β1/IEα2β2 molecules and lacking expression of mouse MHC-II molecules (Abb knockout [KO] mutation, EA0), were previously described (30), and they were purchased from Taconic (Hudson, NY). The chimeric DR0401-IE molecules expressed by DR0401.EA0 mice contain the HLA-DR0401α1β1 peptide binding site, with the remaining domains derived from murine IEα2β2 to allow binding to the mouse CD4 coreceptor. DR0402.EA0 (C57BL/10) mice express full-length human molecules with mutations at positions β110 and β139 to allow mouse CD4 binding (31). DR3.EA0, DQ8.EA0, and DQ6.EA0 (C57BL/10) mice express full-length human molecules that bind to mouse CD4 (32–34). DR0402.EA0, DR3.EA0, DQ8.EA0, and DQ6.EA0 mice were generated at Mayo Clinic, Rochester, MN. The HLA class II molecules expressed in EA0 mice are functional, as they mediate thymic negative (Vβ5, Vβ11, and Vβ12) and positive (Vβ6 and Vβ14) selection, similarly to mouse I-E molecules, and rescue development of CD4 T cells (reviewed in reference 35). BALB/c and C57BL/6 mice were purchased from The Jackson Laboratories (Bar Harbor, ME). EA0 (C57BL/6) mice were generated by breeding DR0401.EA0 mice with C57BL/6 mice, intercrossing the F1 mice, and screening F2 mice for the absence of HLA-DR0401αβ transgenes and the presence of the Aβ KO mutation by PCR. Primers used for screening of the HLA-DR0401αβ transgenes were previously described (36). Primers used to identify the Aβ KO mutation were forward primer CCGCAGGGCATTTCGTGTA and reverse primer GCCGCCGCAGGGAGGTGTG. To generate F1 hybrid mice coexpressing HLA-DR0401 molecules and mouse MHC-II molecules, the DR0401.EA0 mice were crossed with BALB/c or with C57BL/6 mice. F1 control mice (BALB/c.C57BL/6) lacking expression of HLA-DRB1*0401 molecules were generated by crossing EA0 (C57BL/6) mice with BALB/c mice.
Parasites.
Plasmodium yoelii (17XNL strain) sporozoites were obtained by dissecting salivary glands of infected Anopheles stephensi mosquitoes as previously described (37). Py17XNL-infected red blood cells (Py-iRBCs) were obtained from BALB/c.RagKO mice challenged with Py17XNL sporozoites when parasitemia reached >30%. Mice were challenged intravenously with 200 sporozoites or with 5 × 104 Py-iRBCs. Parasitemia was measured using Giemsa-stained thin blood smears by counting an average of 3,000 RBCs.
Measurement of antibody titers by immunofluorescence assay (IFA).
Teflon printed slides (12-well; Electron Microscopy Sciences, Hatfield, PA) were coated with 104 RBCs/well from infected BALB/c.RagKO mice when parasitemia reached >30%. Slides were air dried and stored at −80°C until use. Upon thawing, slides were blocked with phosphate-buffered saline (PBS)–1% bovine serum albumin (BSA) for 30 min at 37°C. Twenty microliters of plasma at various dilutions was added to the wells and incubated for 1 h at 37°C. Slides were washed three times with PBS, incubated with fluorescein isothiocyanate (FITC)-labeled F(ab′)2 goat anti-mouse IgG1, IgG2a, IgG2b, IgG2c, or IgG3 (Southern Biotechnologies, Birmingham, AL) for 30 min at 37°C, washed, and mounted with Vectashield-DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories, Burlingame, CA).
Hematocrit count.
Blood collected with heparin-coated capillaries was diluted 1:5,000 in PBS, and erythrocytes were counted with hemocytometers.
Chloroquine treatment.
Mice were injected intraperitoneally with 25 mg/kg of body weight chloroquine diphosphate (Sigma, St. Louis, MO) in PBS, as described in figure legends.
Immunization with Py17XNL irradiation-attenuated sporozoites.
Py17XNL-infected Anopheles stephensi mosquitoes were irradiated (10,000 rads) by using a cobalt irradiator (gamma irradiator; J. L. Shepherd), and sporozoites were purified from the salivary glands. Mice were injected intravenously with three doses of 30,000 irradiation-attenuated sporozoites (Irrd-spz) at 2-week intervals and challenged 2 weeks after the last immunization with 200 infectious Py17XNL sporozoites.
Fluorescence-activated cell sorter (FACS) analysis.
Isolation and staining of splenocytes were carried out as described previously (38), using anti-human HLA-DR (clone TU39) and anti-mouse IA/IE (clone 2G9), CD3 (clone 145-2C11), CD4 (clone RM4-5), CD25 (clone PC61) (BD Biosciences, San Jose, CA), and Foxp3 (eBiosciences, San Diego, CA). Cells were gated and analyzed on mononuclear forward scatter/side scatter (FSC/SSC).
In vivo depletion of Tregs.
Rat anti-mouse CD25 antibodies were purified from supernatants of PC61 hybridoma cells (ATCC, Manassas, VA), using MAR18 (mouse anti-rat) affinity chromatography columns. Mice were injected intraperitoneally with 250 μg of rat anti-mouse CD25 or the rat IgG isotype control daily for 3 days.
T cell responses.
Splenocytes were cultured ex vivo for 4 days or stimulated with CD3/CD28 Abs (1 μg/ml each; BD Biosciences) or with concanavalin A (ConA) (2.5 μg/ml; Sigma-Aldrich, St. Louis, MO) for 48 h. Cytokine secretion in supernatants was measured by using Luminex kits (Invitrogen, Carlsbad, CA).
B cell responses.
Negatively isolated (untouched) splenic B cells (Dynabeads mouse CD43; Invitrogen) from two mice (pooled) were stimulated with anti-mouse CD40 Abs (3 μg/ml; BD Biosciences), IL-4 (100 U/ml), and IL-2 (15 U/ml) (Invitrogen) or were left unstimulated. CD4+ CD25+ Tregs from 10 to 15 mice (pooled) were positively isolated with Dynabeads Flowcomp mouse CD4+ CD25+ Tregs (Invitrogen). Graded numbers of Tregs were added to B cell cultures in the presence of CD3/CD28 Abs (1 μg/ml each). To prevent cell-cell contact, B cells and Tregs were cultured in 0.4-μm transwells. At day 7 of culture, half of the medium was replaced by fresh medium. Immunoglobulin secretion in supernatants at day 16 of culture was measured by an enzyme-linked immunosorbent assay (ELISA) (mouse IgG ELISA quantitation set; Bethyl Laboratories, Montgomery, TX).
Passive transfer of sera.
Pooled sera from C57BL/6 mice that had self-cured (day 21 postchallenge with Py17XNL sporozoites) or from naive C57BL/6 mice were injected intraperitoneally into infected DR0401.EA0 mice (0.5 ml serum/recipient).
Statistical analysis.
Data were analyzed by using an unpaired (2-tailed) Student t test. For multiple comparisons, we used 2-way analysis of variance (ANOVA) followed by Tukey's post hoc comparisons of multiple conditions. For protection experiments, we used the Fisher exact test.
RESULTS
Mice expressing HLA-DR0401 molecules in the EA0 background (DR0401.EA0) have impaired antibody responses to Py17XNL blood stage parasites and cannot self-cure infection.
Consistent with previous reports (39, 40), we found that C57BL/6 and BALB/c mice challenged with 200 infectious Py17XNL sporozoites develop mild blood stage parasitemia, and they self-cure the infection within 14 to 21 days postchallenge (Fig. 1A, left and middle). In contrast, humanized DR0401.EA0 (C57BL/6) mice developed hyperparasitemia and succumbed to infection within 25 to 30 days (Fig. 1A, right). The cause of death was most likely severe anemia, since the infected DR0401.EA0 mice had a 47% hematocrit reduction at day 21 postchallenge compared to hematocrit at prechallenge (4.5 million ± 1.6 million versus 8.5 million ± 1.4 million red blood cells [RBCs]/μl, respectively; P < 0.0001).
FIG 1.

Mice expressing HLA-DR0401 as the only MHC class II molecule fail to self-cure Py17XNL malaria infection. (A and B) Groups of C57BL/6 (n = 7), BALB/c (n = 5), and DR0401.EA0 (n = 10) mice were challenged intravenously with 200 Py17XNL sporozoites. (A) Parasitemia (iRBCs among 3,000 total RBCs) in individual mice, counted in Giemsa-stained thin blood smears. (B) Kinetics of IgG antibodies to Py17XNL-infected red blood cells (Py-iRBCs), as measured by IFA. Data represent means ± standard deviations for mice analyzed individually. (C) DR0401.EA0 mice were challenged with 200 Py17XNL sporozoites, and 6 days later, they were injected intraperitoneally with immune sera (500 μl/mouse) from self-cured C57BL/6 mice or with an equal volume of sera from naive C57BL/6 mice or were left untreated. Mice were monitored for parasitemia by using thin blood smears. Data represent means ± standard deviations for three mice analyzed individually (∗, P < 0.05 [determined by Student's t test]). (D) DR0401.EA0 mice (n = 5) that were challenged with Py-iRBCs (5 × 104 per mouse) developed very high parasitemia and succumbed to infection. Data show parasitemia values for individual mice. (E) Kinetics of parasite clearance in BALB/c mice that were injected with Py-iRBCs (5 × 104) from infected DR0401.EA0 mice (n = 5 recipients) or with the same number of Py-iRBCs from infected BALB/c.RagKO mice (n = 3 recipients). Both groups of recipient BALB/c mice cleared parasitemia similarly (P = 0.4 [determined by Student's t test]).
C57BL/6 mice have a deletion of the IgG2a gene but a functional IgG2c gene, whereas BALB/c mice have a deletion in the IgG2c gene and a functional IgG2a gene (41, 42). As measured by IFA, C57BL/6 and BALB/c mice developed high titers of antibodies to the infected RBCs (Py-iRBCs) (Fig. 1B, left and middle). The isotype profile of Py-iRBC antibodies differed between C57BL/6 and BALB/c mice, since C57BL/6 mice elicited mainly IgG2c antibodies, while BALB/c mice elicited IgG1 and IgG2a antibodies. Considering that IgG2a and IgG2c isotypes represent the signature of Th1-mediated antibody responses, whereas IgG1 represents the signature of Th2-mediated responses, these results indicated that C57BL/6 mice mounted a predominant Th1 response to the malaria parasites, whereas BALB/c mice mounted a mixed Th1/Th2 response. In contrast, infected DR0401.EA0 mice elicited very low titers of specific antibodies (Fig. 1B, right), which may explain the lethality of Py17XNL infection in these mice. To further determine the protective role of antibodies in Py17XNL malaria, we carried out a passive transfer experiment. As illustrated in Fig. 1C, transfer of immune sera (0.5 ml) from self-cured C57BL/6 mice into infected DR0401.EA0 mice significantly reduced the level of parasitemia compared to that of DR0401.EA0 mice receiving sera from malaria-naive C57BL/6 mice, which indicated that antibodies are important for clearance of Py17XNL parasites. The DR0401.EA0 mice also succumbed to infection when challenged with Py-iRBCs instead of sporozoites (Fig. 1D), which ruled out a possible tolerogenic effect on the blood stage parasites induced during liver stage infection.
Since the virulence of malaria parasites is controlled by genetic and epigenetic factors that can be modulated within the host (43), we also investigated whether the Py17XNL parasites in DR0401.EA0 mice could have undergone increased virulence from a nonlethal to a lethal stage. For this, recipient BALB/c mice were injected with 5 × 104 Py-iRBCs isolated from infected DR0401.EA0 mice, and the recipients were monitored for parasitemia. As a control, BALB/c mice were injected with the same number of Py-iRBCs isolated from infected BALB/c.RagKO mice. As illustrated in Fig. 1E, both groups of BALB/c recipients cleared parasitemia within the same period of time. Finally, we also investigated whether Py17XNL parasites, which infect immature red blood cells (reticulocytes) (44), could have evolved to infect mature red blood cells (erythrocytes) in DR0401.EA0 mice. As illustrated in Fig. S1 in the supplemental material, the Py17XNL parasites in DR0401.EA0 mice infected reticulocytes. In aggregate, our results indicated that lethality of Py17XNL infection in DR0401.EA0 mice is not due to an increased virulence of Py17XNL parasites or a change in tropism from reticulocytes to erythrocytes but rather to the inability of DR0401.EA0 mice to elicit specific antibodies needed to clear the infection.
Chloroquine treatment allows Py17XNL-infected DR0401.EA0 mice to elicit antibodies.
Chloroquine is a potent schizonticide commonly used to treat malaria infection (reviewed in reference 45). Chloroquine treatment also facilitates immune responses to blood stage parasites, since low-dose injection of iRBCs under chloroquine cover induces protective immunity in mice (46). We thus investigated whether chloroquine treatment of infected DR0401.EA0 mice enables antibody responses to blood stage parasites. For this, DR0401.EA0 mice were challenged with Py17XNL sporozoites and treated with three doses of chloroquine (25 mg/kg of body weight) when parasitemia reached >40%. As illustrated in Fig. 2A, treatment with chloroquine led to a drastic decrease in parasitemia, and mice were cured by day 7 posttreatment. Interestingly, by day 20 after treatment with chloroquine, all mice developed high titers of Py-iRBCs antibodies (Fig. 2B) that were comparable to those elicited by C57BL/6 and BALB/c mice upon first-time infection (Fig. 1C).
FIG 2.

Chloroquine treatment allows Py17XNL-infected DR0401.EA0 mice to elicit antibodies. (A to C) DR0401.EA0 mice (n = 5) were challenged with 200 Py17XNL sporozoites and treated with chloroquine (CQ) (three doses of 25 mg/kg body weight) at days 23, 24, and 25 postchallenge (arrows). (A and B) Kinetics of parasitemia in individual mice (A) and mean titers ± standard deviations of Py-iRBC antibodies before and after treatment with chloroquine (B). (C) Mice infected and treated with chloroquine (n = 4) were protected against a second challenge with 200 Py17XNL sporozoites (Pyspz), whereas noninfected mice treated with chloroquine (n = 4) or infectivity controls (n = 4) were unprotected. (D) BALB/c (n = 3) and DR0401.EA0 (n = 14) mice were immunized three times at 2-week intervals with Py17XNL Irrd-spz (30,000/dose) and challenged 2 weeks later with 200 infectious Py17XNL sporozoites. Shown is the percentage of protected (nonparasitemic) mice (P = 1.0 [determined by a Fisher exact test]).
Since chloroquine treatment of infected DR0401.EA0 mice resulted in high titers of specific antibodies, we next investigated whether these mice could be protected against reinfection upon a second challenge with Py17XNL sporozoites. As illustrated in Fig. 2C, none of these mice developed parasitemia. In contrast, control (noninfected) DR0401.EA0 mice treated with chloroquine and challenged with Py17XNL sporozoites became parasitemic, which ruled out that residual levels of chloroquine in blood prevented parasite development. These results demonstrated that chloroquine treatment during infection allowed DR0401.EA0 mice to elicit antibodies to Py17XNL blood stage parasites and protected mice against reinfection.
Immunization with irradiation-attenuated sporozoites (Irrd-spz) protects animals and humans against liver stage malaria (reviewed in reference 47). We next investigated whether DR0401.EA0 mice can mount protective immune responses upon vaccination with Py17XNL Irrd-spz. For this, groups of DR0401.EA0 and BALB/c mice were injected three times with 30,000 Py17XNL Irrd-spz at 2-week intervals and challenged 2 weeks later with 200 infectious Py17XNL sporozoites. As shown in Fig. 2D, immunized-DR0401.EA0 mice were protected to the same extent as immunized-BALB/c mice. Together, these results demonstrated that DR0401.EA0 mice are immunocompetent and can elicit protective immune responses to liver or blood stage parasites upon vaccination, although they are unable to control acute (first-time) Py17XNL malaria infection.
Mice coexpressing HLA-DR0401 and mouse MHC-II molecules self-cure Py17XNL malaria infection.
We next investigated in mice coexpressing HLA-DR0401 and mouse MHC class II molecules whether the HLA-DR0401 molecule plays a dominant negative role in Py17XNL malaria. DR0401.EA0 mice were crossed with C57BL/6 mice (IE0/IAb) or BALB/c mice (IEd/IAd) to generate F1 hybrids expressing DR0401/IE0/IAb (C57BL/6) or DR0401/IEd/IAd (C57BL/6.BALB/c), respectively. Expression of HLA-DR0401 and mouse MHC-II molecules in F1 hybrids is shown in Fig. 3A (left and middle). As a control, we generated F1 hybrids (C57BL/6.BALB/c) expressing IEd/IAd and lacking expression of HLA-DR0401 (Fig. 3A, right) by crossing EA0 (C57BL/6) mice with BALB/c mice. As illustrated in Fig. 3B and C, all F1 hybrid mice challenged with Py17XNL sporozoites elicited high titers of specific antibodies to Py-iRBCs and self-cured the infection by day 21 postchallenge. Of note, F1 hybrid mice lacking expression of HLA-DR0401 molecules (Fig. 3B and C, right) elicited antibodies and cleared the infection as efficiently as mice expressing HLA-DR0401 molecules in the same genetic background (Fig. 3B and C, middle). This indicated that HLA-DR0401 molecules do not play a dominant negative role in malaria when coexpressed with mouse MHC-II molecules.
FIG 3.

Mice coexpressing HLA-DR0401 and mouse MHC-II molecules clear Py17XNL malaria infection. (A) Expression of HLA-DR0401 and mouse MHC-II (IA/IE) molecules in spleens of F1 hybrid mice, as measured by FACS analysis. Data represent means ± standard deviations for three mice analyzed individually. (B and C) Groups of F1 hybrid mice (n = 7) were challenged with 200 infectious Py17XNL sporozoites and monitored for development of specific antibodies (B) and parasitemia (C). Data show that all F1 hybrid mice elicited antibodies and were able to clear infection by day 21 postchallenge.
Py17XNL fosters CD4+ Foxp3+ T regulatory cell expansion and prevents clearance of infection in DR04010.EA0 mice.
CD4+ Foxp3+ T regulatory cells (Tregs) maintain a state of self-tolerance by suppressing immune responses to self-antigens (7). Some pathogens evade the immune system by stimulation of preexistent Tregs (9). To determine whether Tregs could prevent DR0401.EA0 mice from self-curing Py17XNL malaria infection, we carried out a cross-sectional study to compare the splenic frequency of Tregs in naive (uninfected) mice and in mice infected with Py17XNL. As shown in Fig. 4 (top), at day 10 after challenge with Py17XNL sporozoites, the frequency of Tregs in infected C57BL/6, BALB/c, and F1 hybrid mice (all able to elicit antibodies and self-cure infection) significantly decreased compared to that in naive (noninfected) mice. C57BL/6 mice examined at day 16 postchallenge also showed a reduction in Treg frequency similar to that of mice examined at day 10. Despite the decrease in Treg frequency, the total numbers of Tregs per spleen in infected C57BL/6, BALB/c, and F1 hybrid mice (able to self-cure infection) were similar to those in naive mice (Fig. 4, bottom), which relates to the fact that Py17XNL infection induced splenomegaly and hypercellularity (see Table S1 in the supplemental material). In contrast, the frequency of Tregs in infected DR0401.EA0 mice at day 10 or 16 postchallenge was not reduced, and the total numbers of Tregs per spleen were significantly increased compared to those in naive DR0401.EA0 mice or infected C57BL/6, BALB/c, and F1 hybrid mice (Fig. 4, bottom). Thus, these results indicated that Py17XNL infection fostered Treg expansion in DR0401.EA0 mice but not in mice able to self-cure the infection.
FIG 4.
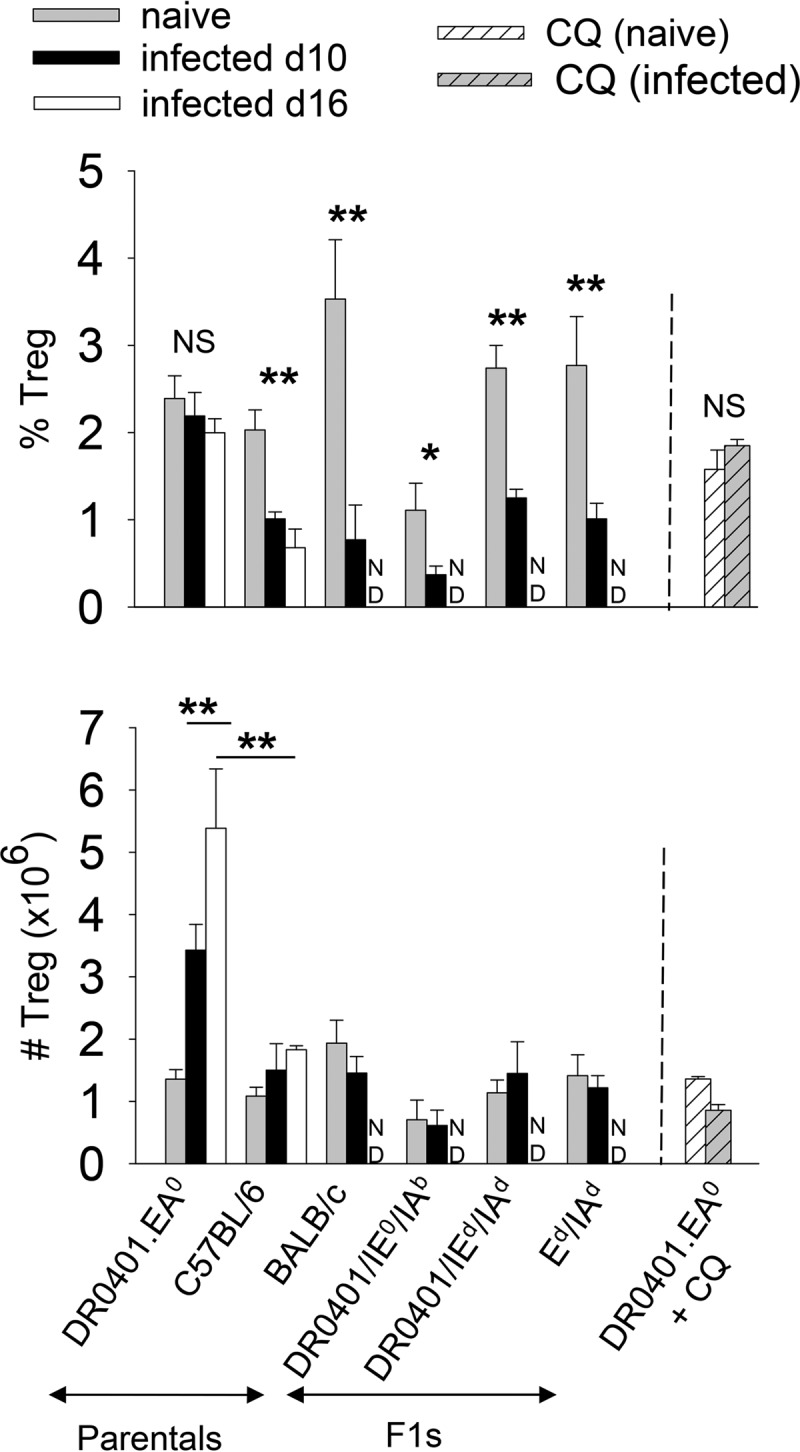
Py17XNL fosters expansion of Foxp3+ regulatory T cells (Tregs) in DR0401.EA0 mice. Groups of naive (noninfected) mice, infected mice at day 10 or 16 after challenge with 200 infectious Py17XNL sporozoites, infected mice treated with chloroquine (CQ) (as in Fig. 2A) at day 20 after chloroquine treatment, or naive mice treated with chloroquine at day 12 posttreatment were examined for the frequency of splenic Tregs (CD3+ CD4+ Foxp3+) by FACS analysis. The top panel shows Treg frequency, and the bottom panel shows total Treg numbers per spleen. Data represent means ± standard deviations for three mice analyzed individually (∗ P < 0.05; ∗∗, P < 0.005; NS, not significant [determined by Student's t test]). ND, not done.
To determine whether Tregs could prevent DR0401.EA0 mice from eliciting antibodies and self-curing the infection, DR0401.EA0 mice were depleted of Tregs by treatment with rat anti-mouse CD25 antibodies (PC61). CD4+ Foxp3+ Tregs express high levels of CD25 (IL-2Rα), as they are dependent on IL-2 for activation (7). Daily injection of CD25 Abs (250 μg) for three consecutive days, but not injection of isotype control Abs, resulted in a significant (4-fold) depletion of splenic Foxp3+ T cells at day 7 postinjection (Fig. 5A). Additional groups of DR0401.EA0 mice (n = 4) were injected with CD25 Abs or control rat isotype Abs as described above, and 3 days later, mice were challenged with Py17XNL sporozoites. As illustrated in Fig. 5B and C, the anti-CD25-treated mice self-cured the infection and elicited antibodies, while the isotype control-treated mice were unable to elicit antibodies and succumbed to the infection. Furthermore, 2 months after the first challenge, the anti-CD25-treated mice were completely protected upon a second challenge with Py17XNL sporozoites, while all infectivity control mice became parasitemic (Fig. 5D). These results demonstrated that Tregs from infected DR0401.EA0 mice suppressed antibody responses to Py17XNL malaria parasites and prevented clearance of first-time (acute) infection.
FIG 5.

Py17XNL-stimulated Foxp3+ regulatory T cells (Tregs) prevent clearance of infection in DR0401.EA0 mice. (A) Groups of DR0401.EA0 mice (n = 3) were injected intraperitoneally three times at 1-day intervals with 250 μg of rat anti-mouse CD25 or the rat isotype control and analyzed 7 days after the last injection for the frequency of CD25+ Foxp3+ Tregs in spleens. Data represent mean values for pooled spleens. (B and C) Parasitemia and kinetics of specific antibodies in groups of DR0401.EA0 mice (n = 4) that were injected with CD25 antibodies or the rat isotype control as in panel B and then challenged with 200 Py17XNL sporozoites 3 days after the last injection. The CD25-treated mice but not the rat isotype-treated mice elicited antibodies and self-cured the infection. Parasitemia values are from individual mice, and antibody titers represent means ± standard deviations for mice analyzed individually. (D) Two months after the first challenge, CD25-treated mice (n = 4) from panels C and D were completely protected against a second challenge with 200 infectious Py17XNL sporozoites. In contrast, all the infectivity control mice (n = 4) became parasitemic.
Py17XNL does not attenuate CD4 T-cell function but stimulates Tregs to directly suppress B cell function.
Tregs can downregulate antibody responses indirectly by suppressing CD4 T helper cells or by direct suppression on B cells (9, 48, 49). Thus, we examined CD4 T cells in infected DR0401.EA0 mice. At day 10 after challenge with Py17XNL sporozoites, the CD4 T cell numbers in spleens of infected DR0401.EA0 mice were significantly higher than those in spleens of noninfected DR0401.EA0 mice and also higher than those in spleens of infected C57BL/6 mice, BALB/c mice, and F1 hybrid mice (Fig. 6A), which indicated that CD4 T cells in DR0401.EA0 mice rapidly expanded during Py17XNL infection. Ex vivo culture of splenic T cells from infected DR0401.EA0 or C57BL/6 mice resulted in secretion of gamma interferon (IFN-γ), whereas IFN-γ was not detectable in cultures of naive (uninfected) mice (Fig. 6B). Depletion of CD4 T cells from spleens of infected DR0401.EA0 mice abrogated the IFN-γ response, which indicated that this cytokine was secreted by activated CD4 T cells during infection. No detectable levels of IL-4 or IL-10 were detected in the ex vivo cultures from infected DR0401.EA0 or C57BL/6 mice (data not shown). Upon polyclonal stimulation with CD3/CD28 Abs (Fig. 6C) or ConA (Fig. 6D), splenic T cells from infected DR0401.EA0 mice were proficient at secreting IFN-γ, IL-4, and IL-10 compared to splenic T cells from naive DR0401.EA0 mice or to splenic T cells from infected C57BL/6 and F1 hybrid mice. In aggregate, these results indicated that the effector CD4 T cell function in DR0401.EA0 mice was not suppressed during acute Py17XNL infection.
FIG 6.

CD4 T cells from infected DR0401.EA0 mice are not suppressed during Py17XNL malaria infection. (A) Total numbers of CD4 T cells in spleens of naive and infected mice. (B) Ex vivo IFN-γ response (pg/ml) of splenic T cells from infected DR0401.EA0 and C57BL/6 mice at day 10 after challenge with Py17XNL sporozoites or from naive (noninfected) mice. No IL-4 or IL-10 secretion was detected in cell culture supernatants (data not shown). Depletion of CD4 T cells from spleens of infected DR0401.EA0 mice abrogated the IFN-γ response. No CD4 T cell depletion was carried out in spleens of naive (uninfected) DR0401.EA0 mice (ND, not done). (C) Cytokine response (pg/ml) of splenic T cells from infected (day 10 after challenge with sporozoites) and naive DR0401.EA0 mice stimulated with CD3/CD28 Abs. (D) Cytokine response (pg/ml) upon stimulation with ConA. Data represent means ± standard deviations for three mice analyzed individually (∗, P < 0.05; ∗∗, P < 0.005; ∗∗∗, P < 0.0005; NS, not significant [determined by Student's t test]). ND, not done.
We next investigated whether Tregs from Py17XNL-infected DR0401.EA0 mice could directly inhibit B cells to secrete antibodies. For this, purified B cells from naive DR0401.EA0 mice were stimulated with CD40 Abs, IL-2, and IL-4 in the absence or presence of graded numbers of purified Tregs from infected DR0401.EA0 mice, and the level of IgG secreted into cell culture supernatants was measured by ELISA. As illustrated in Fig. 7A, the IgG response was proportionally inhibited by increasing numbers of Tregs. To determine whether B cell suppression was mediated by direct cell-cell contact with Tregs, cells were cocultured in a transwell setting. The secretion of IgG was not inhibited in the transwell cultures (Fig. 7A), indicating that B cell suppression was mediated by direct cell contact with Tregs. Interestingly, the Tregs from naive (noninfected) DR0401.EA0 mice failed to inhibit B cells to secrete IgG (Fig. 7B, right), which indicated that Py17XNL parasites stimulate Tregs in DR0401.EA0 mice to suppress B cell function.
FIG 7.

Py17XNL stimulates Tregs to suppress B cell function by direct cell contact with B cells. Splenic B cells (3 × 105) from naive DR0401.EA0 mice were stimulated with CD40 Abs plus IL-2 and IL-4 or left unstimulated (see Materials and Methods) and cultured in the absence or presence of graded numbers of Tregs isolated from DR0401.EA0 mice at day 10 after challenge with Py17XNL sporozoites (A) or Tregs isolated from naive DR0401.EA0 mice (B, right). To prevent direct cell-cell interactions, cultures were also carried out in 0.4-μm transwell plates (A). Likewise, splenic B cells (3 × 105) from naive C57BL/6 mice were stimulated, as described above, in the presence of Tregs isolated from infected C57BL/6 mice (B, left). IgG secretion in cell culture supernatants was measured by ELISA at day 16 of culture in triplicate wells. Data were analyzed by 2-way ANOVA followed by Tukey's test (∗∗, P < 0.005; NS, not significant).
To further determine whether Py17XNL infection also stimulates B cell-suppressogenic Tregs in control mouse strains able to self-cure infection, we carried out a similar experiment using Tregs from infected C57BL/6 mice. As shown in Fig. 7B (left), the Tregs from infected C57BL/6 mice also suppressed B cells to secrete IgG. The fact that the B cell-suppressogenic Tregs expanded in infected DR0401.EA0 mice but not in infected C57BL/6 mice (Fig. 4, bottom) accounts for the inability of DR0401.EA0 mice to elicit specific antibodies and resolve Py17XNL infection.
Humanized mice expressing HLA-II molecules other than HLA-DR0401 elicit specific antibodies and clear Py17XNL malaria infection.
Considering our findings that humanized DR0401.EA0 mice cannot self-cure first-time (acute) Py17XNL infection, it was important to investigate the ability of humanized mice expressing other HLA class II molecules to elicit antibody responses and to clear Py17XNL infection. For this, groups of DR0301.EA0 (C57BL/10), DQ6.EA0 (C57BL/10), DQ8.EA0 (C57BL/10), and DR0402.EA0 (C57BL/10) mice were challenged with 200 sporozoites and monitored for parasitemia and development of specific antibodies. As illustrated in Fig. 8, DR0301.EA0, DQ6.EA0, DQ8.EA0, and DR0402.EA0 mice were able to elicit specific antibodies and to clear the infection. The DR0402.EA0 and DQ8.EA0 mice were more efficient in clearing parasitemia than the DQ6.EA0 mice, since they self-cured by day 28 after challenge, whereas 5 out of 8 DQ6.EA0 mice were still parasitemic by day 28 and needed an additional 12 days to self-cure. On the other hand, the DR0301.EA0 mice showed a more heterogeneous response to the infection, since 8 out of 11 mice elicited antibodies and cleared the infection by day 21, whereas the remaining 3 mice were unable to elicit antibodies and succumbed to the infection. Thus, unlike the HLA-DR0401 molecules, HLA-DR0402, HLA-DQ6, HLA-DQ8, and, to some extent, HLA-DR0301 molecules are proficient at supporting antibody responses to Py17XNL malaria parasites and self-curing acute (first-time) infection.
FIG 8.

Humanized mice expressing HLA-II molecules other than HLA-DR0401 elicit specific antibodies and self-cure Py17XNL infection. Groups of DR0301.EA0 (n = 11), DQ6.EA0 (n = 8), DQ8.EA0 (n = 11), and DR0402.EA0 (n = 5) mice were challenged with 200 Py17XNL sporozoites and monitored for parasitemia and development of specific antibodies to Py-iRBCs. (A) Parasitemia values for individual mice. (B) Mean antibody titers ± standard deviations for mice analyzed individually (∗, P < 0.05; ∗∗, P < 0.005 [determined by Student's t test]).
DISCUSSION
This study shows that expression of HLA-DR0401 molecules in mice results in lethal malaria infection by Py17XNL, a parasite that otherwise induces a self-curing disease in inbred mouse strains. DR0401.EA0 mice developed hyperparasitemia and severe anemia and succumbed to infection, thus resembling the lethal infection induced by Py17XL in inbred mouse strains. The lethality of Py17XNL infection in DR0401.EA0 mice was not due to an increased virulence of Py17XNL malaria parasites but rather to the inability of DR0401.EA0 mice to support antibody responses needed to clear the blood stage infection. These mice were, however, able to mount antibody responses and to clear the infection by in vivo depletion of Tregs, treatment with chloroquine, or coexpression with mouse MHC-II molecules.
CD4 T cells are critical for resolving Py17XNL malaria infection, as demonstrated in mice depleted of CD4 T cells and in nude (T cell-deficient) mice that cannot resolve the infection (50, 51). Our results are consistent with those observations, since the ability of mice to self-cure or succumb to Py17XNL malaria was related solely to the expression of MHC-II (HLA-II) molecules, and the primary role of MHC-II (HLA-II) molecules is to present peptides to CD4 T cells. HLA-II molecules are highly polymorphic and differ in their ability to present different sets of peptides to CD4 T cells. For example, within the HLA-DR4 group, there are about 50 different allelic variants that differ in only 3 amino acids in the peptide binding groove. The HLA-DR0401 but not the HLA-DR0402 molecules predispose to rheumatoid arthritis by their differential ability to present arthritogenic peptides to autoreactive CD4 T cells (52, 53). Here we showed that HLA-DR0402, HLA-DQ6, HLA-DQ8, and, to some extent, HLA-DR0301 molecules sufficed for supporting antibody responses and self-curing Py17XNL infection, while HLA-DR0401 molecules failed to do so. Our results thus indicate that HLA class II restriction of malaria epitopes and, hence, CD4 T cells play a critical role in the clearance of Py17XNL parasites.
Since erythrocytes do not express MHC (HLA) molecules, and infected erythrocytes cannot be eliminated by CD4 T cells in a MHC (HLA) class II-restricted manner, the critical role of CD4 T cells in resolving Py17XNL infection is likely mediated by the ability of CD4 T cells to provide help for B cell differentiation and secretion of antibodies. The contribution of antibodies to the clearance of Py17XNL malaria has been demonstrated by the therapeutic and antiparasitic effects of hyperimmune sera (54, 55) as well as in B cell-deficient mice and mice depleted of B cells with anti-IgM antibodies that failed to resolve Py17XNL malaria infection (24, 46). In agreement with those observations, we found that passive transfer of immune sera from infected C57BL/6 mice (able to elicit antibodies and clear infection) significantly reduced the level of parasitemia in DR0401.EA0 mice. Thus, our study demonstrates a critical role of HLA-II molecules in supporting antibody responses and dictating susceptibility or resistance to Py17XNL malaria.
The Tregs from infected DR0401.EA0 and C57BL/6 mice suppressed B cells to secrete antibodies through a cell-cell interaction, but Tregs from naive (uninfected) mice were unable to suppress B cell function. Thus, our study provides the first evidence for a new sidearm immune evasion mechanism by which Py17XNL malaria parasites stimulate Tregs that suppress the protective B cell function. During the second week of infection, C57BL/6 mice and other mouse strains that self-cure the infection, such as BALB/c and F1 hybrid mice, had total numbers of splenic Tregs comparable to those in spleens of uninfected mice, although the Treg frequency was reduced, since the infection induced splenomegaly. Our results for C57BL/6, BALB/c, and F1 hybrid mice are in agreement with results from previous studies by Couper et al. (12) and Berretta et al. (20), using nonlethal Py17XNL and P. chabaudi AS strains, indicating that Tregs expand during the first week of infection and that the Tregs are then deleted by apoptosis in such a way that the total numbers of Tregs return to basal levels. In contrast, the numbers of Tregs in spleens of infected DR04010.EA0 mice expanded to reach a 5-fold increase over the Treg numbers in spleens of uninfected DR0401.EA0 mice. The ability of Py17XNL parasites to stimulate and expand B cell-suppressogenic Tregs accounts for the inability of DR0401.EA0 mice to elicit antibodies and to self-cure the infection. The role of B cell-inhibitory Tregs in preventing clearance of infection in DR0401.EA0 mice was evident, since in vivo Treg depletion enabled the DR0401.EA0 mice to elicit antibodies and to self-cure the infection.
In humans, B cell-suppressogenic Tregs have been found in T-B area borders within the germinal centers of lymphoid tissues and have been associated with development of systemic lupus erythematosus (48, 49), an autoimmune disorder mediated by exacerbated production of autoantibodies. When it comes to human malaria, the role of Tregs is supported by studies in volunteers challenged with P. falciparum, in which the expansion of CD4+ FOXP3+ T cells in blood was associated with enhanced parasite growth (21), as well as by studies in children and adults from areas where malaria is endemic which indicated an association between Treg frequency in blood and hyperparasitemia (22, 23). However, the ability of Tregs to suppress antibody responses to P. falciparum has not been investigated.
Interestingly, the Tregs from Py17XNL-infected DR0401.EA0 mice did not alter the effector CD4 T helper compartment, since effector CD4 T cells were able to expand during infection, and they were proficient at secreting cytokines either ex vivo or upon polyclonal stimulation with ConA or CD3/CD28 Abs. Although there is overwhelming evidence that Tregs are able to directly suppress CD4 T helper function (reviewed in reference 9), to our knowledge, only studies by Lim et al. (48) and Iikuni et al. (49) and this study demonstrate the existence of a new Treg subset that is able to directly suppress B cells. The mechanisms by which Tregs can selectively suppress B cell function remain to be investigated.
Infected DR0401.EA0 mice treated with chloroquine were able to elicit antibodies and were protected against reinfection. Chloroquine is a potent antimalaria drug able to inhibit parasite development in vitro and in vivo (45), although chloroquine can also enhance immune responses to malaria parasites, as indicated by the following: (i) immunization with low-dose blood stage parasites under chloroquine cover protects mice against blood stage malaria (46), (ii) immunodeficient mice need significantly higher doses of chloroquine and longer treatment to clear parasitemia than immunocompetent mice (46), and (iii) parasitemic immunocompetent but not immunodeficient mice treated with chloroquine are protected against subsequent challenges with malaria parasites (46). However, there is debate on whether the ability of chloroquine to enhance immune responses to malaria parasites is due to its immunomodulatory effects or whether it may simply act by reducing the parasite mass, since blood stage parasites induce dendritic cell dysfunction and tolerance (56, 57). Infected DR0401.EA0 mice developed splenomegaly (see Table S1 in the supplemental material), but upon treatment with chloroquine, their spleens returned to normal size and cellularity by day 20 posttreatment and contained similar numbers of Tregs as those in spleens of uninfected mice. To determine whether chloroquine can deplete Tregs, we treated naive (noninfected) DR0401.EA0 mice with chloroquine (n = 3; three doses of 25 mg/kg at 1-day intervals), and mice were examined at day 12 posttreatment. The uninfected chloroquine-treated mice had Treg numbers similar to those in untreated mice (P = 0.9) (Fig. 4A, bottom), indicating that in the absence of infection, chloroquine does not deplete Tregs. However, at present, we cannot rule out whether chloroquine treatment could have had a direct immunomodulatory effect on the infected DR0401.EA0 mice.
F1 hybrid mice coexpressing HLA-DR0401 and mouse MHC class II molecules elicited antibodies and resolved the infection, which indicated that HLA-DR0401 molecules do not play a dominant negative role in Py17XNL malaria. Studies with ethnic populations in Ghana and Gabon showed an association between HLA-DR4 expression and severe P. falciparum malaria (58, 59), although the HLA-DR4 alleles were not subtyped. The overall frequency of HLA-DR4 in sub-Saharan African populations is 10 times lower than that in Europe or North America (59), and it was undetectable in some other areas where malaria is endemic (60). This has been attributed to negative selection of HLA-DR4 by P. falciparum malaria. The results with the Py17XNL model indicating that coexpression of HLA-DR0401 molecules with other MHC-II molecules allowed mice to elicit antibodies and to resolve infection cannot account for an association between HLA-DR0401 and severe P. falciparum malaria, as most humans express at least two different HLA-II molecules. However, whether or not HLA-DR0401 molecules play a recessive or dominant role when it comes to P. falciparum malaria infection remains to be investigated.
In aggregate, our results demonstrate a differential role of MHC (HLA) class II molecules in supporting antibody responses to Py17XNL malaria and revealed a new immune evasion mechanism whereby Py17XNL parasites activate B cell-suppressogenic Tregs. Humanized HLA-II mice may represent a valuable tool to identify immunogenic or suppressogenic HLA class II-restricted CD4 T cell epitopes expressed by Py17XNL and P. falciparum and for testing of malaria vaccine candidates.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by work unit number 6000.RAD1.F under grants from the Military Infectious Diseases Research Program (MIDRP) and the U.S. Agency for International Development (USAID) to S.C.
We thank Cara Olsen for statistical analyses. We thank Urszula Krzych for helpful discussions.
S.C., T.-D.B., and E.F.V. are U.S. Government employees. The work of these individuals was prepared as part of official government duties. Title 17 U.S.C. §105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person's official duties. The views expressed are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, or the U.S. Government.
Footnotes
Published ahead of print 28 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00272-13.
REFERENCES
- 1.Murray CJL, Rosenfeld C, Lim SS, Andrews K, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. 2012. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379:413–431. 10.1016/S0140-6736(12)60034-8 [DOI] [PubMed] [Google Scholar]
- 2.Stanway RR, Mueller N, Zobiak B, Graewe S, Froehlke U, Zessin PJ, Aepfelbacher M, Heussler VT. 2011. Organelle segregation into Plasmodium liver stage merozoites. Cell. Microbiol. 13:1768–1782. 10.1111/j.1462-5822.2011.01657.x [DOI] [PubMed] [Google Scholar]
- 3.Casares S, Richie TL. 2009. Immune evasion by malaria parasites: a challenge for vaccine development. Curr. Opin. Immunol. 21:321–330. 10.1016/j.coi.2009.05.015 [DOI] [PubMed] [Google Scholar]
- 4.Nogaro SI, Hafalla JC, Walther B, Remarque EJ, Tetteh KK, Conway DJ, Riley EM, Walther M. 2011. The breadth, but not the magnitude, of circulating memory B cell responses to P. falciparum increases with age/exposure in an area of low transmission. PLoS One 6:e25582. 10.1371/journal.pone.0025582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothbard JB, Gefter ML. 1991. Interactions between immunogenic peptides and MHC proteins. Annu. Rev. Immunol. 9:527–565. 10.1146/annurev.iy.09.040191.002523 [DOI] [PubMed] [Google Scholar]
- 6.Ma CS, Deenick EK, Batten M, Tangye SG. 2012. The origins, function, and regulation of T follicular helper cells. J. Exp. Med. 209:1241–1253. 10.1084/jem.20120994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Issa F, Robb RJ, Wood KJ. 2013. The where and when of T cell regulation in transplantation. Trends Immunol. 34:107–113. 10.1016/j.it.2012.11.003 [DOI] [PubMed] [Google Scholar]
- 8.Surls J, Nazarov-Stoica C, Kehl M, Casares S, Brumeanu TD. 2010. Differential effect of CD4+Foxp3+ T-regulatory cells on the B and T helper cell responses to influenza virus vaccination. Vaccine 28:7319–7330. 10.1016/j.vaccine.2010.08.074 [DOI] [PubMed] [Google Scholar]
- 9.Maizels RM, Smith KA. 2011. Regulatory T cells in infection. Adv. Immunol. 112:73–136. 10.1016/B978-0-12-387827-4.00003-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haque A, Best SE, Amante FH, Mustafah S, Desbarrieres L, de Labastida F, Sparwasser T, Hill GR, Engwerda CR. 2010. CD4+ natural regulatory T cells prevent experimental cerebral malaria via CTLA-4 when expanded in vivo. PLoS Pathog. 6:e1001221. 10.1371/journal.ppat.1001221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hisaeda H, Tetsutani K, Imai T, Moriya C, Tu L, Hamano S, Duan X, Chou B, Ishida H, Aramaki A, Shen J, Ishii KJ, Coban C, Akira S, Takeda K, Yasutomo K, Torii M, Himeno K. 2008. TLR9 signaling for immune evasion by activating regulatory T cells. J. Immunol. 180:2496–2503 [DOI] [PubMed] [Google Scholar]
- 12.Couper KN, Blount DG, Wilson MS, Hafalla JC, Belkaid Y, Kamanaka M, Flavell RA, de Souza JB, Riley EM. 2008. IL-10 from CD4CD25Foxp3CD127 adaptive regulatory T cells modulates parasite clearance and pathology during malaria infection. PLoS Pathog. 4:e1000004. 10.1371/journal.ppat.1000004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Y, Wang QH, Zheng L, Feng H, Liu J, Ma SH, Cao YM. 2007. Plasmodium yoelii: distinct CD4(+)CD25(+) regulatory T cell responses during the early stages of infection in susceptible and resistant mice. Exp. Parasitol. 115:301–304. 10.1016/j.exppara.2006.09.015 [DOI] [PubMed] [Google Scholar]
- 14.Long TT, Nakazawa S, Onizuka S, Huaman MC, Kanbara H. 2003. Influence of CD4+ CD25+ T cells on Plasmodium berghei NK65 infection in BALB/c mice. Int. J. Parasitol. 33:175–183. 10.1016/S0020-7519(02)00261-8 [DOI] [PubMed] [Google Scholar]
- 15.Randall LM, Amante FH, McSweeney KA, Zhou Y, Stanley AC, Haque A, Jones MK, Hill GR, Boyle GM, Engwerda CR. 2008. Common strategies to prevent and modulate experimental cerebral malaria in mouse strains with different susceptibilities. Infect. Immun. 76:3312–3320. 10.1128/IAI.01475-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vigario AM, Gorgette O, Dujardin HC, Cruz T, Cazenave PA, Six A, Bandeira A, Pied S. 2007. Regulatory CD4+ CD25+ Foxp3+ T cells expand during experimental Plasmodium infection but do not prevent cerebral malaria. Int. J. Parasitol. 37:963–973. 10.1016/j.ijpara.2007.01.004 [DOI] [PubMed] [Google Scholar]
- 17.Cambos M, Bélanger B, Jacques A, Roulet A, Scorza T. 2008. Natural regulatory (CD4+ CD25+ FOXP+) T cells control the production of pro-inflammatory cytokines during Plasmodium chabaudi adami infection and do not contribute to immune evasion. Int. J. Parasitol. 38:229–238. 10.1016/j.ijpara.2007.07.006 [DOI] [PubMed] [Google Scholar]
- 18.Nie CQ, Bernard NJ, Schofield L, Hansen DS. 2007. CD4+ CD25+ regulatory T cells suppress CD4+ T-cell function and inhibit the development of Plasmodium berghei-specific TH1 responses involved in cerebral malaria pathogenesis. Infect. Immun. 75:2275–2282. 10.1128/IAI.01783-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hisaeda H, Maekawa Y, Iwakawa D, Okada H, Himeno K, Kishihara K, Tsukumo S, Yasutomo K. 2004. Escape of malaria parasites from host immunity requires CD4+ CD25+ regulatory T cells. Nat. Med. 10:29–30. 10.1038/nm975 [DOI] [PubMed] [Google Scholar]
- 20.Berretta F, St-Pierre J, Piccirillo CA, Stevenson MM. 2011. IL-2 contributes to maintaining a balance between CD4+ Foxp3+ regulatory T cells and effector CD4+ T cells required for immune control of blood-stage malaria infection. J. Immunol. 186:4862–4871. 10.4049/jimmunol.1003777 [DOI] [PubMed] [Google Scholar]
- 21.Walther M, Tongren JE, Andrews L, Korbel D, King E, Fletcher H, Andersen RF, Bejon P, Thompson F, Dunachie SJ, Edele F, de Souza JB, Sinden RE, Gilbert SC, Riley EM, Hill AV. 2005. Upregulation of TGF-beta, FOXP3, and CD4+CD25+ regulatory T cells correlates with more rapid parasite growth in human malaria infection. Immunity 23:287–296. 10.1016/j.immuni.2005.08.006 [DOI] [PubMed] [Google Scholar]
- 22.Walther M, Jeffries D, Finney OC, Njie M, Ebonyi A, Deininger S, Lawrence E, Ngwa-Amambua A, Jayasooriya S, Cheeseman IH, Gomez-Escobar N, Okebe J, Conway DJ, Riley EM. 2009. Distinct roles for FOXP3 and FOXP3 CD4 T cells in regulating cellular immunity to uncomplicated and severe Plasmodium falciparum malaria. PLoS Pathog. 5:e1000364. 10.1371/journal.ppat.1000364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minigo G, Woodberry T, Piera KA, Salwati E, Tjitra E, Kenangalem E, Price RN, Engwerda CR, Anstey NM, Plebanski M. 2009. Parasite-dependent expansion of TNF receptor II-positive regulatory T cells with enhanced suppressive activity in adults with severe malaria. PLoS Pathog. 5:e1000402. 10.1371/journal.ppat.1000402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinbaum FI, Evans CB, Tigelaar RE. 1976. Immunity to Plasmodium berghei yoelii in mice. I. The course of infection in T cell and B cell deficient mice. J. Immunol. 117:1999–2005 [PubMed] [Google Scholar]
- 25.Spence PJ, Langhorne J. 2012. T cell control of malaria pathogenesis. Curr. Opin. Immunol. 24:444–448. 10.1016/j.coi.2012.05.003 [DOI] [PubMed] [Google Scholar]
- 26.Geluk A, Taneja V, van Meijgaarden KE, Zanelli E, Abou-Zeid C, Thole JE, de Vries RR, David CS, Ottenhoff TH. 1998. Identification of HLA class II-restricted determinants of Mycobacterium tuberculosis-derived proteins by using HLA-transgenic, class II-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 95:10797–10802. 10.1073/pnas.95.18.10797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DaSilva L, Welcher BC, Ulrich RG, Aman MJ, David CS, Bavari S. 2002. Human like immune response of human leukocyte antigen-DR3 transgenic mice to staphylococcal enterotoxins: a novel model for superantigen vaccines. J. Infect. Dis. 185:1754–1760. 10.1086/340828 [DOI] [PubMed] [Google Scholar]
- 28.Taneja V, David CS. 2010. Role of HLA class II genes in susceptibility/resistance to inflammatory arthritis: studies with humanized mice. Immunol. Rev. 233:62–78. 10.1111/j.0105-2896.2009.00858.x [DOI] [PubMed] [Google Scholar]
- 29.National Research Council 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC [Google Scholar]
- 30.Ito K, Bian HJ, Molina M, Han J, Magram J, Saar E, Belunis C, Bolin DR, Arceo R, Campbell R, Falcioni F, Vidović D, Hammer J, Nagy ZA. 1996. HLA-DR4-IE chimeric class II transgenic, murine class II-deficient mice are susceptible to experimental allergic encephalomyelitis. J. Exp. Med. 183:2635–2644. 10.1084/jem.183.6.2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taneja V, Behrens M, Basal E, Sparks J, Griffiths MM, Luthra H, David CS. 2008. Delineating the role of the HLA-DR4 “shared epitope” in susceptibility versus resistance to develop arthritis. J. Immunol. 181:2869–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das P, Drescher KM, Geluk A, Bradley DS, Rodriguez M, David CS. 2000. Complementation between specific HLA-DR and HLA-DQ genes in transgenic mice determines susceptibility to experimental autoimmune encephalomyelitis. Hum. Immunol. 61:279–289. 10.1016/S0198-8859(99)00135-4 [DOI] [PubMed] [Google Scholar]
- 33.Bradley DS, Nabozny GH, Cheng S, Zhou P, Griffiths MM, Luthra HS, David CS. 1997. HLA-DQB1 polymorphism determines incidence, onset, and severity of collagen-induced arthritis in transgenic mice. Implications in human rheumatoid arthritis. J. Clin. Invest. 100:2227–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strauss G, Vignali DA, Schönrich G, Hämmerling GJ. 1994. Negative and positive selection by HLA-DR3 (DRw17) molecules in transgenic mice. Immunogenetics 40:104–108 [PubMed] [Google Scholar]
- 35.Mangalam AK, Rajapopalan G, Taneja V, David CS. 2008. HLA class II transgenic mice mimic human inflammatory diseases. Adv. Immunol. 97:65–147. 10.1016/S0065-2776(08)00002-3 [DOI] [PubMed] [Google Scholar]
- 36.Danner R, Chaudhari S, Rosenberger J, Surls J, Richie TL, Brumeanu TD, Casares S. 2011. Expression of HLA class II molecules in humanized NOD.Rag1KO.IL2RgcKO mice is critical for development and function of human T and B cells. PLoS One 6:e19826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Limbach K, Aguiar J, Gowda K, Patterson N, Abot E, Sedegah M, Sacci J, Richie T. 2011. Identification of two new protective pre-erythrocytic malaria vaccine antigen candidates. Malar. J. 10:65. 10.1186/1475-2875-10-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Casares S, Hurtado A, McEvoy RC, Sarukhan A, von Boehmer H, Brumeanu TD. 2002. Down-regulation of diabetogenic CD4+ T cells by a soluble dimeric peptide-MHC class II chimera. Nat. Immunol. 3:383–391. 10.1038/ni770 [DOI] [PubMed] [Google Scholar]
- 39.van der Heyde HC, Huszar D, Woodhouse C, Manning DD, Weidanz WP. 1994. The resolution of acute malaria in a definitive model of B cell deficiency, the JHD mouse. J. Immunol. 152:4557–4562 [PubMed] [Google Scholar]
- 40.Soulard V, Roland J, Sellier C, Gruner AC, Leite-de-Moraes M, Franetich JF, Rénia L, Cazenave PA, Pied S. 2007. Primary infection of C57BL/6 mice with Plasmodium yoelii induces a heterogeneous response of NKT cells. Infect. Immun. 75:2511–2522. 10.1128/IAI.01818-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morgado MG, Cam P, Gris-Liebe C, Cazenave PA, Jouvin-Marche E. 1989. Further evidence that BALBrc and C57BLr6 g 2a genes originate from distinct isotypes. EMBO J. 8:3245–8251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jouvin-Marche E, Morgado MG, Leguern C, Voegtle D, Bonhomme F, Cazenave PA. 1989. The mouse Igh-1a and Igh-1b H chain constant regions are derived from two distinct isotypic genes. Immunogenetics 29:92–97. 10.1007/BF00395856 [DOI] [PubMed] [Google Scholar]
- 43.Merrick CJ, Duraisingh MT. 2010. Epigenetics in Plasmodium: what do we really know? Eukaryot. Cell 9:1150–1158. 10.1128/EC.00093-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shear HL, Grinberg L, Gilman J, Fabry ME, Stamatoyannopoulos G, Goldberg DE, Nagel RL. 1998. Transgenic mice expressing human fetal globin are protected from malaria by a novel mechanism. Blood 92:2520–2526 [PubMed] [Google Scholar]
- 45.Steinhardt LC, Magill AJ, Arguin PM. 2011. Malaria chemoprophylaxis for travelers to Latin America. Am. J. Trop. Med. Hyg. 85:1015–1024. 10.4269/ajtmh.2011.11-0464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belnoue E, Voza T, Costa FT, Grüner AC, Mauduit M, Rosa DS, Depinay N, Kayibanda M, Vigário AM, Mazier D, Snounou G, Sinnis P, Rénia L. 2008. Vaccination with live Plasmodium yoelii blood stage parasites under chloroquine cover induces cross-stage immunity against malaria liver stage. J. Immunol. 181:8552–8558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Epstein JE, Giersing B, Mullen G, Moorthy V, Richie TL. 2007. Malaria vaccines: are we getting closer? Curr. Opin. Mol. Ther. 9:12–24 [PubMed] [Google Scholar]
- 48.Lim HW, Hillsamer P, Banham AH, Kim CH. 2005. Direct suppression of B cells by CD4+ CD25+ regulatory T cells. J. Immunol. 175:4180–4183 http://www.jimmunol.org/content/175/7/4180.long [DOI] [PubMed] [Google Scholar]
- 49.Iikuni N, Lourenço EV, Hahn BH, La Cava A. 2009. Regulatory T cells directly suppress B cells in systemic lupus erythematosus. J. Immunol. 183:1518–1522. 10.4049/jimmunol.0901163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar S, Miller LH. 1990. Cellular mechanisms in immunity to blood stage infection. Immunol. Lett. 25:109–114. 10.1016/0165-2478(90)90100-5 [DOI] [PubMed] [Google Scholar]
- 51.Clark IA, Allison AC. 1974. Babesia microti and Plasmodium berghei yoelii infections in nude mice. Nature 252:328–329. 10.1038/252328a0 [DOI] [PubMed] [Google Scholar]
- 52.Gourraud PA, Boyer JF, Barnetche T, Abbal M, Cambon-Thomsen A, Cantagrel A, Constantin A. 2006. A new classification of HLA-DRB1 alleles differentiates predisposing and protective alleles for rheumatoid arthritis structural severity. Arthritis Rheum. 54:593–599. 10.1002/art.21630 [DOI] [PubMed] [Google Scholar]
- 53.Weyand CM, Hicok KC, Conn DL, Goronzy JJ. 1992. The influence of HLA-DRB1 genes on disease severity in rheumatoid arthritis. Ann. Intern. Med. 117:801–806. 10.7326/0003-4819-117-10-801 [DOI] [PubMed] [Google Scholar]
- 54.Diggs CL, Osler AG. 1969. Humoral immunity in rodent malaria. II. Inhibition of parasitemia by serum antibody. J. Immunol. 102:298–305 [PubMed] [Google Scholar]
- 55.Golenser JL, Spira DT, Zuckerman A. 1975. Neutralizing antibody in rodent malaria. Trans. R. Soc. Trop. Med. Hyg. 69:251–258. 10.1016/0035-9203(75)90164-9 [DOI] [PubMed] [Google Scholar]
- 56.Wykes MN, Liu XQ, Beattie L, Stanisic DI, Stacey KJ, Smyth MJ, Thomas R, Good MF. 2007. Plasmodium strain determines dendritic cell function essential for survival from malaria. PLoS Pathog. 3:e96. 10.1371/journal.ppat.0030096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wykes MN, Liu XQ, Jiang S, Hirunpetcharat C, Good MF. 2007. Systemic tumor necrosis factor generated during lethal Plasmodium infections impairs dendritic cell function. Immunol. 179:3982–3987 [DOI] [PubMed] [Google Scholar]
- 58.May J, Meyer CG, Kun JF, Lell B, Luckner D, Dippmann AK, Bienzle U, Kremsner PG. 1999. HLA class II factors associated with Plasmodium falciparum merozoite surface antigen allele families. J. Infect. Dis. 179:1042–1045. 10.1086/314661 [DOI] [PubMed] [Google Scholar]
- 59.Osafo-Addo AD, Koram KA, Oduro AR, Wilson M, Hodgson A, Rogers WO. 2008. HLA-DRB1*04 allele is associated with severe malaria in northern Ghana. Am. J. Trop. Med. Hyg. 78:251–255 [PubMed] [Google Scholar]
- 60.Hananantachai H, Patarapotikul J, Ohashi J, Naka I, Looareesuwan S, Tokunaga K. 2005. Polymorphisms of the HLA-B and HLA-DRB1 genes in Thai malaria patients. Jpn. J. Infect. Dis. 58:25–28 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.