

Abstract
Antiserum to the Borrelia burgdorferi arthritis-related protein, Arp, has been shown to prevent or reduce arthritis in immunodeficient mice. To directly investigate the requirement for this lipoprotein in the generation of Lyme arthritis, we utilized targeted deletion to generate a B. burgdorferi clone that lacked only the arp gene locus. Infection of Lyme disease-susceptible C3H/HeN mice with the arp deletion mutant demonstrated significantly reduced tibiotarsal joint swelling during the first 6 weeks of infection compared to a wild-type control. The severity of joint swelling was restored to wild-type levels in mice infected with an arp mutant clone complemented in cis. Interestingly, the reduced swelling of joint tissues exhibited by mice infected with the arp deletion mutant did not directly correspond to reduced underlying arthritis. Histopathology data at 2 weeks postinfection showed some reduction in arthritis severity caused by the arp mutant clone; however, by 8 weeks, no significant difference was observed between joint tissues infected by the wild-type or arp mutant clones. The spirochete load in the joint tissues of mice infected with the arp mutant was found to be greater than that exhibited by the wild-type control. Our findings demonstrate that this lipoprotein contributes to the generation of early-onset joint swelling and suggests that arp expression has a negative secondary effect on total spirochete numbers in joint tissues.
INTRODUCTION
Lyme disease is caused by infection with the tick-transmitted spirochete Borrelia burgdorferi. It is currently the most common vector-borne disease in the Northern Hemisphere, occurring in parts of North America, Europe, and Asia (1, 2). The disease affects a wide range of mammals, including humans, horses, and dogs (3–7), and ticks of the genus Ixodes are the primary vectors of the disease (8). A blood meal by an infected tick is followed by a strong immune response, and infection results in a multisystem disease characterized by damage to the central nervous system and various organs, including the heart, eyes, and joints. Despite a robust humoral and cellular response, chronic and persistent infection can often result. Among the afflicted tissue sites, the joints are a major site of inflammation (9), and subacute arthritis occurs in 60% of untreated individuals (10, 11). In humans, this subacute arthritis can often develop into a chronic form characterized by bacterial persistence that is often unresponsive to antibiotics. Important in disease pathology is the large number of plasmid-encoded surface lipoproteins that have the potential to trigger host immune responses (12–14). Previous studies have demonstrated that lipoproteins or their derivatives activate endothelial cells, neutrophils, macrophages, and B lymphocytes in vitro and can introduce localized inflammatory infiltrate into joints and dermal sites in vivo (15–20).
A number of genes coding for B. burgdorferi lipoproteins have been shown to be preferentially upregulated at various times in different tissue sites during infection of the mammalian host (21–24). One such gene, coding for the arthritis-related protein (Arp), resides on linear plasmid 28-1 (lp28-1) (25) and has been shown to be upregulated in the joints of infected immunocompetent mice (21). Previous studies demonstrated that B. burgdorferi-infected severe combined immunodeficient (SCID) mice treated with Arp antiserum exhibit reduced arthritis severity without affecting the status of infection, suggesting Arp as a target for immune-mediated resolution of Lyme arthritis (25). In a recent study, published during the preparation of the manuscript, it was shown that deletion of arp by allelic exchange resulted in reduced arthritis severity and spirochete load in immunocompetent C3H mice (26). In the present study, we generated an arp mutant through telomere-targeted deletion and infected immunocompetent C3H/HeN mice to determine if the absence of Arp had an effect on both joint swelling and immune cell infiltration into the joint tissues of mice. The results show that deletion of arp led to a significant reduction in measurable tibiotarsal (ankle) joint swelling during the early onset of infection by the mutant clone. Interestingly, this reduction in swelling did not correspond to a decrease in overall immune cell infiltration and subsequent joint pathology. Additionally, the spirochete load in infected joint tissues was shown to be higher in mice infected with the mutant clone than in mice infected with the wild type (WT).
MATERIALS AND METHODS
B. burgdorferi strains and culture conditions.
Borrelia burgdorferi B31-5A4 (wild type) was a kind gift from Steve Norris. The clones described in the study were generated from the above-mentioned B31 strain, whose infectivity and plasmid profile had already been determined (Table 1) (27). All B. burgdorferi clones were cultivated in liquid Barbour-Stoenner-Kelly II (BSK-II) medium supplemented with 6% rabbit serum (Cedarlane Laboratories, Burlington, NC) and incubated at 35°C in 2.5% CO2. The mutant strains were grown with kanamycin (200 μg/ml) or gentamicin (100 μg/ml), as indicated. Cell densities and growth phases were monitored by dark-field microscopy and enumerated using a Petroff-Hausser counting chamber.
TABLE 1.
Strains used in this study
| B. burgdorferi B31 clone | arp presencea | Reference or source |
|---|---|---|
| 5A4 WT | + | 27 |
| 5A4Δarp (ΔArp) | − | This study |
| 5A4Δarp::arp (cArp) | + | This study |
+, present; −, absent.
Generation of arp deletion and complement mutant clones.
For the targeted deletion of arp, a 1,059-bp region upstream of the arp gene locus (coordinates 1471 to 2484 of the annotated lp28-1 sequence; NCBI reference sequence NC_001851.2 [http://www.ncbi.nlm.nih.gov/]) was PCR amplified using primers P270 and P271 (Table 2). The resulting DNA product was then cloned into the pGCL47-4 plasmid, which carries a flgBp-driven kanamycin gene and a 70-bp replicated telomere (rtel) from the left end of linear plasmid 17 (lp17) (28) in order to generate pPH12. The resulting plasmid was then transformed into recombination-deficient Escherichia coli Ec19 competent cells (recA recB21 recC22 sbcB15 hsdR F− proA2 his4 thi-1 argE3 lacY1 galK2 ara-14 xyl-5 mtl-1 str-31 tsx-33), which are derivatives of E. coli DB1256 (29). Plasmid DNA isolated from individual E. coli clones was verified for correct size and orientation by restriction digestion, and a functional rtel was assessed using a ResT assay, as previously described (30), before transformation into B. burgdorferi cells.
TABLE 2.
Oligonucleotides used in this study
| Primer | Sequence (5′–3′) | Description |
|---|---|---|
| P54 | CATATGAGCCATATTCAACGGGAAACG | Forward primer for kan screening and probe generation |
| P55 | AAAGCCGTTTCTGTAATGAAGGAG | Reverse primer for kan screening and probe generation |
| P91 | CGCAGCAGCAACGATGTTAC | Forward primer for gent screening |
| P92 | CTTGCACGTAGATCACATAAGC | Reverse primer for gent screening |
| P202 | AGAGGGAAATCGTGCGTGAC | Forward primer for qPCR of mouse actB |
| P203 | CAATAGTGATGACCTGGCCGT | Reverse primer for qPCR of mouse actB |
| P204 | CACTGCCGCATCCTCTTCCTCCC | Probe primer for qPCR of mouse actB |
| P270 | CCGGGTACCCAATCGGATTTTTAACTTAAAGTCG | Forward primer for arp targeted deletion with KpnI site |
| P271 | CCGGAGCTCGACAATCTTGTTACTAAGATTGATAACG | Reverse primer for arp targeted deletion with SacI site |
| P302 | CATGCTCCAAACTCAAAAATTG | Forward primer for arp screening and probe generation |
| P303 | GGGTGTGTAATTTTTTCTTCAACTTC | Reverse primer for arp screening and probe generation |
| P357 | CCGGCTAGCGATGTAGAAAATGATGTAGCCTCTACTAAATAATGTG | Reverse primer for arp complement generation with NheI site |
| P360 | CCGGCTAGCTGCAAAAATTTGTATAATCTAAAATTATACATTAATG | Forward primer for arp complement generation with NheI site |
| P359 | CCGGGATCCTTAACTTAAACCCTTTACACTTTCTTCG | Reverse primer arp recombinant protein with BamHI site |
| P361 | CCGCATATGAAATTTGATAGTCTTAATTTATCTACAAAAAGC | Forward primer arp recombinant protein with NdeI site |
| P411 | GAGTTTCTGGTAAGATTAATGCTC | Forward primer for qPCR of flaB |
| P412 | CATTTAAATTCCCTTCTGTTGTCTGA | Reverse primer for qPCR of flaB |
| P413 | AGAGGTTTGTCACAAGCTTCTAGAAATACTTCAAAGGC | Probe for qPCR of flaB |
The in cis arp complement B. burgdorferi clone was generated by amplifying arp, including 400 bp of upstream sequence (containing the native promoter element), followed by insertion into pPH12 at the NheI restriction site to create the complementation construct pPH25. The kanamycin resistance gene in this construct was replaced by cloning in the aacC1 gene conferring gentamicin resistance at the NgoMIV and NheI restriction sites. The resulting plasmid DNA construct was then transformed into E. coli and cultured under gentamicin selection. The plasmids isolated from verified E. coli clones were then used to transform the arp mutant clone.
B. burgdorferi transformation.
B. burgdorferi B31-5A4 wild-type or arp deletion mutant cells were electroporated and cultured as previously described (28). DNA from culture-positive wells was extracted using a DNeasy Blood and Tissue kit (Qiagen, Germantown, MD) and used for PCR analysis to confirm the presence of the antibiotic resistance gene and the presence or absence of arp (Table 2 lists the primers). The plasmid content for each verified transformant was determined by PCR using plasmid-specific primers, as previously described (27).
Southern blot analysis.
Total plasmid DNA was extracted from B. burgdorferi clones using the Plasmid Midi kit (Qiagen) and separated on a 1% agarose gel at 80 V for 23 h (250 ng of DNA was used per lane). The DNA was then transferred onto a nylon membrane and hybridized with digoxigenin (DIG)-labeled probes following the manufacturer's guidelines (Roche, Indianapolis, IN).
Murine infection.
All animal infections were carried out in accordance with approved protocols from the Institutional Animal Care and Use Committee (IACUC) of Washington State University. Male C3H/HeN mice (Harlan, Indianapolis, IN) at 7 weeks of age were infected by subcutaneous needle inoculation with 105 total spirochetes. Five mice per experimental group were treated at each time point. B. burgdorferi clones were passaged no more than two times in vitro from frozen glycerol stock prior to use in mouse infection studies. Infection was monitored by culturing either blood samples or ear biopsy specimens at the indicated times postinfection. Blood and tissue samples were cultured in BSK-II medium containing Borrelia antibiotic cocktail (0.02 mg/ml phosphomycin, 0.05 mg/ml rifampin, and 2.5 μg/ml amphotericin B). Disease progression was noted weekly by visual examination and digital-caliper measurements. At 2, 4, and 8 weeks postinfection, mice were sacrificed, and ear, heart, bladder, and joint tissues were obtained aseptically and cultured in BSK-II medium containing Borrelia antibiotic cocktail. Dark-field microscopy was used to determine the presence or absence of viable spirochetes for each cultured tissue sample.
Measurement of tibiotarsal joints.
Mice were anesthetized with isoflurane prior to joint measurements taken with a digital metric caliper (Mitutoyo, Tokyo, Japan). Measurements were taken in the anterior-to-posterior position with the knee extended, through the thickest portion of the ankle (31). The ankle diameter increase was obtained by subtracting the preinfection joint diameter from the measurements taken at each week for 4 or 8 weeks.
Histology of tibiotarsal joints.
At 2, 4, and 8 weeks postinfection, 5 mice from each experimental group were sacrificed, and the tibiotarsal joint displaying the greatest swelling was collected for histopathology. The joints were immediately fixed in 10% neutral buffered formalin. Sections from the decalcified and paraffin-embedded joints were then stained with hematoxylin and eosin. Sections were scored blindly from 0 to 4, and the scores were defined as follows: 0, no change (no inflammation); 1, minimal change (less than 1% of the area of the tibiotarsal joint infiltrated with leukocytes); 2, mild change (1 to 25% of the area infiltrated with leukocytes); 3, moderate change (25 to 50% of the area infiltrated with leukocytes, often with synovial hyperplasia, periarticular fibrosis, and/or minimal to mild exudate within the joint and/or tendon sheath); 4, severe change (more than 50% of the area infiltrated as described above plus moderate to severe exudate within the joint and/or tendon sheath, often also with superficial inflammation of the bone). The average score for each group at each time point was obtained, and significant differences were determined.
qPCR analysis.
Mice infected with experimental strains for quantitative-PCR (qPCR) analysis were sacrificed at either 2, 4, or 8 weeks, and the most swollen tibiotarsal joint was collected from each mouse. Four mice per group were used at the 2-week time point and 5 mice per group at the 4- and 8-week time points. The samples were immediately snap-frozen in liquid nitrogen and ground thoroughly with a mortar and pestle. DNA was extracted by using the DNeasy Minikit following the manufacturer's instructions (Qiagen) and stored at −20°C. Plasmid constructs containing the mouse β-actin gene (actB) and the B. burgdorferi flaB gene were generated to create absolute standards. The sequences of primers and internal probes were described previously for flaB (21) and actB (32) (Table 2). Primers for flaB and actB amplified a 115-bp and a 138-bp DNA fragment, respectively. Each of these DNA fragments was separately cloned into pJET2.1 (Fermentas) and verified by DNA sequencing. DNA concentrations were determined by measuring the optical density at 260-nm wavelength and converted to the respective copy numbers.
qPCR analyses were performed using the CFX96 Touch Real-Time PCR detection system (Bio-Rad Laboratories, Hercules, CA). qPCR was carried out in 20-μl reaction mixtures containing 1× SsoFast Probes Supermix (Bio-Rad). DNA standards containing 104 to 100 copies/well of the B. burgdorferi flaB gene or mouse actB containing 107 to 103 copies/well were run on each plate. Both standards and samples were amplified in triplicate. The amplification program was performed as follows: (i) heating at 95°C for 2 min for polymerase activation and DNA denaturation and (ii) amplification for 40 cycles with denaturation at 95°C for 10 s and extension and annealing at 60°C. Plate reading was done at 60°C. The mean DNA copy numbers of flaB and actB for each DNA sample were calculated from triplicate wells. Tissue spirochete levels were converted to flaB DNA copy numbers per 106 actB DNA copies. The average copy number for each group at each time point was obtained, and significant differences were determined.
Statistical analysis.
SigmaPlot 11.0 software was used for all statistical data analysis. The Student t test was used for the comparison of two groups, while one-way analysis of variance (ANOVA) was used when comparing data sets comprising more than two groups. This was followed by the all-pairwise multiple-comparison procedure (Holm-Sidak method) if there were significant differences among treatment means (P < 0.05). The Kruskal-Wallis ANOVA on ranks was applied in cases where the normality test failed, followed by the all-pairwise multiple-comparison procedure (Tukey test; P < 0.05).
RESULTS
Generation of B. burgdorferi arp deletion and complement clones.
Antiserum to Arp has been shown to prevent or reduce arthritis in immunodeficient mice (25), suggesting involvement of the lipoprotein in the generation of Lyme arthritis. To investigate the requirement for Arp in joint inflammation associated with B. burgdorferi infection, a mutant clone lacking the arp gene locus (ΔArp) was generated. To achieve this, a telomere-mediated targeted-deletion strategy was utilized (Fig. 1A) (33–35). Briefly, this approach involves the introduction of a replicated telomere (rtel) at any position within a linear plasmid via integration of a deletion construct containing a target site for homologous recombination. Following integration of the deletion construct, the internal rtel is recognized and processed by the endogenous B. burgdorferi protein ResT (36, 37). Telomere resolution by ResT leads to the production of a new covalently closed hairpin end, resulting in deletion of the entire plasmid region located upstream or downstream of the target sequence.
FIG 1.
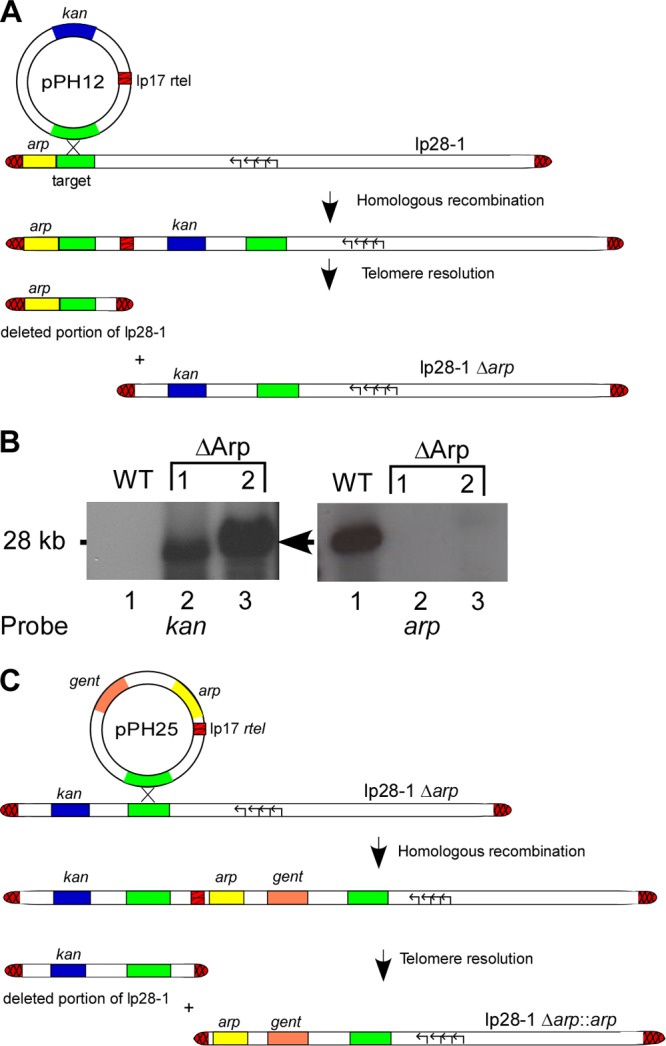
Generation of arp mutant clones. (A) Schematic of the deletion strategy for the arp knockout on lp28-1. The deletion construct (pPH12) was created by cloning a target region upstream of the arp locus (green) into a vector that carries a kanamycin resistance gene (kan) (blue) for selection and a replicated telomere (rtel) (red crosshatched regions) that is specifically recognized by the telomere resolvase, ResT. After transformation of the deletion construct into a fully infectious clone of B. burgdorferi, integration of the plasmid at the homologous target site and resolution of the rtel by endogenous ResT results in the loss of a DNA fragment containing the arp locus. (B) Southern blot confirmation of B. burgdorferi ΔArp mutant clones. Two clones matching the required criteria were selected for Southern blot analysis (lanes 2 and 3). The blot confirmed the presence of kan (arrow) and the absence of arp by the use of the respective probes on lp28-1. (C) Schematic of the complementation strategy of arp onto lp28-1Δarp. To generate the arp replacement construct, the arp locus with the native promoter (yellow) was cloned into pPH12 to yield pPH25, which contained the same DNA target sequence homologous to lp28-1 that was used for targeted deletion of arp, along with a gentamicin resistance marker (orange) and an rtel. The complementation plasmid was then used to transform ΔArp competent cells.
As shown in Fig. 1A, arp is the first open reading frame (ORF) residing on lp28-1 and thus is the only lp28-1-resident gene lost following telomere resolution of the internally introduced rtel. Transformants were PCR screened for the presence of the kanamycin resistance cassette. Three clones meeting this criterion were further analyzed for their total plasmid profile, and two of the three clones contained all of the parental plasmids. DNA isolated from these two mutant B. burgdorferi clones was then subjected to Southern blot analysis to confirm the loss of arp (Fig. 1B).
In order to verify that any potential reduction in joint swelling exhibited by the ΔArp mutant clone was due solely to the loss of the arp gene, a complemented Arp mutant clone was generated. The strategy for this involved reconstituting the arp gene back onto the lp28-1 plasmid harboring the arp deletion (Fig. 1C) (38). Previous studies have shown that transcriptional regulation of some B. burgdorferi genes is dependent on the topology (i.e., linear or circular) of the encoding genetic element (39). In addition, placement of arp back onto its native lp28-1 ensures the proper gene copy number (due to the lp28-1 copy number maintained in the cell), which can also have an effect on total protein expression.
To generate the arp replacement construct, the arp locus with the native promoter was cloned into the plasmid pPH25, which contained the same DNA target sequence homologous to lp28-1 that was used for targeted deletion of arp, along with a gentamicin resistance marker and rtel (Fig. 1C). Plasmid DNA was then transformed into ΔArp competent cells, and transformants were screened for the presence of the gentamicin and arp genes, as well as for all of the parental plasmids. One in cis Arp complement clone (cArp) that met all of the criteria was chosen for Western blot analysis in an attempt to ensure expression and surface localization of Arp. However, similar to previous findings with wild-type B. burgdorferi (40), expression of Arp could not be detected from any of the in vitro-derived B. burgdorferi clones (data not shown).
To assess the individual infectivities of the newly generated ΔArp and complemented B. burgdorferi clones, groups of 5 C3H/HeN (C3H) mice each were needle inoculated with the ΔArp, cArp, or wild-type B. burgdorferi clone. As shown in Table 3, blood samples collected at day 7 postinfection produced cultures positive for spirochetes in all inoculated mouse groups. Moreover, ear biopsy specimens collected at weeks 2 to 7 postinfection were positive for spirochetes in all mouse groups tested. Finally, ear, heart, bladder, and joint tissues harvested from sacrificed mice at week 8 postinfection produced cultures positive for spirochetes, demonstrating that the ΔArp and Arp complemented clones were fully capable of persistent host infection and dissemination.
TABLE 3.
Infectivity of B. burgdorferi clones in C3H mice
| Clone | No. of mice positivea at wk postinfection: |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (blood) | 2 (ear) | 3 (ear) | 4 (ear) | 5 (ear) | 6 (ear) | 7 (ear) | 8 |
||||
| Heart | Ear | Joint | Bladder | ||||||||
| WT | 5/5b | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
| ΔArp | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
| cArp | 5/5 | 3/5 | 4/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
Number of positive cultures/number of mice tested. Five mice were used for each B. burgdorferi clone.
Arp is required for early-onset joint swelling.
Ankle swelling has been shown to reflect the amount of edema and to provide a gross measurement of the inflammatory response (31). To assess the requirement for Arp in ankle joint swelling, 5 C3H mice each were infected with either ΔArp, cArp, or wild-type B. burgdorferi clones. The progress of infection was monitored weekly via blood culture or ear biopsy samples. Gross ankle swelling was evaluated weekly by digital caliper measurements of the tibiotarsal joint thickness for up to 8 weeks postinfection. No significant differences were noted by day 7 postinfection (P = 0.305). Joint swelling in mice infected with the wild type was apparent beginning at 2 weeks postinfection and peaked between weeks 4 and 5 before starting to resolve (Fig. 2). Mice infected with the ΔArp clone exhibited drastically reduced joint swelling that was not statistically different from that in the uninfected-medium control mice throughout the duration of the study, except at the 8-week time point (P = 0.037). In contrast, the cArp complement clone group exhibited significantly greater swelling than the uninfected control group throughout the study period. No statistically significant difference was observed in joint swelling between cArp and the wild type, and both clones caused significantly higher swelling than the ΔArp clone (P < 0.05).
FIG 2.
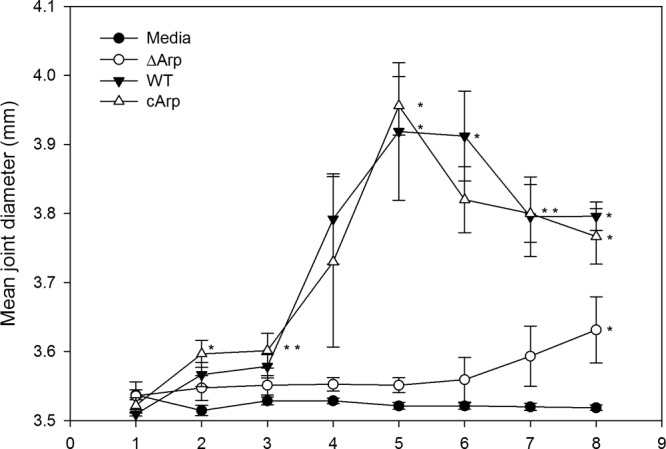
Tibiotarsal joint swelling in C3H mice infected with B. burgdorferi wild-type or arp mutant clones. Five mice were used in each group. The joints were measured with a digital metric caliper at weekly intervals after infection. The values are shown as the differences from uninfected, week zero measurements, and the mean joint diameter for each group is indicated at various weeks after infection. The error bars indicate the standard errors of the mean within the groups. An asterisk indicates a group(s) that was significantly different from one or more groups at a P value of <0.05. The joint swelling caused by infection with the ΔArp clone was not statistically different from that in the uninfected-medium control during the study period, except at week 8 (P = 0.037). There was no significant difference in joint swelling between the cArp- and wild type-infected mice throughout the study period, and both clones caused significantly more swelling than the knockout (P < 0.05).
Overall, the above-described experiments show that mice infected with the ΔArp clone had reduced joint swelling during the first 5 weeks of infection and began to display increased swelling at week 6 postinfection. These results suggest that Arp is required for maximal severity of joint swelling during murine infection. Moreover, the pattern of joint swelling attributed to the ΔArp clone from the 8-week infection study suggests that Arp is specifically important for early-onset joint swelling.
The presence of Arp contributes to efficient immune cell infiltration into the joint during early infection.
Although the results from the above-described infection assays demonstrate the importance of Arp for maximal joint swelling caused by B. burgdorferi during the acute stages of host infection, it was unclear whether a decrease in joint swelling correlated with a reduction in joint tissue pathology. Caliper measurements of ankle swelling have been used to provide a gross measurement of the inflammatory response without the need to sacrifice the animal (41). However, they may correlate poorly with the severity of the underlying arthritis. Histological analysis provides a more complete determination of arthritis severity and characterization of the underlying inflammatory response (31). Our previous infection assay demonstrated that joint swelling levels in mice infected with the ΔArp clone started increasing notably at 6 weeks postinfection. Thus, it is conceivable that a difference in pathology between the ΔArp and wild-type clones could be clearly detected in joint tissues harvested at earlier time points.
To determine histopathological changes during infection, groups of C3H mice were infected with either ΔArp, cArp, or wild-type B. burgdorferi clones and sacrificed at 2, 4, or 8 weeks postinfection. Significantly reduced pathology was observed in mice infected with the ΔArp mutant compared to those infected with the wild type (scores, 1.5 ± 1.0 and 3.3 ± 0.5, respectively; P = 0.034) (Table 4) at 2 weeks postinfection. Mice infected with the cArp clone (score, 3.3 ± 1.0) showed severe joint pathology indistinguishable from that with wild-type B. burgdorferi (score, 3.3 ± 0.5) (Table 4). The tibiotarsal joints of mice infected with the wild-type and cArp clones displayed inflammation that was mainly characterized by infiltration of neutrophils involving the tibiotarsal flexor tendon and sheath (Fig. 3B and E, and C and F, respectively). Synovial hyperplasia and inflammation were also observed, and there was neutrophilic exudate in the lumen. There was also increased interstitial clear space and flocculent eosinophilic edema residue. Though this was recognized in some sections, it was difficult to quantitate histologically. All of these features, especially neutrophilic infiltration, were reduced in mice infected with the ΔArp clone (Fig. 3A and D).
TABLE 4.
Histopathology scores of tibiotarsal joints
| B. burgdorferi clone | Histopathology score (±SD) at wka: |
||
|---|---|---|---|
| 2 | 4 | 8 | |
| WT | 3.3 ± 0.5 | 3.6 ± 0.55 | 2.2 ± 0.84 |
| ΔArp | 1.5 ± 1.0b | 2.6 ± 1.14 | 2.4 ± 1.14 |
| cArp | 3.3 ± 1.0 | 1.2 ± 0.45b | 1.6 ± 0.55 |
Four mice were used for each B. burgdorferi clone at 2 weeks and 5 mice were used for each clone at weeks 4 and 8.
Significantly different group using one-way ANOVA.
FIG 3.

(Top row) Differences in tibiotarsal changes in C3H mice infected with B. burgdorferi clones at 2 weeks postinfection. (A) ΔArp. (B) Wild type. (C) cArp. Reduced cellular infiltration and synovial hyperplasia (black arrow versus white arrows) and minimal to absent neutrophilic infiltration were observed in the tibiotarsal flexor tendons of mice infected with the ΔArp clone. Histopathological changes between the WT and cArp clones were indistinguishable. (Bottom) High-power magnification of histopathologic changes in C3H mice infected with B. burgdorferi clones. (D) Minimal changes observed in the tibiotarsal flexor tendon (T) of ΔArp clone-infected mice with markedly reduced synovial hyperplasia and little to no neutrophilic infiltration in the lumen (L) of the tendon sheath (S) (magnification, ×100). (E) Inflamed hyperplastic tendon sheath in a mouse infected with wild-type B. burgdorferi (magnification, ×60). (F) Synovial lumen with inflammatory exudate comprised of neutrophils, edema residue, and fibrin and hyperplasia of the tendon sheath observed in mice infected with the cArp clone (magnification, ×100).
In contrast to the week 2 histologic samples, increasingly similar levels of inflammatory-cell infiltration were observed in week 4 and 8 joint tissues infected with either the wild-type (scores, 3.6 ± 0.5 and 2.6 ± 1.14, respectively; P = 0.064) or the ΔArp (scores, 2.2 ± 0.84 and 2.4 ± 1.14, respectively; P = 0.760) B. burgdorferi clone (Table 4). Inflammation in both groups was characterized by infiltration of mainly neutrophils, macrophages, and lymphocytes. At 8 weeks, neutrophils were present but were not the predominant cell type. Joint tissues recovered from mice infected with the wild-type and ΔArp clones showed intermediate arthritis at week 8. The reduction in cellular infiltrate indicates early stages of arthritis resolution (9). ΔArp clone-infected mice showed intermediate levels of inflammatory-cell infiltration despite the low levels of ankle edema observed for mice infected with the mutant clone at 2 and 4 weeks postinfection (Fig. 2). Mice infected with cArp had reduced arthritis scores at weeks 4 and 8 (scores, 1.2 ± 0.45 and 1.6 ± 0.55, respectively). At 8 weeks, no significant difference in arthritis scores was observed between the ΔArp, wild-type, and cArp B. burgdorferi clones (P = 0.355). These findings suggest that the joint edema associated with the presence of Arp in B. burgdorferi aids in the efficient recruitment of inflammatory cells to the site of spirochete colonization during early-onset Lyme arthritis but is dispensable in the later courses of inflammation.
Arp contributes to control of the spirochete burden in joint tissues during infection.
Following observations that deletion of arp reduced joint swelling and immune cell infiltration during early infection, we went on to determine if there would be an effect on spirochete colonization of the joint. To assess the spirochete burden in joints, qPCR analysis was conducted on collected joint tissues of C3H mice infected with either wild-type, ΔArp, or cArp B. burgdorferi clones at 2, 4, and 8 weeks postinfection. These time points were chosen because they represent early, peak, and resolving pathology based on results from the previous joint swelling experiments. At 2 weeks postinfection, there was no significant difference in spirochete burdens among the different B. burgdorferi-inoculated groups of mice (P = 0.532) (Table 5). After 4 weeks of infection, the number of spirochetes in the joints of mice infected with the wild-type clone was significantly lower than that in mice infected with the ΔArp clone (6.23 × 103 versus 2.03 × 103; P = 0.027). Next, it was determined whether the observed change in cellular infiltrate through time and resolution of arthritis corresponded to a decrease in the spirochete burden during infection with the experimental clones at 8 weeks postinfection. A reduction in the spirochete burden was observed in joints from mice infected with all three clones (Table 5). Though there was no statistically significant difference, mice infected with ΔArp consistently had higher numbers of spirochetes than mice infected with the wild type. Spirochete numbers from the joints of mice infected with the cArp clone compared to those infected with the ΔArp clone also showed no statistical difference at selected time points. From these results, the presence of Arp seems to equate to a generally lower burden of spirochetes in the joint, while the absence of Arp results in an overall increased spirochete burden in the joints. The results from the complement clone may hint that proper regulation of Arp expression in the clone is lacking, which could explain the high spirochete burden in the joints of mice infected with the clone at 4 weeks. Overall, the above-mentioned results suggest that the presence of the arp gene is associated with decreased spirochete burdens and increased pathology within the joint, and these phenotypes are more pronounced during early infection.
TABLE 5.
B. burgdorferi quantification in tibiotarsal joints
| B. burgdorferi clonea | No. of spirochetes/106 host cells (±SD) at wk: |
||
|---|---|---|---|
| 2 | 4 | 8 | |
| WT | 151 ± 113 | 2,033 ± 4,165 | 1,470 ± 1,188 |
| ΔArp | 206 ± 285 | 6,233 ± 4986b | 2,167 ± 1,772 |
| cArp | 62 ± 87 | 6,569 ± 7,343 | 1,521 ± 1,859 |
Five mice were used for each B. burgdorferi clone.
Significantly different group using one-way ANOVA.
DISCUSSION
Studies with the murine model of Lyme disease have provided evidence that joint inflammation is a result of interaction of host immune responses with spirochete constituents (15–20). B. burgdorferi spirochetes are highly invasive and possess several lipoproteins that have been shown to have potent proinflammatory properties. These lipoproteins are encoded by genes that are regulated by diverse metabolic and immune microenvironments within mammalian tissues (21–24). The joint is thought to provide a protective niche for B. burgdorferi colonization, which may partially explain spirochete persistence in the tissue (42). In this study, we set out to determine if deletion of the arp gene has an effect on the pathogenesis of Lyme arthritis. The results reported here show a significant reduction in joint swelling in ΔArp clone-infected mice compared to those infected with wild-type B. burgdorferi, but only during the early onset of arthritis. The difference in the grossly measurable ankle swelling observed between the Arp mutant and wild-type B. burgdorferi during early host infection strongly suggests that Arp is required for the periarticular edema and inflammation associated with Lyme arthritis. We also observed that in the absence of arp, the spirochete load within the joint tissue remained high, suggesting that Arp expression may be somewhat detrimental to spirochete survival in that tissue.
The presence of Arp affects influx of inflammatory cells into joint tissue during early stages of infection.
Although substantial reduction in joint swelling was observed, analysis of histopathology data on the joints of mice infected with the ΔArp clone at 4 and 8 weeks postinfection did not show any significant reduction in arthritis severity compared to those infected with the wild type. However, a significant difference in histopathology was observed when mice infected with the wild type (score, 3.3 ± 0.5) were compared to mice infected with the ΔArp clone (score, 1.5 ± 1.0; P = 0.034) at 2 weeks postinfection (Fig. 3), highlighting the importance of the lipoprotein in the early stages of infection. The edema associated with ankle swelling is typically correlated with infiltration of neutrophils and macrophages in response to the presence of spirochetes in the subcutaneous tissue (9). Our results suggest that the significant reduction of joint swelling observed after infection with ΔArp corresponds to decreased influx of inflammatory cells into joint tissue during early murine infection with B. burgdorferi. Several inflammatory cytokines and chemokines have been shown to influence the infiltration of neutrophils into B. burgdorferi-infected joint tissue (43, 44). Recently, BBA57 was shown to be a major trigger of Lyme arthritis, with its deficiency resulting in decreased neutrophil chemotaxis (45). The results of this study suggest that Arp may stimulate recruitment of neutrophils or other immune components, allowing the production of cytokines that lead to periarticular edema. Further studies will be needed in order to determine if the cytokine profile differs between mice infected with wild-type B. burgdorferi and those infected with the Arp deletion mutant.
In agreement with the findings reported here, a study by Imai et al. published during the preparation of the manuscript showed joint arthritis to be significantly reduced in C3H mice infected with an arp mutant clone at day 14 postinfection (26). However, the Imai et al. study also observed that their B. burgdorferi arp mutant displayed reduced joint pathology for the entire 42-day infection period, which contrasts with the present study, which found a significant difference in pathology only at day 14. Although mouse experiments in both studies involved an infectious-dose inoculum of 105 spirochetes or higher, it is possible that the ΔArp clone used here, if diluted, may have demonstrated an attenuated phenotype relative to the wild type at later time periods. Several additional factors might also explain the discrepancies between the two studies. First, the methods of gene deletion utilized differed between the two studies (allelic exchange [26] versus telomere-mediated deletion [this study]). It is possible that differences in these methodologies could have resulted in subtle genetic alterations or polar effects on flanking genes. A second factor is that complementation typically involves reintroduction of a wild-type copy of only the gene of interest (46). However, the Imai et al. study complemented arp through displacement of the altered lp28-1 plasmid via transformation with a wild-type version of lp28-1, which has the potential to mask any additional (non-arp-related) genetic changes that might have contributed to the observed phenotype (26). Finally, this study set out to specifically characterize how joint cell infiltration and edema were affected by the deletion of arp, which was not explicitly analyzed in the recent publication. Our results show that Arp contributes greatly to the generation of neutrophilic inflammation and joint edema during the early phase of infection, as shown by joint swelling experiments. This suggests that Arp may play a role in cytokine changes responsible for recruitment of neutrophils to the B. burgdorferi-colonized joint tissue and subsequent edema. Elucidating the mechanisms, cells, and cytokines involved will require further investigation.
Arp is immunopotent and may contribute to control of the spirochete burden in the joint.
Pathology during B. burgdorferi infection has been primarily associated with host inflammatory responses (11, 19), and accordingly, invasion of joint tissues by B. burgdorferi has been linked with the pathogenesis of arthritis (46–48). Arp has been shown to be highly immunogenic, and antibody against the lipoprotein has been detected from day 7 postinfection up to day 90 (25). In this study, deletion of arp resulted in a burden of spirochetes within joint tissues at 4 weeks postinfection significantly higher than that exhibited in mice infected by the wild type (6.23 × 103 versus 2.03 × 103, respectively; P = 0.027) and was comparatively high at additional time points. Immune recognition of Arp in mice infected by wild-type B. burgdorferi may explain the observed decrease in histopathology scores obtained at early and peak infection compared to late infection (scores, 3.3 ± 0.5 at 2 weeks and 3.6 ± 0.5 at 4 weeks compared to 2.2 ± 0.84 at week 8; P = 0.111 and P = 0.014, respectively). No statistically significant differences were noted in the histopathology scores of mice infected with the ΔArp clone at similar time points. This suggests that expression of Arp may be detrimental to B. burgdorferi survival, which may hint at an underlying important biological role for the lipoprotein during host infection. Moreover, the finding that ΔArp spirochetes are present at higher numbers in joint tissues suggests that Arp may not be involved in the tropism of B. burgdorferi to this tissue site. In addition, there was no statistically significant difference between the spirochete burdens in the joints of mice infected with cArp and ΔArp at week 4, despite the measured difference in tibiotarsal swelling at this time point and beyond. This raises the question of whether the difference in spirochete numbers at week 4 between the wild type and the ΔArp clone is biologically significant. It would also argue that arthritis is not directly correlated with bacterial levels and suggests that there is no fitness defect associated with the loss of arp.
Again, contrary to the findings reported here, the recently published study by Imai et al. (26) found that the spirochete burden was lower in mice infected with the arp mutant clone. This may be due to the observed impaired dissemination reported by the authors, which we did not observe in this study. A possible explanation for this discrepancy is that genetic manipulation of lp28-1 in the Imai et al. study may have had an effect on a yet to be identified gene residing on the plasmid that has been suggested to express a putative regulator of the outer surface protein OspC (49). It has been recently shown that OspC may be an important dissemination factor of B. burgdorferi during mammalian infection (50). Once again, because the method of complementation reported in the Imai et al. study involved total plasmid replacement, any potential genetic alterations of lp28-1 affecting OspC regulation that may have led to impaired dissemination by the arp mutant could have been masked in the complemented clone.
Overall, the findings presented here demonstrate the requirement for Arp in B. burgdorferi-induced joint swelling and pathology. Further studies directed toward elucidating the specific role that Arp has in pathogenesis and, possibly, immune manipulation by the Lyme disease spirochete will likely provide insight into the mechanisms responsible for B. burgdorferi-associated joint inflammation and aid in the understanding of Lyme disease pathogenesis.
ACKNOWLEDGMENTS
We thank Tim Casselli, Allison James, and Yvonne Tourand for critical reading of the manuscript.
This work was supported by an intramural grant from the College of Veterinary Medicine at Washington State University.
Footnotes
Published ahead of print 7 October 2013
REFERENCES
- 1.Ligor M, Olszowy P, Buszewski B. 2012. Application of medical and analytical methods in Lyme borreliosis monitoring. Anal. Bioanal Chem. 402:2233–2248. 10.1007/s00216-011-5451-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhate C, Schwartz RA. 2011. Lyme disease. Part I. Advances and perspectives. J. Am. Acad. Dermatol. 64:619–636. 10.1016/j.jaad.2010.03.046 [DOI] [PubMed] [Google Scholar]
- 3.Parker JL, White KK. 1992. Lyme borreliosis in cattle and horses: a review of the literature. Cornell Vet. 82:253–274 [PubMed] [Google Scholar]
- 4.Appel MJ, Allan S, Jacobson RH, Lauderdale TL, Chang YF, Shin SJ, Thomford JW, Todhunter RJ, Summers BA. 1993. Experimental Lyme disease in dogs produces arthritis and persistent infection. J. Infect. Dis. 167:651–664. 10.1093/infdis/167.3.651 [DOI] [PubMed] [Google Scholar]
- 5.Steere AC. 1989. Lyme disease. N. Engl. J. Med. 321:586–596. 10.1056/NEJM198908313210906 [DOI] [PubMed] [Google Scholar]
- 6.Humair PF, Postic D, Wallich R, Gern L. 1998. An avian reservoir (Turdus merula) of the Lyme borreliosis spirochetes. Zentralbl. Bakteriol. 287:521–538 [PubMed] [Google Scholar]
- 7.Wagner B, Freer H, Rollins A, Garcia-Tapia D, Erb HN, Earnhart C, Marconi R, Meeus P. 2012. Antibodies to Borrelia burgdorferi OspA, OspC, OspF, and C6 antigens as markers for early and late infection in dogs. Clin. Vaccine Immunol. 19:527–535. 10.1128/CVI.05653-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. 1982. Lyme disease—a tick-borne spirochetosis? Science 216:1317–1319. 10.1126/science.7043737 [DOI] [PubMed] [Google Scholar]
- 9.Barthold SW, de Souza MS, Janotka JL, Smith AL, Persing DH. 1993. Chronic Lyme borreliosis in the laboratory mouse. Am. J. Pathol. 143:959–971 [PMC free article] [PubMed] [Google Scholar]
- 10.Wooten RM, Weis JJ. 2001. Host-pathogen interactions promoting inflammatory Lyme arthritis: use of mouse models for dissection of disease processes. Curr. Opin. Microbiol. 4:274–279. 10.1016/S1369-5274(00)00202-2 [DOI] [PubMed] [Google Scholar]
- 11.Steere AC, Glickstein L. 2004. Elucidation of Lyme arthritis. Nat. Rev. Immunol. 4:143–152. 10.1038/nri1267 [DOI] [PubMed] [Google Scholar]
- 12.Haake DA. 2000. Spirochaetal lipoproteins and pathogenesis. Microbiology 146:1491–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, Weis JJ. 2002. Toll-like receptor 2 plays a pivotal role in host defense and inflammatory response to Borrelia burgdorferi. Vector Borne Zoonotic Dis. 2:275–278. 10.1089/153036602321653860 [DOI] [PubMed] [Google Scholar]
- 14.Alexopoulou L, Thomas V, Schnare M, Lobet Y, Anguita J, Schoen RT, Medzhitov R, Fikrig E, Flavell RA. 2002. Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in TLR1- and TLR2-deficient mice. Nat. Med. 8:878–884. 10.1038/nm732 [DOI] [PubMed] [Google Scholar]
- 15.Wooten RM, Modur VR, McIntyre TM, Weis JJ. 1996. Borrelia burgdorferi outer membrane protein A induces nuclear translocation of nuclear factor-kappa B and inflammatory activation in human endothelial cells. J. Immunol. 157:4584–4590 [PubMed] [Google Scholar]
- 16.Ma Y, Seiler KP, Tai KF, Yang L, Woods M, Weis JJ. 1994. Outer surface lipoproteins of Borrelia burgdorferi stimulate nitric oxide production by the cytokine-inducible pathway. Infect. Immun. 62:3663–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norgard MV, Arndt LL, Akins DR, Curetty LL, Harrich DA, Radolf JD. 1996. Activation of human monocytic cells by Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides proceeds via a pathway distinct from that of lipopolysaccharide but involves the transcriptional activator NF-kappa B. Infect. Immun. 64:3845–3852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morrison TB, Weis JH, Weis JJ. 1997. Borrelia burgdorferi outer surface protein A (OspA) activates and primes human neutrophils. J. Immunol. 158:4838–4845 [PubMed] [Google Scholar]
- 19.Ma Y, Weis JJ. 1993. Borrelia burgdorferi outer surface lipoproteins OspA and OspB possess B-cell mitogenic and cytokine-stimulatory properties. Infect. Immun. 61:3843–3853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norgard MV, Riley BS, Richardson JA, Radolf JD. 1995. Dermal inflammation elicited by synthetic analogs of Treponema pallidum and Borrelia burgdorferi lipoproteins. Infect. Immun. 63:1507–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang FT, Yan J, Mbow ML, Sviat SL, Gilmore RD, Mamula M, Fikrig E. 2004. Borrelia burgdorferi changes its surface antigenic expression in response to host immune responses. Infect. Immun. 72:5759–5767. 10.1128/IAI.72.10.5759-5767.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher MA, Grimm D, Henion AK, Elias AF, Stewart PE, Rosa PA, Gherardini FC. 2005. Borrelia burgdorferi sigma54 is required for mammalian infection and vector transmission but not for tick colonization. Proc. Natl. Acad. Sci. U. S. A. 102:5162–5167. 10.1073/pnas.0408536102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang FT, Nelson FK, Fikrig E. 2002. Molecular adaptation of Borrelia burgdorferi in the murine host. J. Exp. Med. 196:275–280. 10.1084/jem.20020770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narasimhan S, Caimano MJ, Liang FT, Santiago F, Laskowski M, Philipp MT, Pachner AR, Radolf JD, Fikrig E. 2003. Borrelia burgdorferi transcriptome in the central nervous system of non-human primates. Proc. Natl. Acad. Sci. U. S. A. 100:15953–15958. 10.1073/pnas.2432412100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng S, Hodzic E, Barthold SW. 2000. Lyme arthritis resolution with antiserum to a 37-kilodalton Borrelia burgdorferi protein. Infect. Immun. 68:4169–4173. 10.1128/IAI.68.7.4169-4173.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imai D, Holden K, Velazquez EM, Feng S, Hodzic E, Barthold SW. 2013. Influence of arthritis-related protein (BBF01) on infectivity of Borrelia burgdorferi B31. BMC Microbiol. 13:100. 10.1186/1471-2180-13-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purser JE, Norris SJ. 2000. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. U. S. A. 97:13865–13870. 10.1073/pnas.97.25.13865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bankhead T, Chaconas G. 2007. The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol. Microbiol. 65:1547–1558. 10.1111/j.1365-2958.2007.05895.x [DOI] [PubMed] [Google Scholar]
- 29.DeLange AM, Reddy M, Scraba D, Upton C, McFadden G. 1986. Replication and resolution of cloned poxvirus telomeres in vivo generates linear minichromosomes with intact viral hairpin termini. J. Virol. 59:249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tourand Y, Deneke J, Moriarty TJ, Chaconas G. 2009. Characterization and in vitro reaction properties of 19 unique hairpin telomeres from the linear plasmids of the lyme disease spirochete. J. Biol. Chem. 284:7264–7272. 10.1074/jbc.M808918200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma Y, Seiler KP, Eichwald EJ, Weis JH, Teuscher C, Weis JJ. 1998. Distinct characteristics of resistance to Borrelia burgdorferi-induced arthritis in C57BL/6N mice. Infect. Immun. 66:161–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Liu X, Beck DS, Kantor FS, Fikrig E. 2006. Borrelia burgdorferi lacking BBK32, a fibronectin-binding protein, retains full pathogenicity. Infect. Immun. 74:3305–3313. 10.1128/IAI.02035-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaconas G, Stewart PE, Tilly K, Bono JL, Rosa P. 2001. Telomere resolution in the Lyme disease spirochete. EMBO J. 20:3229–3237. 10.1093/emboj/20.12.3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beaurepaire C, Chaconas G. 2005. Mapping of essential replication functions of the linear plasmid lp17 of B. burgdorferi by targeted deletion walking. Mol. Microbiol. 57:132–142. 10.1111/j.1365-2958.2005.04688.x [DOI] [PubMed] [Google Scholar]
- 35.Casselli T, Tourand Y, Bankhead T. 2012. Altered murine tissue colonization by Borrelia burgdorferi following targeted deletion of linear plasmid 17-carried genes. Infect. Immun. 80:1773–1782. 10.1128/IAI.05984-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobryn K, Chaconas G. 2002. ResT, a telomere resolvase encoded by the Lyme disease spirochete. Mol. Cell 9:195–201. 10.1016/S1097-2765(01)00433-6 [DOI] [PubMed] [Google Scholar]
- 37.Chaconas G. 2005. Hairpin telomeres and genome plasticity in Borrelia: all mixed up in the end. Mol. Microbiol. 58:625–635. 10.1111/j.1365-2958.2005.04872.x [DOI] [PubMed] [Google Scholar]
- 38.Rogovskyy AS, Bankhead T. 2013. Variable VlsE is critical for host reinfection by the Lyme disease spirochete. PLoS One 8:e61226. 10.1371/journal.pone.0061226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beaurepaire C, Chaconas G. 2007. Topology-dependent transcription in linear and circular plasmids of the segmented genome of Borrelia burgdorferi. Mol. Microbiol. 63:443–453. 10.1111/j.1365-2958.2006.05533.x [DOI] [PubMed] [Google Scholar]
- 40.Fikrig E, Chen M, Barthold SW, Anguita J, Feng W, Telford SR, III, Flavell RA. 1999. Borrelia burgdorferi erpT expression in the arthropod vector and murine host. Mol. Microbiol. 31:281–290. 10.1046/j.1365-2958.1999.01171.x [DOI] [PubMed] [Google Scholar]
- 41.Yang L, Ma Y, Schoenfeld R, Griffiths M, Eichwald E, Araneo B, Weis JJ. 1992. Evidence for B-lymphocyte mitogen activity in Borrelia burgdorferi-infected mice. Infect. Immun. 60:3033–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang FT, Brown EL, Wang T, Iozzo RV, Fikrig E. 2004. Protective niche for Borrelia burgdorferi to evade humoral immunity. Am. J. Pathol. 165:977–985. 10.1016/S0002-9440(10)63359-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown CR, Blaho VA, Loiacono CM. 2004. Treatment of mice with the neutrophil-depleting antibody RB6-8C5 results in early development of experimental lyme arthritis via the recruitment of Gr-1-polymorphonuclear leukocyte-like cells. Infect. Immun. 72:4956–4965. 10.1128/IAI.72.9.4956-4965.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ritzman AM, Hughes-Hanks JM, Blaho VA, Wax LE, Mitchell WJ, Brown CR. 2010. The chemokine receptor CXCR2 ligand KC (CXCL1) mediates neutrophil recruitment and is critical for development of experimental Lyme arthritis and carditis. Infect. Immun. 78:4593–4600. 10.1128/IAI.00798-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang X, Qin J, Promnares K, Kariu T, Anderson JF, Pal U. 2013. Novel microbial virulence factor triggers murine lyme arthritis. J. Infect. Dis. 207:907–918. 10.1093/infdis/jis930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radolf JD, Samuels DS. 2010. Borrelia: molecular biology, host interaction, and pathogenesis, p 202 Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 47.Barthold SW, Persing DH, Armstrong AL, Peeples RA. 1991. Kinetics of Borrelia burgdorferi dissemination and evolution of disease after intradermal inoculation of mice. Am. J. Pathol. 139:263–273 [PMC free article] [PubMed] [Google Scholar]
- 48.Steere AC. 1995. Lyme arthritis: the joint lesions in Lyme borreliosis in the USA. Ter Arkh. 67:43–45 (In Russian.) [PubMed] [Google Scholar]
- 49.Embers ME, Alvarez X, Ooms T, Philipp MT. 2008. The failure of immune response evasion by linear plasmid 28-1-deficient Borrelia burgdorferi is attributable to persistent expression of an outer surface protein. Infect. Immun. 76:3984–3991. 10.1128/IAI.00387-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seemanapalli SV, Xu Q, McShan K, Liang FT. 2010. Outer surface protein C is a dissemination-facilitating factor of Borrelia burgdorferi during mammalian infection. PLoS One 5:e15830. 10.1371/journal.pone.0015830 [DOI] [PMC free article] [PubMed] [Google Scholar]