

Abstract
During infection of the mammalian host, Histoplasma capsulatum yeasts survive and reside within macrophages of the immune system. Whereas some intracellular pathogens escape into the host cytosol, Histoplasma yeasts remain within the macrophage phagosome. This intracellular Histoplasma-containing compartment imposes nutritional challenges for yeast growth and replication. We identified and annotated vitamin synthesis pathways encoded in the Histoplasma genome and confirmed by growth in minimal medium that Histoplasma yeasts can synthesize all essential vitamins with the exception of thiamine. Riboflavin, pantothenate, and biotin auxotrophs of Histoplasma were generated to probe whether these vitamins are available to intracellular yeasts. Disruption of the RIB2 gene (riboflavin biosynthesis) prevented growth and proliferation of yeasts in macrophages and severely attenuated Histoplasma virulence in a murine model of respiratory histoplasmosis. Rib2-deficient yeasts were not cleared from lung tissue but persisted, consistent with functional survival mechanisms but inability to replicate in vivo. In addition, depletion of Pan6 (pantothenate biosynthesis) but not Bio2 function (biotin synthesis) also impaired Histoplasma virulence. These results indicate that the Histoplasma-containing phagosome is limiting for riboflavin and pantothenate and that Histoplasma virulence requires de novo synthesis of these cofactor precursors. Since mammalian hosts do not rely on vitamin synthesis but instead acquire essential vitamins through diet, vitamin synthesis pathways represent druggable targets for therapeutics.
INTRODUCTION
The fungal pathogen Histoplasma capsulatum infects and resides within phagocytic cells of the mammalian immune system. Histoplasma is endemic to regions of North, Central, and South America, where it causes respiratory and systemic disease. Infections are not limited to immunocompromised individuals, although the severity and progression of disease are increased in the absence of cell-mediated immunity (1). The yeast form of Histoplasma is the pathogenic morphotype found within macrophages, which serve as the primary host cell (2). Infection of macrophages by yeast cells is facilitated by binding of yeasts to complement receptors and internalization into phagosomes (3, 4). Survival of the initial encounter with microbicidal phagocytes is enhanced by elimination of phagocyte-produced reactive oxygen through yeast-expressed extracellular superoxide dismutase and catalase (5, 6).
Once initial survival of immune defenses is achieved, Histoplasma yeast must obtain sufficient nutrition to enable yeast cell growth and replication within the macrophage host cell. Proliferation of yeasts intracellularly ultimately leads to lysis of the host cell and release of yeasts for infection of new phagocytes. Research on the intraphagosomal growth of intracellular pathogens suggests that the phagosome is limited for many nutrients (7–10). Although the exact composition of the pathogen-containing intracellular compartment differs for each pathogen, gene expression studies and infections with mutant strains consistently show that the intracellular environment encountered by the pathogen is nutritionally unlike the rich growth media routinely used for laboratory culture (11–22). From these studies of bacterial, fungal, and parasite pathogens of phagocytes, some general features of the intracellular compartment emerge showing that intracellular pathogens must have mechanisms for utilization of nonglucose carbon sources, transport and metabolism of amino acids, and acquisition mechanisms for magnesium, phosphate, and/or iron (7–10, 23–27). Because intracellular nutritional sources are more limited, pathogen growth requires ample biosynthetic capacity to supply molecules that the pathogen cannot scavenge from the lumen of the vacuole, phagosome, or phagolysosome.
Limited information currently exists regarding the nutritional requirements for intracellular Histoplasma growth. Early studies of yeast growth in culture indicated that yeasts but not mycelia of most Histoplasma species are auxotrophic for cysteine due to temperature-dependent expression of sulfite reductase and the consequent inability to incorporate inorganic sulfate into cysteine (28–32). Organic sulfhydryls, such as cysteine, also reduce the redox potential, which contributes to yeast phase differentiation (33–35). Growth of yeasts in macrophages requires cysteine to be present in the culture medium, consistent with yeast-phase auxotrophy (36). An undefined cysteine auxotroph, derived by mutagenesis of a cysteine-prototrophic yeast strain, remains virulent in mice, suggesting that cysteine is available to yeast in vivo (37, 38). In contrast, yeast virulence requires synthesis of uracil, since deletion of the gene encoding orotidine-5′-monophosphate pyrophosphorylase (39) attenuates virulence in macrophages and in vivo (40). Full virulence of yeasts also depends on acquisition of iron. Histoplasma yeasts produce hydroxamate siderophores which can steal iron from transferrin (41, 42), the most likely source of iron within the phagosome. Without siderophore production, intracellular Histoplasma growth is hampered (43, 44). In addition, Histoplasma produces extracellular iron reductases (41, 42), including a γ-glutamyltransferase (Ggt1) which causes a pH-independent release of iron from transferrin that is necessary for full Histoplasma virulence in phagocytes (45). Beyond iron acquisition and pyrimidine biosynthesis, little is known about the nutritional growth requirements of intracellular Histoplasma yeasts.
To identify additional factors that enable intracellular growth, we performed a genetic screen for Histoplasma insertion mutants that are unable to replicate in macrophages. Characterization of an isolated riboflavin auxotroph whose intramacrophage growth is impaired indicates that riboflavin is limiting in the Histoplasma-containing phagosome. Further exploration of additional candidate vitamin pathways suggests not only that Histoplasma is capable of synthesizing most essential vitamins but that de novo vitamin biosynthesis is necessary for full virulence of Histoplasma yeasts.
MATERIALS AND METHODS
Histoplasma strains and culture.
Histoplasma capsulatum strains were derived from the wild-type strain G217B (ATCC 26032) and are listed in Table 1. Histoplasma yeasts were grown in Histoplasma-macrophage medium (HMM) (46) or modified 3M medium (6, 46) lacking all vitamins except thiamine. Yeast cultures were grown with continuous shaking (200 rpm) at 37°C until late log/early stationary phase. For growth of uracil auxotrophs, media were supplemented with 100 μg/ml uracil. For growth of vitamin auxotrophs, 3M basal medium was supplemented with 200 nM riboflavin, 2.5 μM calcium pantothenate, 400 nM biotin, or a cocktail of all vitamins (400 nM biotin, 2.5 μM calcium pantothenate, 500 μM myoinositol, 5 μM nicotinic acid, 200 nM riboflavin, and 2 μM thiamine-HCl). For testing of biotin auxotrophs, it was necessary to fully deplete cells of residual biotin by growth in biotin-lacking medium until cells reached saturation, followed by dilution of cultures into fresh medium with and without supplemental biotin. Growth rates of yeasts in liquid culture were determined by measurement of culture turbidity (absorbance at 595 nm). Precise enumeration of yeasts was done by hemacytometer counts. For growth on solid medium, HMM was solidified with 0.6% agarose and supplemented with 25 μM FeSO4 and additional vitamins where appropriate.
TABLE 1.
Histoplasma strains
| Straina | Genotype | Other description |
|---|---|---|
| WU15b | ura5-42Δ | WT |
| OSU11 | ura5-42Δ rib2::T-DNA [hph] | rib2 |
| OSU32c | ura5-42Δzzz::T-DNA [hph PTEF1-gfp] | WT |
| OSU75 | ura5-42Δ/pCR468 [URA5 PH2B-gfp:FLAG] | RIB2 |
| OSU73 | ura5-42Δ rib2::T-DNA [hph]/pCR468 [URA5 PH2B-gfp:FLAG] | rib2 |
| OSU83 | ura5-42Δ rib2::T-DNA [hph]/pCR543 [URA5 PH2B-RIB2:FLAG] | rib2/RIB2 |
| OSU180 | ura5-42Δzzz::T-DNA [hph PTEF1-gfp]/pCR473 [URA5 gfp-RNAi] | gfp-RNAi |
| OSU181 | ura5-42Δzzz::T-DNA [hph PTEF1-gfp]/pAG09 [URA5 gfp:PAN6-RNAi] | PAN6-RNAi |
| OSU182 | ura5-42Δzzz::T-DNA [hph PTEF1-gfp]/pAG12 [URA5 gfp:BIO2-RNAi] | BIO2-RNAi |
Generation and mapping of Histoplasma T-DNA insertional mutants.
Histoplasma yeasts were mutagenized using Agrobacterium-mediated transformation (47–49). Agrobacterium tumefaciens strain LBA1100 harboring plasmid pCM41 was cocultured with WU15 Histoplasma yeasts for 48 h and transferred to HMM plus uracil medium containing 100 μg/ml hygromycin to select for Histoplasma transformants. Individual transformants were picked into HMM plus uracil medium in wells of a 96-well plate. After growth to saturation, the average yeast culture density was estimated by counting four representative wells of the 96-well plate by hemacytometer. Monolayers of P388D1 lacZ-expressing macrophage cells (48) were then inoculated with approximately 4 × 104 yeasts per well (representing a multiplicity of infection [MOI] of 1:1). After 7 days, surviving macrophages were lysed, and the β-galactosidase activity was determined to identify Histoplasma mutants deficient in macrophage killing. Mutants failing to lyse at least 50% of the macrophages were retained as candidate attenuated strains. Before further analysis, mutants were single colony purified.
The location of the T-DNA insertion was determined by thermal asymmetric interlaced PCR (TAIL-PCR) (47–50). Two hundred nanograms of genomic DNA was used as the template for the primary PCR, with a T-DNA left border (LB)- or right border (RB)-specific primer (LB11 or RB9) and one of four semirandom primers (LAD1 to -4). The primary PCR was diluted 1,000-fold and used as the template for the secondary PCR with nested LB or RB primers (LB12 or RB10) and AC1 primer. PCR products were sequenced and aligned to the Histoplasma genome sequence. T-DNA insertion at the RIB2 locus was confirmed by PCR and sequencing using RIB2-specific primers in conjunction with LB11 and RB9. Primer sequences are listed in Table 2.
TABLE 2.
Primers used in this work
| Primer | Primer sequence (5′ → 3′) | Directiona |
|---|---|---|
| TEF1-4 | CTGCCACACTGCCCACATTGCTTGC | Forward |
| TEF1-5 | GCCTTGGTGACCTTGCCAGCAG | Reverse |
| GDH1-10 | CTGTGGGAGGGCGTATTGACTG | Forward |
| GDH1-11 | GAAGCTTTCTTTGCCGTCCGTG | Reverse |
| RIB1-1 | GGAATGACACCGTTGAGGCCA | Forward |
| RIB1-2 | TTCTGTCCGCCTGATCGCCAT | Reverse |
| RIB2-10 | GGGCAGTCGGTGGGTTGTAAG | Forward |
| RIB2-11 | CCACGCTCCTCATCTCCCACC | Reverse |
| RIB2-1 | AGGCGCGCCAATGACCAACCCGTCCCTGCC | Forward |
| RIB2-2 | GCACTAGTAATATCAGCCTCCATCATCCTTTTC | Reverse |
| RIB3-1 | GAGGATGGAGAGGAGGTAGAAGGG | Forward |
| RIB3-2 | CCACCTTCCCTTCCAATCCCT | Reverse |
| RIB4-7 | GAAGGCGTTGAGAGAGGCGG | Forward |
| RIB4-8 | GGTCAGGGAGGCGGCATAGAT | Reverse |
| RIB5-7 | GCGGTGGCGGAACGTCTTTG | Forward |
| RIB5-8 | TAAGTTGGTGCGGCGTAGGGT | Reverse |
| RIB7-1 | GAACTGCCAAGGAAGGGAAGGG | Forward |
| RIB7-2 | CGCTGGTGATTCCCTCGGTTC | Reverse |
| PAN5-7 | ACCCTTGCCGTGAGTGTGATGG | Forward |
| PAN5-8 | GAGCCAACACCATCTGAACCACC | Reverse |
| PAN6-7 | CAAGGAAGCGGAAAGGGCGT | Forward |
| PAN6-5 | CAGCACCACTCAAGACGGCAC | Reverse |
| ECM31-1 | CGAGTGAGGTGTGCTTGGACGAG | Forward |
| ECM31-2 | GAGACGAATAGCGGAAGCAACG | Reverse |
| TDNA-15 | GAACTCTCAAGCCTACAGGACACACATTCATCG | Forward |
| LB11 | CCAAAATCCAGTACTAAAATCCAGATCCCCCGA | |
| LB12 | CGGCGTTAATTCAGTACATTAAAAACGTCCGCA | |
| LB14 | ACGTCCGCAATGTGTTATTAAG | |
| RB9 | CCGCACCGATCGCCCTTCCCAACAG | |
| RB10 | GCCTGAATGGCGAATGCTAGAGCAGCTTG | |
| RB11 | GCTAGAGCAGCTTGAGCTTGGATCAGATTGTC | |
| LAD-1 | ACGATGGACTCCAGAGCGGCCGCVNVNNNGGAA | |
| LAD-2 | ACGATGGACTCCAGAGCGGCCGCBNBNNNGGTT | |
| LAD-3 | ACGATGGACTCCAGAGCGGCCGCVVNVNNNCCAA | |
| LAD-4 | ACGATGGACTCCAGAGCGGCCGCBDNBNNNCGGT | |
| AC1 | ACGATGGACTCCAGAG |
Direction relative to gene transcription.
Biosynthetic pathway predictions and identification of enzyme homologs.
Pathway mapping and identification of biosynthetic enzyme homologs encoded in the Histoplasma G217B genome were determined using the KEGG Automatic Annotation Server (KAAS) (51). Metabolic pathways of Histoplasma were predicted based on the KEGG database of Saccharomyces cerevisiae KEGG modules, and the predicted orthologs in Histoplasma were assigned a KEGG orthology entry. Riboflavin, pantothenate, and biotin biosynthesis genes in Histoplasma were further investigated and confirmed by top BLAST search results to the Saccharomyces cerevisiae or Aspergillus nidulans genome from the National Center for Biotechnology Information database. Reciprocal top-hit BLAST searches were performed using the Saccharomyces or Aspergillus proteins as queries against the Histoplasma genome. Orthology was inferred if the same genes emerged as the top hits in reciprocal BLAST searches. Precise Histoplasma gene structures were determined by extraction of the orthologous gene from a G217B transcriptome sequencing (RNA-seq) database (52).
Complementation of the rib2 mutation.
The wild-type RIB2 coding sequence was amplified by PCR from reverse-transcribed RNA from Histoplasma G217B using RIB2-specific primers (RIB2-1 and RIB2-2) (Table 2). The RIB2 PCR product was cloned into the URA5-based plasmid pCR468 (6), replacing the gfp transgene and fusing the RIB2 coding sequence (CDS) with sequence encoding a C-terminal FLAG epitope. The rib2::T-DNA mutant (OSU11) was transformed with either linearized gfp (pCR468) or RIB2 (pCR543) expression plasmids by electroporation (39). Ura+ transformants were selected by plating on solid HMM.
Depletion of Pan5 and Bio2 functions by RNAi.
Pan5 and Bio2 functions were depleted from Histoplasma yeasts by transformation of the gfp-expressing sentinel strain OSU32 (53) with appropriate RNA interference (RNAi) vectors. RNAi vectors for PAN6 and BIO2 were created by amplification of a 778-bp region of PAN5 (nucleotides [nt] 164 to 942) or an 884-bp region of BIO2 (nt 165 to 1049) by PCR. Forward and inverted copies were cloned into the sentinel-RNAi vector pCR473 (53). Linearized plasmids were transformed into Histoplasma strain OSU32, and Ura+ transformants with reduced fluorescence of the green fluorescent protein (GFP) sentinel were selected on HMM supplemented with pantothenate and biotin. Transformants were patched, and GFP fluorescence was quantified using a modified gel documentation system (53) and the Image J software program (v1.44p; http://imagej.nih.gov/ij). PAN5- and BIO2-RNAi line auxotrophies were validated by growth in media lacking vitamins and rescued in medium supplemented with pantothenate or biotin.
qRT-PCR.
Histoplasma RNA was isolated from wild-type G217B yeasts in vitamin-lacking or vitamin-supplemented 3M medium. Yeast cells in exponential-phase growth were collected by centrifugation (2,000 relative centrifugal force [RCF]) and lysed by mechanical disruption with 0.5-μm-diameter glass beads in Ribozol reagent (Amresco, Inc.). RNA was recovered from lysates by CHCl3 extraction and precipitation of the aqueous phase with isopropanol. Total RNA was digested twice with DNase, and removal of residual genomic DNA was validated by lack of PCR amplification when used as the template. Five micrograms of total RNA was reverse transcribed with Superscript III reverse transcriptase (Invitrogen) using 250 ng random pentadecamers for priming reverse transcription. Vitamin synthesis gene expression was determined by quantitative PCR (qPCR) using a SYBR green-based PCR amplification mix (Bioline) and 1 μl of a 1:5 dilution of reverse-transcribed RNA (RT-RNA) as the template. Changes in transcript levels relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (54) in vitamin-containing or vitamin-deficient medium were determined using the ΔΔCT method (55) after normalization of cycle thresholds to expression of the transcription elongation factor 1 (TEF1) gene and comparison to GAPDH cycle thresholds. Gene-specific quantitative reverse-transcription PCR (qRT-PCR) primer sequences are listed in Table 2.
Macrophage culture and infection.
The lacZ-transgenic derivative of the macrophage cell line P388D1 was used for host cells for Histoplasma yeasts. Cells were maintained in Ham's F-12 medium with 10% fetal bovine serum (FBS) and 2 mM l-glutamine at 37°C under 5% CO2–95% air. For infections, macrophages were seeded in wells of a 96-well plate at 4 × 104 macrophages per well. After adherence, the medium was replaced with suspensions of yeasts in HMM-M (HMM buffered to pH 7.2 with 25 mM bicarbonate) at an MOI of 1:1. Plates were agitated for 60 s at 1,000 rpm twice daily during the time course of the assay. At 7 days postinfection, surviving macrophages were quantified by measurement of the residual β-galactosidase activity (48). To determine yeast cell viability and growth in macrophages, P388D1 cells were infected in 96-well plates at an MOI of 1:1. At 24, 48, 72, and 96 h postinfection, macrophages were lysed by addition of H2O and serial dilutions of the lysate were plated on solid HMM with riboflavin to measure viable yeast CFU. To test rescue of the mutant phenotype by riboflavin, medium was supplemented with 500 nM riboflavin 5 h after initial infection.
Yeast association index determination.
To assess association of yeast with macrophages, 1 × 105 P388D1 cells were adhered onto 12-mm acid washed coverslips, and 1 × 105 yeasts were added for 2 h at 37°C. Unattached yeasts were removed by washing with phosphate-buffered saline (PBS). Cells and adherent yeasts were fixed in 3% paraformaldehyde and permeabilized with 0.2% saponin in 3% bovine serum albumin (BSA), and yeasts were stained with Uvitex 3BSA in PBS (Ciba-Geigy). Macrophages and yeasts were imaged by collecting a Z-stack of differential interference contrast (DIC) and Uvitex fluorescence images, respectively, using an Eclipse Ti epifluorescence microscope (Nikon) with a 1.4-megapixel charge-coupled-device (CCD) camera (CoolSnap Hq2; Photometrics). Approximately 300 macrophages were scored, and the total number of associated yeasts was tallied. The association index is reported as the ratio of yeasts to macrophages. Three separate experiments were performed to derive the mean association index.
Determination of in vivo virulence.
C57BL/6 mice (NCI) were infected with Histoplasma yeast by intranasal delivery of approximately 1 × 104 yeast cells in PBS collected from exponentially growing liquid cultures. For the rib2 mutant experiment, strains were grown in HMM before infection. For infections with RNAi strains, yeasts were grown in 3M with pantothenate (PAN6-RNAi strain) or basal 3M (BIO2-RNAi strain), in which the biotin auxotroph can grow for a single passage. Actual inocula delivered were determined by plating of serial dilutions of the inoculum suspensions for enumeration of CFU. At day 4 and day 8 postinfection, mice were euthanized and lungs and spleens collected. Lung and spleen tissues were homogenized in 5 ml and 3 ml HMM, respectively, and serial dilutions of the homogenates were plated on solid HMM supplemented with vitamins to determine the fungal burden in each organ. Mean CFU counts were compared between infections by using a one-tailed Student t test for statistical significance. Potential reversion of PAN6-RNAI or BIO2-RNAi yeasts to vitamin prototrophy following infection was determined by patching individual colonies recovered from mice onto solid 3M medium lacking pantothenate or biotin. For histology, lungs were removed from mice after 8 days of infection with RIB2, rib2, and rib2/RIB2 yeasts and fixed in 10% neutral buffered formalin. Tissue sections were stained with hematoxylin and eosin (H&E) and interpreted by a board-certified pathologist (Comparative Pathology and Mouse Phenotyping Facility, Ohio State University College of Veterinary Medicine). All animal experiments were performed in compliance with the National Research Council's Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Ohio State University (protocol 2007A0241).
Immunoblotting.
Yeast cellular lysates were prepared by mechanical disruption of yeasts with 0.5-μm-diameter glass beads (4 rounds of 30 s [each] interspersed with 30-s rests on ice). Lysates were clarified by centrifugation (10 min at 14,000 RCF), and the protein yield was determined by Bradford assay. Twenty micrograms of denatured total protein was separated by electrophoresis through 10% polyacrylamide and transferred to a nitrocellulose membrane. The membrane was blocked with 4% nonfat milk in Tris-buffered saline (TBS) with 0.05% Tween 20 followed by addition of anti-FLAG antibody (F1804; Sigma). Antibody detection was performed by incubation with horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody and addition of HRP chemiluminescent substrate (Millipore).
RESULTS
Isolation of a Histoplasma riboflavin auxotroph from a genetic screen for attenuated mutants.
To identify Histoplasma factors required for virulence in macrophages, Histoplasma yeasts were mutagenized and screened for impaired ability to lyse macrophages. Agrobacterium-mediated transformation was used as the insertional mutagen, transferring and integrating into the Histoplasma genome a T-DNA element carrying a hygromycin resistance cassette (pCM41 [56]). Individual hygromycin-resistant transformants were used to infect monolayers of lacZ-expressing P388D1 macrophages, with which LacZ activity can be used to quantify surviving macrophages after infection (48). Seven days postinfection, LacZ activity was measured to identify wells with significant numbers of remaining macrophages, which indicates impairment of the yeasts' capacity to lyse host cells. Mutant 9E2 had an 84% decrease in macrophage killing (86% remaining macrophages, versus 14% remaining macrophages when infected with wild-type yeasts). This mutant was selected for further characterization and designated OSU11.
The T-DNA insertion in OSU11 was mapped to identify the disrupted locus. TAIL-PCR was used to obtain genomic sequences flanking the T-DNA insertion left and right borders. Sequencing of the TAIL-PCR product revealed that the T-DNA element was inserted into the gene encoding a homolog of the S. cerevisiae Rib2 protein. The Histoplasma RIB2 locus consists of a single 873-bp exon whose product has 26.1% amino acid identity (33.3% similarity) to the C-terminal 171 amino acids of the S. cerevisiae Rib2 protein (Table 3). The S. cerevisiae Rib2 protein is a dual-function protein with an N-terminal domain (amino acids 140 to 431) containing a pseudouridine synthase function and a C-terminal domain (amino acids 439 to 591) containing diaminohydroxyphosphoribosylaminopyrimidine (DRAP) deaminase activity. The C-terminal domain is essential for the synthesis of riboflavin (57). Unlike the gene corresponding to the S. cerevisiae protein, the Histoplasma RIB2 gene encodes only a DRAP deaminase, while a separate gene in the Histoplasma genome shows similarity to that encoding the Saccharomyces Rib2 pseudouridine synthase domain. The T-DNA element in OSU11 is located 194 bp downstream of the initiation codon and deletes 7 bp of the RIB2 coding region (Fig. 1A). Characterization of the T-DNA element ends revealed truncations of 10 bp and 2,477 bp from the left and right borders of the T-DNA element, respectively (Fig. 1A).
TABLE 3.
Riboflavin and pantothenate biosynthesis pathway enzymes in Histoplasma
| Protein | Enzyme name | EC no. | S. cerevisiae GenBank accession no. | BLAST e-value (S. cerevisiae to H. capsulatum) | BLAST e-value (H. capsulatum to S. cerevisiae) | Identitya (%) | Similaritya (%) |
|---|---|---|---|---|---|---|---|
| Rib1 | GTP cylcohydrolase II | 3.5.4.25 | NP_009520.1 | 3E−79 | 9E−98 | 37.4 | 47.0 |
| Rib2 | 2,5-Diamino-6-ribitylamino-4(3H)-pyrimidinone 5′-phosphate deaminase | 5.4.99.- | NP_014575.1 | 5E−23 | 4E−16 | 26.1b | 33.3b |
| Rib3 | 3,4-Dihydroxy-2-butanone 4-phosphate synthase | 4.1.99.12 | NP_010775.1 | 6E−47 | 1E−54 | 41.1 | 57.3 |
| Rib4 | Lumazine synthase | 2.5.1.78 | NP_014498.1 | 9E−37 | 8E−45 | 41.4 | 57.5 |
| Rib5 | Riboflavin synthase | 2.5.1.9 | NP_009815.1 | 6E−63 | 6E−79 | 47.7 | 67.8 |
| Rib7 | 2,5-Diamino-6-ribosylamino-4(3H)-pyrimidinone 5′-phosphate reductase | 1.1.1.302 | NP_009711.3 | 9E−40 | 3E−46 | 32.3 | 42.2 |
| Ecm31 | 3-Methyl-2-oxobutanoate hydroxymethyl-transferase | 2.1.2.11 | NP_009735.3 | 6E−47 | 1E−57 | 35.0 | 55.7 |
| Pan5 | 2-Dehydropantoate 2-reductase | 1.1.1.169 | NP_011930.1 | 9E−10c | 1E−20 | 19.2 | 34.3 |
| Pan6 | Pantothenate synthetase | 6.3.2.1 | NP_012121.2 | 1E−55 | 3E−67 | 38.2 | 49.5 |
Identity and similarity using the BLOSUM92 matrix.
Alignment to the C-terminal deaminase domain of S. cerevisiae Rib2 (amino acids 420 to 591).
Aligned to the G217B genome, since no PAN5 gene was found in the ab initio gene predictions.
FIG 1.
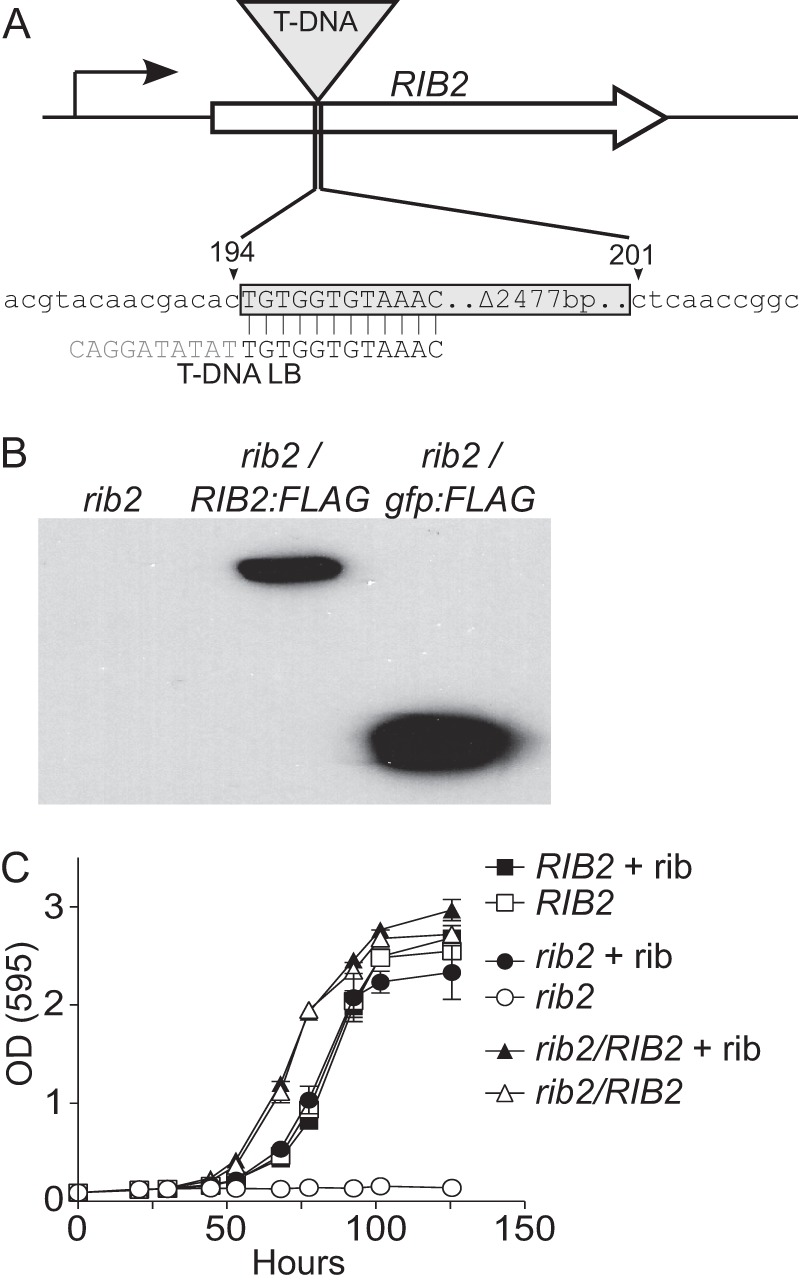
T-DNA disruption of RIB2 causes riboflavin auxotrophy. (A) Schematic representation of mapping results showing that the T-DNA insertion in attenuated mutant OSU11 is located in the coding sequence of the RIB2 gene. The T-DNA element is inserted 194 bp downstream of the RIB2 start codon and deleted 7 bp of RIB2 sequence. T-DNA integration was associated with truncations of 10 bp and 2,477 bp from the left and right borders of the element, respectively. (B) Complementation of the rib2 mutant allele. The rib2::T-DNA mutant (OSU11) was transformed with an empty vector (lane 1), a vector expressing the RIB2 gene fused to sequences encoding a C-terminal FLAG epitope (RIB2:FLAG; OSU83; lane 2), or a vector expressing gfp with a C-terminal FLAG epitope (gfp:FLAG; OSU73; lane 3). RIB2 or gfp transgene expression was confirmed by immunoblotting of Histoplasma cellular lysates with antibodies to the FLAG epitope. (C) Phenotypic confirmation of riboflavin auxotrophy and rib2 complementation. The graph shows the growth of the Rib2 prototroph (OSU75; RIB2), the rib2::T-DNA mutant (OSU73; rib2), and the complemented strain (OSU83; rib2/RIB2) during broth culture in minimal medium. Strains were grown in medium with (+ rib; filled symbols) and without (open symbols) 200 nM riboflavin supplementation. Yeast growth was monitored by increasing culture turbidity at 595 nm over time. Data points represent the mean turbidity measurement ± standard error for replicate cultures (n = 3).
To demonstrate that disruption of the Histoplasma RIB2 locus was responsible for the mutant phenotypes, the OSU11 strain was transformed with a RIB2-complementing plasmid. The entire RIB2 coding sequence was amplified by PCR from wild-type Histoplasma and inserted in frame with sequences for a C-terminal FLAG epitope tag. Constitutive expression of the transgene was provided by the Histoplasma constitutive H2B promoter (53). The RIB2 expression plasmid (pCR543) or the parental plasmid expressing gfp instead of RIB2 (pCR468) was transformed into OSU11. Immunoblotting of cellular lysates of transformants with an antibody to the FLAG epitope confirmed expression of the gfp and RIB2 transgenes (Fig. 1B).
To test if the T-DNA insertion in RIB2 caused an auxotrophy for riboflavin, we examined the growth of the mutant and complemented yeast strains in minimal medium with or without supplemental riboflavin (Fig. 1C). The parental strain that has a wild-type RIB2 allele grows normally in defined minimal medium lacking riboflavin; however, the rib2::T-DNA mutant is unable to grow. Complementation of the rib2 mutant with RIB2 completely restores its ability to grow in the absence of riboflavin, confirming that the mutation of the RIB2 gene is the source of the defect. Supplementation of the medium with riboflavin restores normal growth kinetics to the rib2 mutant, indicating the T-DNA mutation-induced growth defect results from riboflavin auxotrophy. Addition of riboflavin to growth medium shows a dose-response rescue of the mutant growth, with 200 nM riboflavin being sufficient for normal growth (see Fig. S1 in the supplemental material).
Histoplasma virulence requires riboflavin synthesis.
We used the rib2 riboflavin auxotroph to probe whether the phagosomal environment lacked riboflavin and if riboflavin synthesis is necessary for intramacrophage yeast growth. With the riboflavin auxotrophy defined, we examined yeast survival and proliferation within macrophages (Fig. 2A). Unlike the RIB2 parental line or the RIB2-complemented strain, which replicated intracellularly at least 25-fold over 48 h and increased up to 96 h, the rib2 mutant showed only a 2.5-fold increase over 48 h with no increase in recoverable CFU after the first day. To determine if the lack of intracellular proliferation of rib2 mutant yeasts prevents yeast-dependent lysis of the macrophages, we quantified macrophage survival after infection with Histoplasma yeast (Fig. 2B). Consistent with their lack of replication, rib2 mutant yeasts had diminished ability to lyse host cells, since 87% of the macrophages were still surviving. Complementation of the rib2 mutation restored yeast virulence to that of the wild type (ranging from 13 to 21% macrophage survival), confirming that insertion of the T-DNA in the RIB2 gene was responsible for the attenuated virulence of the mutant recovered from the screen. To further demonstrate that riboflavin synthesis is necessary for efficient intracellular replication, excess exogenous riboflavin was added to infected macrophages (Fig. 2B). Addition of riboflavin partially restores virulence of the rib2 mutant yeasts, with 32% of macrophages surviving. Attenuation of the rib2 mutant is not due to failure of yeasts to infect macrophages, since the association indices of RIB2, rib2, and rib2/RIB2 yeasts with macrophages are not significantly different (see Fig. S2 in the supplemental material). Together these tests demonstrate that the intracellular environment of Histoplasma yeasts lacks sufficient riboflavin to support yeast growth and thus intracellular replication requires de novo riboflavin synthesis.
FIG 2.
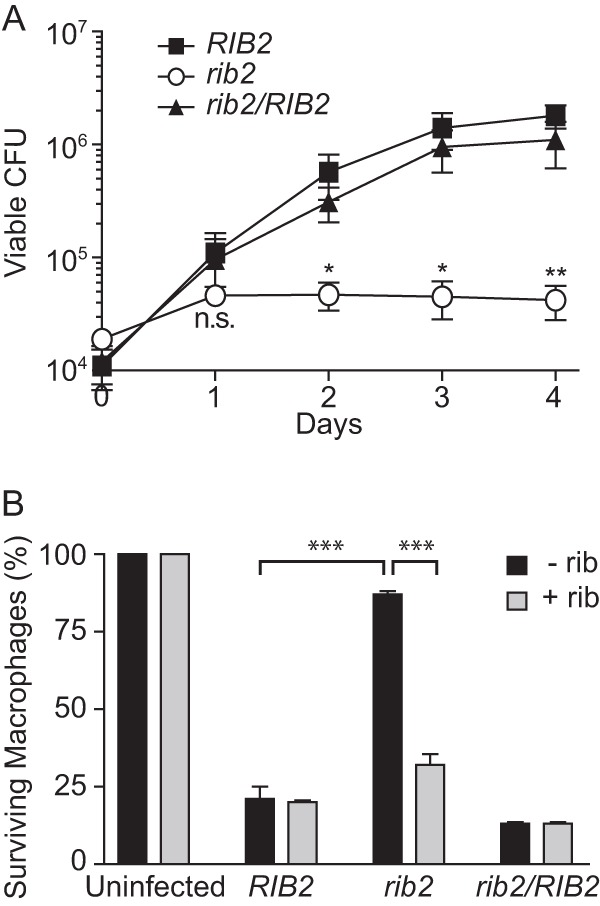
Riboflavin synthesis is required for intracellular proliferation of Histoplasma yeasts. (A) Viability and replication of intracellular Histoplasma yeasts after infection of macrophages. Macrophages were infected with RIB2 (OSU75), rib2 (OSU73), and rib2/RIB2 (OSU83) isogenic strains. Yeasts were recovered from infected macrophages at 1, 2, 3, and 4 days postinfection by lysis of the macrophages, and viability of the intracellular yeasts was determined by plating of lysates and enumerating CFU. Data points represent the means ± standard errors for replicate experiments (n = 4), each performed in triplicate. Statistically significant differences in yeast growth compared to that of the RIB2 strain at each day were determined by Student's t test and are indicated with asterisks (n.s., nonsignificant; ∗, P < 0.05; ∗∗, P < 0.01). (B) Virulence of RIB2 (OSU75), rib2 (OSU73), and rib2/RIB2 (OSU83) yeasts as determined by their ability to cause macrophage lysis. Transgenic lacZ-expressing macrophages infected with Histoplasma yeasts were quantified at 7 days postinfection by measuring the remaining macrophage-expressed LacZ activity. Infections were performed in HMM-M, which contains 0.1 μM riboflavin with supplementation with 0.5 μM riboflavin (+ rib) or without additional riboflavin supplementation (− rib). Data represent the means ± standard errors of the relative percentages of surviving macrophages after normalization to uninfected macrophage populations. Data were averaged from separate experiments (n = 3), each performed in triplicate. Statistically significant differences between infections with the RIB2 and rib2 strains without riboflavin supplementation and between media with and without riboflavin supplementation for the rib2 infection were determined by Student's t test and are indicated by asterisks (∗∗∗, P < 0.001).
To determine if the intramacrophage growth deficiency of the rib2 auxotroph translates into attenuated virulence in vivo, we examined the infection kinetics of the rib2 mutant in mice. Increasing fungal burden in lungs following sublethal respiratory infection was used as the in vivo model of histoplasmosis (Fig. 3A and B). Wild-type Histoplasma yeasts infected and replicated in murine lungs, increasing 181-fold by 8 days. In contrast, the rib2 riboflavin auxotroph burden failed to increase in the lung. Following an initial 10-fold drop in the fungal burden, lung colonization by the rib2 mutant neither increased nor decreased, consistent with a defect in proliferation but not persistence. As an indicator of dissemination to peripheral organs, the fungal burden in spleens was determined at day 8 postinfection (Fig. 3C). Paralleling the lung burdens, the wild-type RIB2 strain shows dissemination to the spleen, but dissemination of the rib2 mutant was virtually absent. Complementation of the rib2 mutant restores proliferation within the lungs, as well as dissemination to the spleen, close to wild-type levels.
FIG 3.

Histoplasma virulence in vivo requires riboflavin biosynthesis. Fungal burdens in lung (A and B) or spleen (C) tissue following respiratory infection with Histoplasma are shown. Wild-type C57BL/6 mice were infected intranasally with RIB2 (OSU75), rib2 (OSU73), and rib2/RIB2 (OSU83) Histoplasma yeasts. The number of viable yeasts in lungs was determined at 4 (A) and 8 (B) days postinfection by plating of lung homogenates for yeast CFU. Extrapulmonary dissemination was determined at 8 days postinfection by plating of spleen homogenates for yeast CFU (C). Data points represent the CFU counts per organ for each mouse (n = 5) at each time point. The dashed line represents the inoculum size. Horizontal bars represent the mean CFU counts, and asterisks denote statistically significant differences (∗, P < 0.05; ∗∗∗, P < 0.001) between the rib2 strain infections and either the RIB2 or rib2/RIB2 strain infections as determined by Student's t test. The lower limit of detection of the assay is 100 CFU. (D and E) Lung tissue pathology after 8 days of infections with RIB2 (OSU75), rib2 (OSU73), and rib2/RIB2 (OSU83) yeasts. Lung sections were stained with hematoxylin and eosin and imaged at low magnification (D) or high magnification (E) to ascertain the inflammatory composition and degree of pathology. Scale bars, 1 mm (D) or 100 μm (E).
Tissue pathology was also dramatically reduced in lungs infected with the rib2 mutant (Fig. 3D). Lungs infected with the RIB2 strain had 60% of the section effaced by multifocal to coalescing nodules of inflammation, including within the alveolar space (Fig. 3D). Inflammation was composed of neutrophils, foamy alveolar macrophages, and lymphocytes. Pleural disruption resulted in fibrin and fluid (edema) within alveolar spaces (Fig. 3E). Lungs from mice infected with the rib2/RIB2 yeasts showed pathology similar to that of those infected by the RIB2 strain with multifocal nodules of inflammation but to a slightly lesser degree (approximately 40% of the lobe). In contrast, only 2 inflammatory nodules (representing approximately 5% of the lung lobe) were noted in the mouse infected with the rib2 mutant strain (Fig. 3D). Inflammation was distributed around bronchioles and blood vessels but had minimal extension into the adjacent alveolar spaces. Thus, pulmonary inflammation mirrors the lung fungal burdens, with severe inflammation present in lungs infected with the RIB2 and rib2/RIB2 strains but with substantially reduced pathology in lungs infected with a rib2 strain that is unable to synthesize riboflavin de novo.
Histoplasma yeasts synthesize most vitamins de novo.
The growth of wild-type yeasts in minimal medium lacking all vitamins (Fig. 1C) suggests that Histoplasma yeasts are able to synthesize all essential vitamins. Essential vitamins for microbes include vitamin B1 (thiamine), vitamin B2 (riboflavin), vitamin B3 (niacin), vitamin B5 (pantothenic acid), vitamin B6 (pyridoxine), vitamin B7 (biotin), and vitamin B9 (folate). Previous studies of the nutritional requirements for Histoplasma yeast growth indicated that most strains do not require nutritional supplementation with essential vitamins with the possible exception of thiamine (58). Some strains were auxotrophic for niacin, pantothenate, and/or biotin, but all were able to grow without added riboflavin, folate, and pyridoxine. Given the potential variability, we explored in more detail the vitamin requirements of the wild-type G217B strain. Inoculation of defined medium lacking all vitamins with wild-type Histoplasma G217B resulted in normal growth kinetics, with the turbidity of the culture reaching a typical density (OD) at 595 nm of >2 (Fig. 4). However, when this vitamin-deprived culture was diluted back into medium without vitamins, yeast growth was poor and what growth was observed was mostly mycelial as determined by microscopy (data not shown). Supplementation of the medium with all essential vitamins or with only thiamine for the second round of growth was sufficient to restore normal growth (Fig. 4). Thus, repeated passage in medium lacking thiamine reveals that Histoplasma G217B yeasts are thiamine auxotrophs in liquid culture, but they are able to synthesize all other vitamins necessary for growth. Similar results showing de novo vitamin biosynthesis capability were found with the G186A strain and three fresh clinical isolates (two North American type 1 lineage isolates and one North American type 2 lineage isolate; data not shown).
FIG 4.
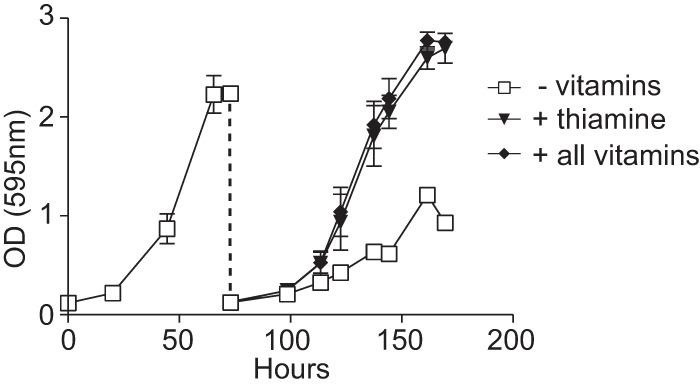
Histoplasma yeasts can synthesize all vitamins essential for growth except thiamine. Growth of Histoplasma yeasts in minimal medium with and without vitamin supplementation. Yeasts were pregrown in rich medium (HMM), washed in 3M medium lacking vitamins, and inoculated into 3M medium containing no vitamins (− vitamins) after pregrowth in HMM and yeast growth measured as increasing culture turbidity (absorbance at 595 nm) over time. At saturation, the culture was diluted into fresh 3M medium without vitamins (open squares), medium with 2 μM thiamine (filled triangles), or medium with all vitamins (filled circles; vitamins include biotin, folate, myoinositol, nicotinic acid, pantothenate, pyridoxine, and thiamine). Data points represent the mean culture turbidities ± standard errors for three biological replicate cultures.
Since Histoplasma yeasts can meet their vitamin requirements through de novo synthesis, we examined select biosynthetic pathways encoded in the genome and their regulation under exogenous vitamin-limiting conditions. Metabolic pathways and biosynthetic enzymes in Histoplasma were annotated using the KEGG Automatic Annotation Server (KAAS). Biosynthetic enzymes were investigated individually by a BLAST search of the G217B genome using well-studied S. cerevisiae enzymes as queries. To be classified as orthologous proteins, we required matches to be the top hit in reciprocal BLAST searches between Histoplasma and S. cerevisiae or Aspergillus nidulans. Orthologous proteins and their respective Enzyme Commission number are given in Table 3. Consistent with our earlier results identifying the RIB2 gene, we were able to identify all riboflavin biosynthetic enzymes in the Histoplasma genome which would produce riboflavin from the central metabolite GTP (see Fig. S3A in the supplemental material). We also identified the key enzymes in the pantothenate (see Fig. S3B) and biotin (see Fig. S3C) biosynthetic pathways. The relative expression levels (54) of the genes in the riboflavin and pantothenate biosynthetic pathways were relatively low, consistent with the small amount of vitamins required for growth (see Fig. S4). There was a small (less than 2-fold) but not statistically significant change in riboflavin, pantothenate, and biotin biosynthesis gene expression when exogenous vitamins were added (see Fig. S4).
Full Histoplasma virulence requires de novo synthesis of vitamins.
To determine whether the vitamin requirement for Histoplasma virulence is limited to riboflavin or if de novo vitamin synthesis is a general necessity for infection, we created strains that were auxotrophic for two additional vitamins. Pantothenate and biotin were selected as targeted pathways, since clear orthologs were identified (see Fig. S3 in the supplemental material) and the PAN6 and BIO2 gene products represent key control points at the terminal end of each pathway. Because construction of targeted gene knockouts in Histoplasma is difficult, we used RNA interference (RNAi) to deplete the Pan6 or Bio2 function (59). Sequences corresponding to the PAN6 or BIO2 gene were inserted into an RNAi vector which includes, in the inverted repeat, sequences for RNAi of gfp that function as a fluorescent sentinel for RNAi knockdown (53, 60). Linearized RNAi plasmids were transformed into the GFP-fluorescent strain OSU32, and transformants with strongly reduced GFP fluorescence (representing knockdown of the gfp sentinel and the cotargeted PAN6 or BIO2 gene) were selected. A PAN6-RNAi isolate and a BIO2-RNAi isolate showed 7.8-fold and 7.7-fold reduction in GFP fluorescence, respectively, similar to the level of GFP fluorescence of a gfp-RNAi line or a GFP-negative background, indicating good knockdown of the targeted PAN6 and BIO2 genes (Fig. 5A).
FIG 5.
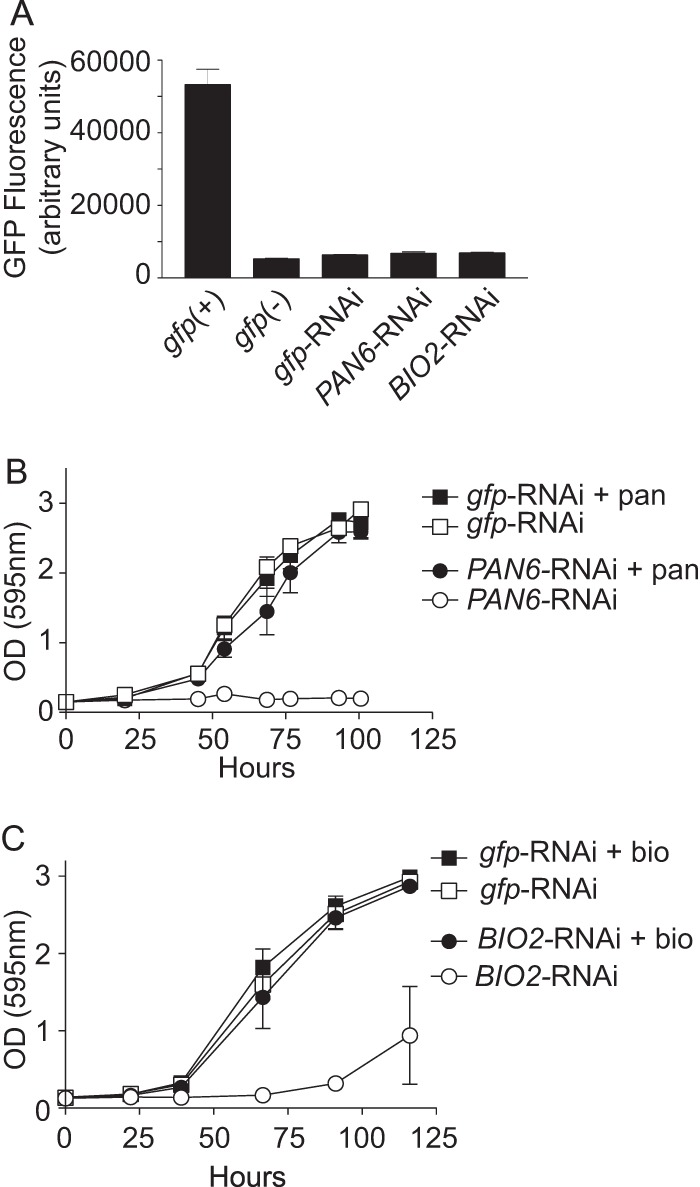
RNAi-based depletion of PAN6 and BIO2 creates pantothenate and biotin auxotrophies. (A) Knockdown of the GFP sentinel indicating knockdown of the cotargeted PAN6 or BIO2 gene. The graph shows the GFP fluorescence of gfp-RNAi (OSU180), gfp:PAN6-RNAi (OSU181), and gfp:BIO2-RNAi (OSU182) yeasts relative to the GFP+ parental background [gfp(+); OSU32] and GFP− [gfp(-); WU15] strains. Fluorescence values represent the mean GFP fluorescences ± standard errors for replicate patches (n = 3) on solid vitamin-containing medium. (B) Confirmation of pantothenate auxotrophy caused by PAN6-RNAi. The graph shows the growth of the pantothenate prototroph (gfp-RNAi; OSU180) and the gfp:PAN6-RNAi strain (PAN6-RNAi; OSU181) in minimal 3M medium lacking pantothenate (open symbols) or medium supplemented with 2.5 μM pantothenate (+ pan; filled symbols). Growth was monitored by measurement of culture turbidity at 595 nm over time. Data points represent the mean turbidities ± standard errors for replicate cultures (n = 3). (C) Confirmation of biotin auxotrophy caused by BIO2-RNAi. The graph shows the growth of the biotin prototroph (gfp-RNAi; OSU180) and the gfp:BIO2-RNAi strain (BIO2-RNAi; OSU181) in minimal 3M medium lacking biotin (open symbols) or minimal medium with 400 nM biotin (+ bio; filled symbols). For the biotin auxotrophy tests, yeasts were pregrown in 3M medium lacking all vitamins prior to inoculation. Yeast growth was monitored by measurement of culture turbidity (595 nm) over time. Data points represent the mean turbidities ± standard errors for replicate cultures (n = 3).
To confirm that RNAi of the PAN6 and BIO2 target genes sufficiently depleted the Pan6 and Bio2 functions, the RNAi lines were tested for pantothenate and biotin auxotrophy, respectively. In the absence of exogenous pantothenate, growth of the PAN6-RNAi line was virtually eliminated, whereas RNAi of gfp alone did not cause any pantothenate auxotrophy (Fig. 5B). Supplementation of the medium with 2.5 μM pantothenate completely rescued growth, confirming that PAN6-RNAi specifically depleted de novo pantothenate synthesis. In defined medium lacking biotin, growth of the BIO2-RNAi line was not impaired (data not shown); however, a second passage in medium lacking biotin revealed that the BIO2-RNAi line was unable to grow (Fig. 5C). The small amount of growth that was observed in the second passage without biotin was mostly mycelial, suggesting strong nutritional stress. Supplementation of the medium with biotin during the second round of growth rescued the growth of the BIO2-RNAi strain, indicating that the proliferation defect resulted from a biotin auxotrophy caused by BIO2-RNAi.
The successful creation and validation of strains unable to synthesize pantothenate or biotin enabled us to test if Histoplasma virulence in vivo requires de novo synthesis of pantothenate or biotin. Before infection of mice, the PAN6-RNAi line was grown in liquid medium containing 2.5 μM pantothenate and the BIO2-RNAi line was grown in liquid medium lacking biotin to deplete cells of residual biotin. Following respiratory infection, Histoplasma yeast harboring the gfp-RNAi plasmid increased to 2.5 × 105 yeasts/lung at 8 days postinfection (Fig. 6A). Histoplasma yeast unable to synthesize pantothenate due to the PAN6 knockdown increased only to 4.8 × 104 yeasts/lung, representing a 5-fold defect in lung infection compared to results for the gfp-RNAi control (Fig. 6A). The BIO2-RNAi biotin auxotroph was not statistically different from the gfp-RNAi control for lung infection (Fig. 6A). Infections with all strains led to dissemination, as evidenced by fungal burdens in the spleen at 8 days postinfection (Fig. 6B). Loss of pantothenate synthesis (PAN6-RNAi) caused the most profound defect, with 28.8-fold less yeast in the spleen than with control infections (gfp-RNAi). Despite equivalent lung burdens between the control and the BIO2-RNAi line, deficient biotin synthesis reduced the number of yeasts in the spleen by 3.6-fold.
FIG 6.
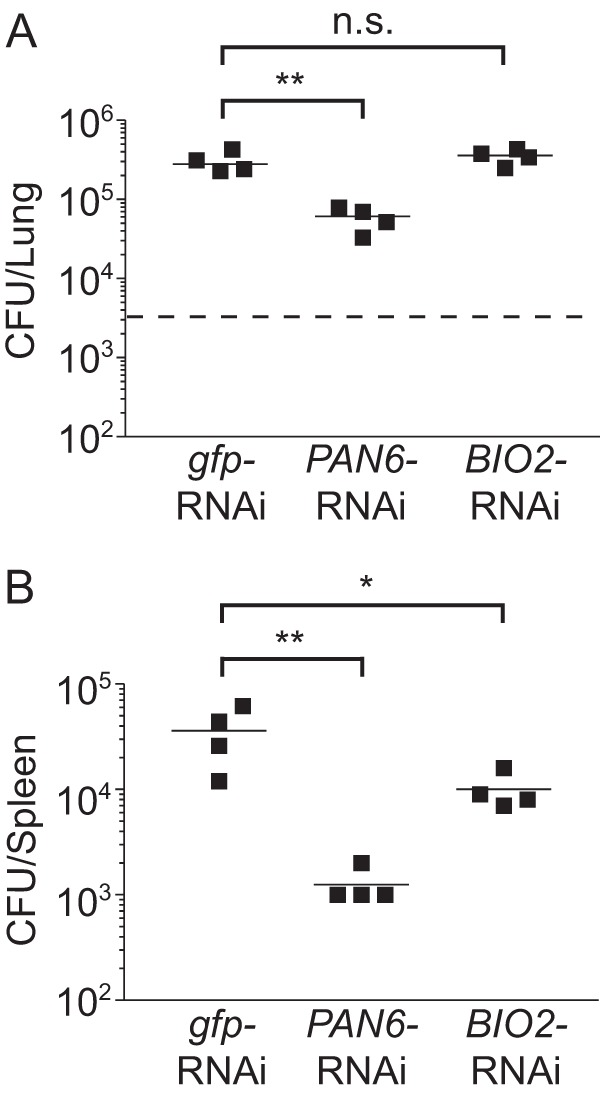
Full virulence of Histoplasma in vivo requires synthesis of pantothenate but not biotin. Fungal burdens in lung (A) or spleen (B) tissue at 8 days postinfection are shown. Wild-type C57BL/6 mice were infected intranasally with prototrophic yeast (gfp-RNAi; OSU180), the pantothenate auxotroph (PAN6-RNAi; OSU181), or the biotin auxotroph (BIO2-RNAi; OSU182). The number of viable Histoplasma yeasts in lung tissue (A) or spleen tissue (B) was determined at 8 days postinfection by plating of lung and spleen homogenates on vitamin-supplemented rich medium for yeast CFU. The dashed line represents the inoculum size. Data points represent the CFU counts per organ for each mouse (n = 4) at each time point. Horizontal bars represent the mean CFU counts, and asterisks denote statistically significant differences between the gfp-RNAi infections and the PAN6-RNAi or BIO2-RNAi infections as determined by Student's t test (n.s., nonsignificant; ∗, P < 0.05; ∗∗, P < 0.01). The lower limit of detection of the assay is 100 CFU.
Infection and replication in lungs by the PAN6-RNAi and BIO2-RNAi lines cannot be explained by loss of auxotrophy (i.e., due to loss of the RNAi effect). Inoculation of liquid medium lacking pantothenate or biotin using the same yeast suspensions used for murine infections confirmed that the PAN6-RNAi and BIO2-RNAi yeasts were still auxotrophic for pantothenate and biotin at the time of infection, respectively (data not shown). Passage of PAN6-RNAi or BIO2-RNAi yeasts in medium with vitamins shows a less than 2% unselected reversion rate (0 of 52) to vitamin prototrophy. Testing of individual yeast colonies recovered from the lungs of infected mice likewise showed minimal rates of reversion to pantothenate or biotin prototrophy through loss of target gene silencing. For the PAN6-RNAi line, 4.7% of the recovered colonies (2 out of 43) had regained pantothenate prototrophy. All prototrophic colonies had also regained fluorescence of the sentinel GFP reporter, indicating that the reversion to prototrophy stems from the loss of the RNAi effect. Similar results were seen for the BIO2-RNAi line, with a less than 2.2% reversion rate (0 of 44) after murine infection. Thus, the vast majority of yeasts recovered from murine infections maintain the pantothenate or biotin auxotrophy caused by RNAi knockdown of PAN6 or BIO2, respectively. These data show that full Histoplasma virulence in vivo requires de novo synthesis of pantothenate and that spleen infection is enhanced by de novo synthesis of biotin.
DISCUSSION
Pathogens that infect host cells and replicate within them face unique challenges for intracellular growth and replication. Successful pathogens of phagocytes are often characterized by expression of multiple defense systems designed to negate phagocyte reactive oxygen (6, 61–66). However, these strategies only provide for survival, and intracellular pathogens must acquire sufficient nutrition to provide for growth and replication. For intracellular pathogens that escape to the nutrient-rich cytosol, this is generally not a problem. However, those pathogens that establish their intracellular niche within a phagosome or vacuole-like compartment face significant nutritional challenges because the phagosome has limited sources for energy production and anabolic precursor molecules. Thus, intracellular growth requires acquisition of nutritional substrates and biosynthetic enzymes to synthesize those nutrients that are not readily obtained from the phagosome/vacuole lumen.
In this study, we showed that the Histoplasma-containing phagosome is limiting for some essential vitamins through isolation and characterization of vitamin auxotroph strains. Histoplasma yeasts grew in minimal medium lacking vitamins, consistent with an ability to synthesize the necessary vitamins to support growth in a vitamin-poor phagosome. The only exception was the inability to grow without thiamine, although this required repeated passage of cells in vitro to discover the thiamine auxotrophy. This thiamine auxotrophy suggests availability of host thiamine to Histoplasma yeasts during infection and/or the operation of sufficient thiamine scavenging pathways.
Riboflavin is essential for but unavailable to intracellular Histoplasma yeasts. Consequently, Histoplasma yeasts rely upon de novo riboflavin synthesis during infection of macrophages. Riboflavin is the precursor molecule of flavin nucleotides, which act as electron carriers for many metabolic and energy-generating processes of the cell. Consistent with the central role of flavins, Histoplasma yeasts unable to synthesize riboflavin are severely attenuated in vivo. The Histoplasma riboflavin auxotroph is not cleared, but the fungal burden also does not increase over time, consistent with an inability to generate energy and to synthesize cellular components necessary for yeast cell growth and replication. Even with full complementation in vitro, growth of the RIB2-complemented mutant is not fully restored in vivo. We suspect that expression of the complementing RIB2 gene from a constitutive promoter rather than the native RIB2 promoter may account for this discrepancy. Alternatively, other unknown changes to the genetic background that impact virulence may have occurred during the construction of the complemented strain. Nonetheless, in vivo growth of the rib2/RIB2 strain is increased by several logs over that of the rib2 mutant, indicating that the loss of de novo riboflavin synthesis attenuates virulence in vivo. This attenuation is also seen with macrophage infections in culture, since numbers of intracellular Histoplasma CFU do not increase without the ability to synthesize riboflavin. Thus, intramacrophage growth requires de novo riboflavin biosynthesis, despite the fact that the extracellular growth medium contains sufficient riboflavin to rescue a yeast riboflavin auxotrophy in vitro. If the growth medium on the macrophages is supplemented with a large amount of riboflavin, the intracellular growth of the Histoplasma RIB2 mutant is partially restored, likely due to some leakage into or trafficking of extracellular medium to the Histoplasma-containing phagosome. Riboflavin synthesis is also required for Candida albicans virulence, as evidenced by identification of conditional rib1 and rib2 mutants that have attenuated virulence in vivo (67). In contrast, Mycobacterium tuberculosis virulence does not require riboflavin biosynthesis (68).
Like riboflavin biosynthesis, the ability to synthesize pantothenate is required for full Histoplasma virulence. The decrease in virulence of the pantothenate knockdown strain is not as severe as that of the riboflavin mutant. This could result from some de novo pantothenate synthesis in the PAN6-RNAi line due to incomplete depletion of Pan6 by RNAi. However, RNAi of PAN6 causes a robust pantothenate auxotrophy in vitro, arguing that Pan6 function is adequately depleted by RNAi. Alternatively, PAN6-RNAi yeasts may be recovered from infected lungs, not because they are not attenuated but because they have lost the RNAi silencing and reverted to pantothenate prototrophy. While growth in vivo increases the selection pressure for pantothenate prototrophy, the reversion rate is still low, indicating that the majority of yeasts recovered from infected lungs are still pantothenate auxotrophs. Since the PAN6-RNAi auxotrophy is largely maintained, the less severe attenuation of the PAN6-RNAi strain in vivo may reflect reduced demand for de novo pantothenate synthesis. This may result either from the availability of a limited pantothenate supply in the host which can be scavenged by Histoplasma yeasts or a reduced requirement for pantothenate-dependent reactions. Since pantothenate is a precursor of coenzyme A and acyl carrier protein, cofactors that are central to many essential metabolic reactions, the most likely explanation is the availability of small amounts of pantothenate in vivo.
In contrast to the case with riboflavin and pantothenate, de novo biotin synthesis does not seem to be required by Histoplasma in vivo. Biotin serves as a cofactor for pyruvate carboxylase (leading to gluconeogenesis) and fatty acid synthesis enzymes. The in vitro biotin auxotrophy phenotype indicates sufficient knockdown of BIO2 by RNAi, but the phenotype required repeated passage of the BIO2-RNAi strain, suggesting that small amounts of biotin are sufficient for Histoplasma growth. In support of this, we found that as little as 3 nM biotin is sufficient to restore in vitro growth to the biotin auxotroph (see Fig. S1C in the supplemental material). We suspect that Histoplasma encounters small amounts of biotin in vivo and the efficiency with which biotin is used or scavenged frees Histoplasma yeast from the need for de novo biotin synthesis.
Similar to our results in Histoplasma, pantothenate auxotrophs of Mycobacterium tuberculosis and Candida albicans also have attenuated virulence (67, 69). This suggests that pantothenate limitation is a common feature of intracellular pathogens. Pantothenate auxotrophs of Francisella tularensis do not have decreased virulence (70), and Listeria, which is incapable of synthesizing some essential vitamins, also is not impaired in intracellular proliferation (71). However, these intracellular pathogens escape into the host cell cytosol, where nutrients are plentiful, eliminating the need for biosynthesis of pantothenate and other vitamins. In contrast to the results in Histoplasma, both Mycobacterium tuberculosis and Candida biotin auxotrophs are impaired in intracellular replication (11, 67, 68, 72). Furthermore, initial escape from the phagosome for Francisella also requires biotin biosynthesis (73). On the other hand, Salmonella biotin and pantothenate auxotrophs are not attenuated (74). These results indicate the diversity of phagosomal compositions for different pathogens and/or different metabolic requirements for pantothenate and biotin-derived cofactors. Nonetheless, the necessity of de novo vitamin biosynthesis appears to characterize compartmentalized pathogens and reflects the paucity of different vitamins in the phagosome.
The generation of mutant Histoplasma strains deficient for vitamin synthesis not only provides tools for probing the nutrient composition of the phagosome but also indicates that vitamin biosynthetic enzymes can be exploited as drug targets for histoplasmosis.
Due to the eukaryotic similarities between host cells and yeast cells, the number of druggable targets for fungal pathogens is low. Although vitamins are essential for both fungal and host cells, mammalian cells acquire vitamins through diet and do not rely on de novo synthesis. While the host environment has ample vitamins, these same nutrients are unavailable to intraphagosomal yeasts, and thus de novo synthetic capability could be exploited as a selective target for intraphagosomal pathogens without detrimental effects on the hosts. The variation in the vitamins available and required by different pathogens highlights the need to firmly establish whether a specific auxotrophy impairs pathogen growth in vivo. Our data with riboflavin and pantothenate auxotrophs confirm that these two biosynthetic pathways represent attractive possibilities as druggable targets for Histoplasma.
The ability of vitamin auxotroph mutants to still successfully infect host cells combined with their inability to proliferate also suggests they can be exploited as candidate vaccine strains. This has the advantage over dead microorganism vaccines, since the vitamin auxotrophs cannot replicate and lead to disease progression but still provide enough persistence of the infection to elicit protective immunity. This has been shown for Mycobacterium tuberculosis pantothenate auxotrophs, which are being investigated as tuberculosis vaccine backgrounds (69). Thus, vitamin pathways represent attractive possibilities for therapeutic avenues. The necessity for de novo vitamin synthesis imposed by the phagosomal environment may constitute a potential Achilles heel for pathogens that defeat innate immunity by infecting and surviving within host phagocytic cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank William E. Goldman for the T-DNA vector used in Agrobacterium-mediated insertional mutagenesis. We thank Charles J. Daniels for assistance in metabolic pathway analysis of the Histoplasma genome and construction of the KEGG-annotated Histoplasma database.
This research was supported in part by grant 0865450D from the American Heart Association.
Footnotes
Published ahead of print 4 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00824-13.
REFERENCES
- 1.Kauffman CA. 2007. Histoplasmosis: a clinical and laboratory update. Clin. Microbiol. Rev. 20:115–132. 10.1128/CMR.00027-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen VQ, Sil A. 2008. Temperature-induced switch to the pathogenic yeast form of Histoplasma capsulatum requires Ryp1, a conserved transcriptional regulator. Proc. Natl. Acad. Sci. U. S. A. 105:4880–4885. 10.1073/pnas.0710448105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bullock WE, Wright SD. 1987. Role of the adherence-promoting receptors, CR3, LFA-1, and p150,95, in binding of Histoplasma capsulatum by human macrophages. J. Exp. Med. 165:195–210. 10.1084/jem.165.1.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin J-S, Huang J-H, Hung L-Y, Wu S-Y, Wu-Hsieh BA. 2010. Distinct roles of complement receptor 3, Dectin-1, and sialic acids in murine macrophage interaction with Histoplasma yeast. J. Leukoc. Biol. 88:95–106. 10.1189/jlb.1109717 [DOI] [PubMed] [Google Scholar]
- 5.Holbrook ED, Smolnycki KA, Youseff BH, Rappleye CA. 2013. Redundant catalases detoxify phagocyte reactive oxygen and facilitate Histoplasma pathogenesis. Infect. Immun. 81:2334–2346. 10.1128/IAI.00173-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youseff BH, Holbrook ED, Smolnycki KA, Rappleye CA. 2012. Extracellular superoxide dismutase protects Histoplasma yeast cells from host-derived oxidative stress. PLoS Pathog. 8:e1002713. 10.1371/journal.ppat.1002713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorenz MC, Fink GR. 2002. Life and death in a macrophage: role of the glyoxylate cycle in virulence. Eukaryot. Cell 1:657–662. 10.1128/EC.1.5.657-662.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naderer T, McConville MJ. 2008. The Leishmania-macrophage interaction: a metabolic perspective. Cell. Microbiol. 10:301–308. 10.1111/j.1462-5822.2007.01096.x [DOI] [PubMed] [Google Scholar]
- 9.Muñoz-Elías EJ, McKinney JD. 2006. Carbon metabolism of intracellular bacteria. Cell. Microbiol. 8:10–22. 10.1111/j.1462-5822.2005.00648.x [DOI] [PubMed] [Google Scholar]
- 10.Rohde K, Yates RM, Purdy GE, Russell DG. 2007. Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 219:37–54. 10.1111/j.1600-065X.2007.00547.x [DOI] [PubMed] [Google Scholar]
- 11.Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 100:12989–12994. 10.1073/pnas.2134250100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan W, Kraus PR, Boily M-J, Heitman J. 2005. Cryptococcus neoformans gene expression during murine macrophage infection. Eukaryot. Cell 4:1420–1433. 10.1128/EC.4.8.1420-1433.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu G, Cheng P-Y, Sham A, Perfect JR, Kronstad JW. 2008. Metabolic adaptation in Cryptococcus neoformans during early murine pulmonary infection. Mol. Microbiol. 69:1456–1475. 10.1111/j.1365-2958.2008.06374.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faucher SP, Mueller CA, Shuman HA. 2011. Legionella pneumophila transcriptome during intracellular multiplication in human macrophages. Front. Microbiol. 2:60. 10.3389/fmicb.2011.00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sauer J-D, Bachman MA, Swanson MS. 2005. The phagosomal transporter A couples threonine acquisition to differentiation and replication of Legionella pneumophila in macrophages. Proc. Natl. Acad. Sci. U. S. A. 102:9924–9929. 10.1073/pnas.0502767102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price MS, Betancourt-Quiroz M, Price JL, Toffaletti DL, Vora H, Hu G, Kronstad JW, Perfect JR. 2011. Cryptococcus neoformans requires a functional glycolytic pathway for disease but not persistence in the host. mBio 2(3):e00103–00111. 10.1128/mBio.00103-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorenz MC, Bender JA, Fink GR. 2004. Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryot. Cell 3:1076–1087. 10.1128/EC.3.5.1076-1087.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barelle CJ, Priest CL, Maccallum DM, Gow NAR, Odds FC, Brown AJP. 2006. Niche-specific regulation of central metabolic pathways in a fungal pathogen. Cell. Microbiol. 8:961–971. 10.1111/j.1462-5822.2005.00676.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198:693–704. 10.1084/jem.20030846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stewart GR, Patel J, Robertson BD, Rae A, Young DB. 2005. Mycobacterial mutants with defective control of phagosomal acidification. PLoS Pathog. 1:269–278. 10.1371/journal.ppat.0010033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKinney JD, Höner zu Bentrup K, Muñoz-Elías EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Jr, Russell DG. 2000. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406:735–738. 10.1038/35021074 [DOI] [PubMed] [Google Scholar]
- 22.Janakiraman A, Slauch JM. 2000. The putative iron transport system SitABCD encoded on SPI1 is required for full virulence of Salmonella typhimurium. Mol. Microbiol. 35:1146–1155. 10.1046/j.1365-2958.2000.01783.x [DOI] [PubMed] [Google Scholar]
- 23.Boshoff HIM, Barry CE., III 2005. Tuberculosis—metabolism and respiration in the absence of growth. Nat. Rev. Microbiol. 3:70–80. 10.1038/nrmicro1065 [DOI] [PubMed] [Google Scholar]
- 24.Brown AJP, Odds FC, Gow NAR. 2007. Infection-related gene expression in Candida albicans. Curr. Opin. Microbiol. 10:307–313. 10.1016/j.mib.2007.04.001 [DOI] [PubMed] [Google Scholar]
- 25.Jules M, Buchrieser C. 2007. Legionella pneumophila adaptation to intracellular life and the host response: clues from genomics and transcriptomics. FEBS Lett. 581:2829–2838. 10.1016/j.febslet.2007.05.026 [DOI] [PubMed] [Google Scholar]
- 26.Kronstad J, Saikia S, Nielson ED, Kretschmer M, Jung W, Hu G, Geddes JMH, Griffiths EJ, Choi J, Cadieux B, Caza M, Attarian R. 2012. Adaptation of Cryptococcus neoformans to mammalian hosts: integrated regulation of metabolism and virulence. Eukaryot. Cell 11:109–118. 10.1128/EC.05273-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McConville MJ, de Souza D, Saunders E, Likic VA, Naderer T. 2007. Living in a phagolysosome; metabolism of Leishmania amastigotes. Trends Parasitol. 23:368–375. 10.1016/j.pt.2007.06.009 [DOI] [PubMed] [Google Scholar]
- 28.Salvin SB. 1949. Cysteine and related compounds in the growth of the yeast like phase of Histoplasma capsulatum. J. Infect. Dis. 84:275–283. 10.1093/infdis/84.3.275 [DOI] [PubMed] [Google Scholar]
- 29.Howard DH, Dabrowa N, Otto V, Rhodes J. 1980. Cysteine transport and sulfite reductase activity in a germination-defective mutant of Histoplasma capsulatum. J. Bacteriol. 141:417–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boguslawski G, Akagi JM, Ward LG. 1976. Possible role for cysteine biosynthesis in conversion from mycelial to yeast form of Histoplasma capsulatum. Nature 261:336–338. 10.1038/261336a0 [DOI] [PubMed] [Google Scholar]
- 31.Stetler DA, Boguslawski G. 1979. Cysteine biosynthesis in a fungus, Histoplasma capsulatum. Sabouraudia 17:23–34. 10.1080/00362177985380041 [DOI] [PubMed] [Google Scholar]
- 32.Boguslawski G, Stetler DA. 1979. Aspects of physiology of Histoplasma capsulatum. (A review). Mycopathologia 67:17–24. 10.1007/BF00436235 [DOI] [PubMed] [Google Scholar]
- 33.McVeigh I, Houston WE. 1972. Factors affecting mycelial to yeast phase conversion and growth of the yeast phase of Histoplasma capsulatum. Mycopathol. Mycol. Appl. 47:135–151. 10.1007/BF02126161 [DOI] [PubMed] [Google Scholar]
- 34.Garrison RG, Dodd HT, Hamilton JW. 1970. The uptake of low molecular weight sulfur-containing compounds by Histoplasma capsulatum and related dimorphic fungi. Mycopathol. Mycol. Appl. 40:171–180. 10.1007/BF02051995 [DOI] [PubMed] [Google Scholar]
- 35.Rippon JW. 1968. Monitored environment system to control cell growth, morphology, and metabolic rate in fungi by oxidation-reduction potentials. Appl. Microbiol. 16:114–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu-Hsieh BA, Howard DH. 1992. Intracellular growth inhibition of Histoplasma capsulatum induced in murine macrophages by recombinant gamma interferon is not due to a limitation of the supply of methionine or cysteine to the fungus. Infect. Immun. 60:698–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobson ES, Harrell AC. 1982. Cysteine-independent and cysteine-requiring yeast-strains of Histoplasma capsulatum. Mycopathologia 77:69–73. 10.1007/BF00437386 [DOI] [PubMed] [Google Scholar]
- 38.Jacobson ES, Harrell AC. 1982. A prototrophic yeast-strain of Histoplasma capsulatum. Mycopathologia 77:65–68. 10.1007/BF00437385 [DOI] [PubMed] [Google Scholar]
- 39.Woods JP, Heinecke EL, Goldman WE. 1998. Electrotransformation and expression of bacterial genes encoding hygromycin phosphotransferase and β-galactosidase in the pathogenic fungus Histoplasma capsulatum. Infect. Immun. 66:1697–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Retallack DM, Heinecke EL, Gibbons R, Deepe GS, Jr, Woods JP. 1999. The URA5 gene is necessary for Histoplasma capsulatum growth during infection of mouse and human cells. Infect. Immun. 67:624–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Timmerman MM, Woods JP. 1999. Ferric reduction is a potential iron acquisition mechanism for Histoplasma capsulatum. Infect. Immun. 67:6403–6408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Timmerman MM, Woods JP. 2001. Potential role for extracellular glutathione-dependent ferric reductase in utilization of environmental and host ferric compounds by Histoplasma capsulatum. Infect. Immun. 69:7671–7678. 10.1128/IAI.69.12.7671-7678.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hwang LH, Mayfield JA, Rine J, Sil A. 2008. Histoplasma requires SID1, a member of an iron-regulated siderophore gene cluster, for host colonization. PLoS Pathog. 4:31000044. 10.1371/journal.ppat.1000044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hilty J, Smulian AG, Newman SL. 2008. The Histoplasma capsulatum vacuolar ATPase is required for iron homeostasis, intracellular replication in macrophages, and virulence in a murine model of histoplasmosis. Mol. Microbiol. 70:127–139. 10.1111/j.1365-2958.2008.06395.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zarnowski R, Cooper KG, Brunold LS, Calaycay J, Woods JP. 2008. Histoplasma capsulatum secreted γ-glutamyltransferase reduces iron by generating an efficient ferric reductant. Mol. Microbiol. 70:352–368. 10.1111/j.1365-2958.2008.06410.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Worsham PL, Goldman WE. 1988. Quantitative plating of Histoplasma capsulatum without addition of conditioned medium or siderophores. Med. Mycol. 26:137–143. 10.1080/02681218880000211 [DOI] [PubMed] [Google Scholar]
- 47.Zemska O, Rappleye CA. 2012. Agrobacterium-mediated insertional mutagenesis in Histoplasma capsulatum, p 51–66 In Brand AC, MacCallum DM. (ed), Host-fungus interactions. Humana Press, New York, NY: [DOI] [PubMed] [Google Scholar]
- 48.Edwards JA, Zemska O, Rappleye CA. 2011. Discovery of a role for Hsp82 in histoplasma virulence through a quantitative screen for macrophage lethality. Infect. Immun. 79:3348–3357. 10.1128/IAI.05124-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kemski MM, Stevens B, Rappleye CA. 2013. Spectrum of T-DNA integrations for insertional mutagenesis of Histoplasma capsulatum. Fungal Biol. 117:41–51. 10.1016/j.funbio.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y-G, Chen Y. 2007. High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. Biotechniques 43:649–650, 652, 654 passim. 10.2144/000112601 [DOI] [PubMed] [Google Scholar]
- 51.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35:W182–W185. 10.1093/nar/gkm321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edwards JA, Chen C, Kemski MM, Hu J, Mitchell TK, Rappleye CA. 2013. Histoplasma yeast and mycelial transcriptomes reveal pathogenic-phase and lineage-specific gene expression profiles. BMC Genomics 14:695. 10.1186/1471-2164-14-695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Youseff BH, Rappleye CA. 2012. RNAi-based gene silencing using a GFP sentinel system in Histoplasma capsulatum, p 151–164 In Brand AC, MacCallum DM. (ed), Host-fungus interactions. Humana Press, New York, NY: [DOI] [PubMed] [Google Scholar]
- 54.Gavrilin MA, Bouakl IJ, Knatz NL, Duncan MD, Hall MW, Gunn JS, Wewers MD. 2006. Internalization and phagosome escape required for Francisella to induce human monocyte IL-1β processing and release. Proc. Natl. Acad. Sci. U. S. A. 103:141–146. 10.1073/pnas.0504271103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- 56.Marion CL, Rappleye CA, Engle JT, Goldman WE. 2006. An α-(1,4)-amylase is essential for α-(1,3)-glucan production and virulence in Histoplasma capsulatum. Mol. Microbiol. 62:970–983. 10.1111/j.1365-2958.2006.05436.x [DOI] [PubMed] [Google Scholar]
- 57.Oltmanns O, Bacher A, Lingens F, Zimmermann FK. 1969. Biochemical and genetic classification of riboflavine deficient mutants of Saccharomyces cerevisiae. Mol. Gen. Genet. 105:306–313. 10.1007/BF00277585 [DOI] [PubMed] [Google Scholar]
- 58.McVeigh I, Morton K. 1965. Nutritional studies of Histoplasma capsulatum. Mycopathol. Mycol. Appl. 25:294–308. 10.1007/BF02049917 [DOI] [PubMed] [Google Scholar]
- 59.Rappleye CA, Engle JT, Goldman WE. 2004. RNA interference in Histoplasma capsulatum demonstrates a role for α-(1,3)-glucan in virulence. Mol. Microbiol. 53:153–165. 10.1111/j.1365-2958.2004.04131.x [DOI] [PubMed] [Google Scholar]
- 60.Edwards JA, Alore EA, Rappleye CA. 2011. The yeast-phase virulence requirement for α-glucan synthase differs among Histoplasma capsulatum chemotypes. Eukaryot. Cell 10:87–97. 10.1128/EC.00214-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Piddington DL, Fang FC, Laessig T, Cooper AM, Orme IM, Buchmeier NA. 2001. Cu,Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect. Immun. 69:4980–4987. 10.1128/IAI.69.8.4980-4987.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fradin C, De Groot P, MacCallum D, Schaller M, Klis F, Odds FC, Hube B. 2005. Granulocytes govern the transcriptional response, morphology and proliferation of Candida albicans in human blood. Mol. Microbiol. 56:397–415. 10.1111/j.1365-2958.2005.04557.x [DOI] [PubMed] [Google Scholar]
- 63.Melillo AA, Mahawar M, Sellati TJ, Malik M, Metzger DW, Melendez JA, Bakshi CS. 2009. Identification of Francisella tularensis live vaccine strain CuZn superoxide dismutase as critical for resistance to extracellularly generated reactive oxygen species. J. Bacteriol. 191:6447–6456. 10.1128/JB.00534-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Groote MA, Ochsner UA, Shiloh MU, Nathan C, McCord JM, Dinauer MC, Libby SJ, Vazquez-Torres A, Xu Y, Fang FC. 1997. Periplasmic superoxide dismutase protects Salmonella from products of phagocyte NADPH-oxidase and nitric oxide synthase. Proc. Natl. Acad. Sci. U. S. A. 94:13997–14001. 10.1073/pnas.94.25.13997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martchenko M, Alarco A-M, Harcus D, Whiteway M. 2004. Superoxide dismutases in Candida albicans: transcriptional regulation and functional characterization of the hyphal-induced SOD5 gene. Mol. Biol. Cell 15:456–467. 10.1091/mbc.E03-03-0179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fang FC, DeGroote MA, Foster JW, Bäumler AJ, Ochsner U, Testerman T, Bearson S, Giárd JC, Xu Y, Campbell G, Laessig T. 1999. Virulent Salmonella typhimurium has two periplasmic Cu, Zn-superoxide dismutases. Proc. Natl. Acad. Sci. U. S. A. 96:7502–7507. 10.1073/pnas.96.13.7502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Becker JM, Kauffman SJ, Hauser M, Huang L, Lin M, Sillaots S, Jiang B, Xu D, Roemer T. 2010. Pathway analysis of Candida albicans survival and virulence determinants in a murine infection model. Proc. Natl. Acad. Sci. U. S. A. 107:22044–22049. 10.1073/pnas.1009845107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rengarajan J, Bloom BR, Rubin EJ. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. U. S. A. 102:8327–8332. 10.1073/pnas.0503272102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sambandamurthy VK, Wang X, Chen B, Russell RG, Derrick S, Collins FM, Morris SL, Jacobs WR. 2002. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat. Med. 8:1171–1174. 10.1038/nm765 [DOI] [PubMed] [Google Scholar]
- 70.Miller CN, LoVullo ED, Kijek TM, Fuller JR, Brunton JC, Steele SP, Taft-Benz SA, Richardson AR, Kawula TH. 2013. PanG, a new ketopantoate reductase involved in pantothenate synthesis. J. Bacteriol. 195:965–976. 10.1128/JB.01740-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsai H-N, Hodgson DA. 2003. Development of a synthetic minimal medium for Listeria monocytogenes. Appl. Environ. Microbiol. 69:6943–6945. 10.1128/AEM.69.11.6943-6945.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu J, Niu C, Wang D, Li M, Teo W, Sun G, Wang J, Liu J, Gao Q. 2011. MMAR_2770, a new enzyme involved in biotin biosynthesis, is essential for the growth of Mycobacterium marinum in macrophages and zebrafish. Microbes Infect. Inst. Pasteur 13:33–41. 10.1016/j.micinf.2010.08.010 [DOI] [PubMed] [Google Scholar]
- 73.Napier BA, Meyer L, Bina JE, Miller MA, Sjöstedt A, Weiss DS. 2012. Link between intraphagosomal biotin and rapid phagosomal escape in Francisella. Proc. Natl. Acad. Sci. U. S. A. 109:18084–18089. 10.1073/pnas.1206411109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bacon GA, Burrows TW, Yates M. 1950. The effects of biochemical mutation on the virulence of Bacterium typhosum: the virulence of mutants. Br. J. Exp. Pathol. 31:714–724 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.