

Abstract
Upon entry of Legionella pneumophila into amoebas and macrophages, host-mediated farnesylation of the AnkB effector enables its anchoring to the Legionella-containing vacuole (LCV) membrane. On the LCV, AnkB triggers docking of K48-linked polyubiquitinated proteins that are degraded by the host proteasomes to elevate cellular levels of amino acids needed for intracellular proliferation. Interference with AnkB function triggers L. pneumophila to exhibit a starvation response and differentiate into the nonreplicative phase in response to the basal levels of cellular amino acids that are not sufficient to power intracellular proliferation of L. pneumophila. Therefore, we have determined whether the biological function of AnkB is temporally and spatially triggered upon bacterial attachment to the host cell to circumvent a counterproductive bacterial differentiation into the nonreplicative phase upon bacterial entry. Here, we show that upon attachment of L. pneumophila to human monocyte-derived macrophages (hMDMs), the host farnesylation and ubiquitination machineries are recruited by the Dot/Icm system to the plasma membrane exclusively beneath sites of bacterial attachment. Transcription and injection of ankB is triggered by attached extracellular bacteria followed by rapid farnesylation and anchoring of AnkB to the cytosolic side of the plasma membrane beneath bacterial attachment, where K48-linked polyubiquitinated proteins are assembled and degraded by the proteasomes, leading to a rapid rise in the cellular levels of amino acids. Our data represent a novel strategy by an intracellular pathogen that triggers rapid nutritional remodeling of the host cell upon attachment to the plasma membrane, and as a result, a gratuitous surplus of cellular amino acids is generated to support proliferation of the incoming pathogen.
INTRODUCTION
The Legionnaires' disease-causing bacterium, Legionella pneumophila, replicates within alveolar macrophages, causing pneumonia (1). The organism is transmitted to humans from the aquatic environment, where L. pneumophila replicates within amoebas and ciliates (for recent reviews, see references 2, 3, 4, and 5). Coevolution and adaptation of L. pneumophila to the intracellular lifestyle within amoebas in the aquatic environment is believed to have played a major role in its ability to exploit evolutionarily conserved eukaryotic processes, which enables its proliferation within human alveolar macrophages (2–4, 6, 7). Within both evolutionarily distant host cells, L. pneumophila evades endocytic fusion and intercepts endoplasmic reticulum (ER)-to-Golgi vesicle traffic to remodel its phagosome into an ER-derived vacuole, designated the Legionella-containing vacuole (LCV) (1, 3, 8, 9). During late stages of intracellular proliferation, the bacteria escape from the LCV into the cytosol, where they finish the last 1 or 2 rounds of proliferation, during which the bacteria exhibit a starvation response, presumably in response to the depletion of nutrients (10–13).
Modulation of various cellular processes by L. pneumophila is dependent on a functional Dot/Icm type IVB secretion system (14, 15). This system injects into the host cell a cadre of ∼300 effectors to modulate a myriad of cellular processes to reprogram the host cell into a proliferation niche (1, 8, 9, 16). However, the roles of most of the Dot/Icm-translocated effectors in the intracellular infection remain unknown, and only few have a detectable role in intracellular replication (1). The AnkB effector, which is found in all genome-sequenced L. pneumophila strains and in 211/217 tested strains (17), is essential for proliferation of L. pneumophila within mammalian and protozoan cells and for intrapulmonary bacterial proliferation and manifestation of pulmonary disease in the mouse model (18–23). Most of the structure of AnkB is composed of eukaryotic domains or motifs, including an F-box domain involved in polyubiquitination, two Ankyrin protein-protein interaction domains (24), and a C-terminal “CaaX” farnesylation motif (3, 25). In contrast to what is seen in the AA100/130b strain and other genome-sequenced strains of L. pneumophila, the AnkB homologue of the Paris strain is missing the farnesylation motif due to truncation of the C terminus, which likely explains functional differences and the various levels of attenuation of the ankB mutant among the different strains (26).
Host-mediated prenylation by farnesylation of AnkB is essential for anchoring AnkB of strain AA100 to the LCV membrane, which is essential for biological function of the effector in macrophages and amoebas and for virulence in mice (22, 27). Prenylation is a eukaryotic posttranslational modification that covalently links a 15-carbon farnesyl or 20-carbon geranyl-geranyl lipid moiety to a conserved cysteine residue within the C terminus “CaaX” motif of a protein, which enables anchoring of hydrophilic proteins into the lipid bilayer of membranes (25). Farnesylation (15-carbon farnesyl addition) of AnkB allows its anchoring into the outer leaflet of the LCV membrane (22). Many other C terminus CaaX motif-containing effectors of L. pneumophila have been shown to be anchored to host membranes through host-mediated prenylation (28, 29). In silico genomic analyses have shown the presence of the “CaaX” motif in numerous proteins of unknown function or protein effectors of other pathogens (22, 25), suggesting the potential of a general paradigm of exploiting host prenylation to anchor bacterial effectors into host membranes. The eukaryotic enzymes necessary for farnesylation, i.e., farnesyl transferase (FTase), Ras-converting enzyme 1 (RCE1), and isoprenyl cysteine carboxyl methyltransferase (IcmT), are all recruited to the LCV in a Dot/Icm-dependent fashion (22). Anchoring of AnkB into the LCV membrane via farnesylation is indispensable for its biological activity, since substitution of the cysteine residue within the CaaX motif, RNA interference (RNAi) knockdown, or chemical inhibition of the host FTase abolishes intracellular replication of L. pneumophila (22, 27).
The host SCF1 E3 ubiquitin ligase complex contains the RING domain protein RBX1 (RING-box 1), Cul1 (cullin 1), and Skp1 (S-phase-kinase associated protein 1), which bind to the F-box domain within F-box proteins directly through Skp1 (24, 30–32). The ubiquitination substrate binds to the F-box protein through interaction with a protein-protein interaction domain such as leucine-rich repeat (LRR), WD40, or ankyrin domain. The AnkB effector of L. pneumophila is a bona fide F-box protein that interacts with the SCF1 ubiquitin ligase of amoebas and mammals (21, 23, 27, 31). Deletion of the F-box domain of AnkB or substitution of two of its conserved residues abolishes AnkB-Skp1 interaction (21). Furthermore, knockdown of Skp1 expression by RNAi restricts intracellular proliferation of L. pneumophila (21).
Based on nutrient availability, growth of L. pneumophila cycles between two phases, which are the motile nonreplicative and the nonmotile replicative phases (33). In response to nutrient depletion during late stages of infection within macrophages and amoebas, L. pneumophila exhibits a dramatic starvation response manifested by triggering expression of RelA and SpoT (32), both of which synthesize the alarmone ppGpp (34). The elevated ppGpp levels in L. pneumophila trigger a complex network of regulatory cascades that leads to differentiation into the nonreplicative phase associated with dramatic phenotypic modulation, such as flagellation (12, 33–38).
Within amoebas and mammalian cells, the LCV membrane-anchored AnkB functions as a platform for the assembly of K48-linked polyubiquitinated proteins (21, 23, 39) that are degraded by the host proteasomal machinery (32), which is required for intracellular proliferation (32, 39). This generates short peptides (2 to 24 amino acids) that are rapidly degraded by host cytosolic oligo- and aminopeptidases into free amino acids. This increases the cellular levels of amino acids, particularly the limiting ones, such as Cys (40), which is semiessential in mammals and is essential for amoebas (32). Cysteine is an essential amino acid for L. pneumophila, which converts it to pyruvate and metabolizes it through the tricarboxylic acid (TCA) cycle as a major and metabolically preferable source of carbon and energy to power intracellular replication (32). Upon entry of the wild-type (WT) strain into proteasome-inhibited cells or entry of the ankB mutant into untreated cells, the bacteria exhibit a dramatic starvation response manifested by triggering expression of RelA and SpoT (32), both of which synthesize the alarmone ppGpp (34). However, the starvation response and the associated differentiation exhibited by the ankB mutant upon entry into untreated host cells or by the wild-type strain upon entry into proteasome-inhibited cells are totally circumvented by amino acid supplementation (32). Therefore, to prevent a starvation response and differentiation into a nonreplicative phase by L. pneumophila, we hypothesize that AnkB is likely to function rapidly upon contact to the host cell to generate a gratuitous surplus of cellular amino acids, which are the major sources of carbon and energy generation for L. pneumophila.
To test our hypothesis, we have determined the early temporal and spatial manipulation of the host cell by AnkB to circumvent such an early starvation response and differentiation into the nonreplicative phase by L. pneumophila. Here, we show that upon initial intimate contact with human monocyte-derived macrophages (hMDMs), L. pneumophila injects native AnkB, which becomes anchored into the cytosolic side of the plasma membrane exclusively beneath sites of bacterial attachment. Membrane anchoring of the injected AnkB effector is mediated by the host farnesylation machinery, which is rapidly recruited to the plasma membrane, in a Dot/Icm-dependent manner, beneath sites of bacterial attachment. The host SCF1 ubiquitin ligase is also recruited to the plasma membrane beneath attached extracellular L. pneumophila, with subsequent AnkB-dependent rapid proteasomal degradation of K48-linked polyubiquitinated proteins leading to a rapid rise in the cellular levels of amino acids. Taken together, attached extracellular L. pneumophila utilizes the AnkB effector to trigger rapid nutritional preparation of the host cell through a rapid rise in the cellular levels of amino acids upon bacterial entry. This suppresses a potential bacterial starvation response, triggers differentiation into the replicative phase, and provides a gratuitous source of carbon and energy needed for robust intracellular proliferation.
MATERIALS AND METHODS
Bacterial strains, cell cultures, and plasmids.
L. pneumophila strain AA100/130b (ATCC BAA-74) and the isogenic ankB and dotA mutants were grown on buffered charcoal yeast extract (BCYE) agar plates for 3 days at 37°C prior to use in infections as described previously (20). hMDMs and U937 cells were cultured using RPMI 1640 medium as we described previously (21). The plasmid PXDC61M, which contains the blaM gene, encoding the mature form of TEM-1 beta-lactamase, was obtained from Zhao-Qing Luo at Purdue University. The ankB gene was PCR amplified with restriction enzymes and cloned in frame with the beta-lactamase at the BamHI-XbaI sites to generate a transcriptional fusion protein. The resulting plasmid was introduced into L. pneumophila strain AA100/130b and the dotA mutant. To verify expression of the fusion proteins in L. pneumophila, strains harboring pXDC61M were grown on BCYE containing chloramphenicol and IPTG (isopropyl-β-d-thiogalactopyranoside) (0.5 mM) and analyzed by Western blotting. Protein from 1 × 109 bacteria were transferred to nitrocellulose membranes and detected by Western blotting with a primary monoclonal antibody specific to TEM-1 β-lactamase (QED Bioscience) and antimouse peroxide conjugate as a secondary antibody.
Real-time qPCR.
Quantitative real-time PCR (qPCR) on attached bacteria was performed as we described previously (11, 20). Briefly, hMDMs were plated at a density of 5 × 105 in 24-well plates and treated with 1 μM cytochalasin D for 30 min prior to infection. The hMDMs were then infected with WT bacteria at a multiplicity of infection (MOI) of 10 for 0, 7.5, or 15 min, and intimate and synchronized attachment was achieved by centrifugation at 1,000 rpm for 3 min. To assess RNA expression levels of ankB, mompS, and 16S RNA gene in response to attachment to hMDMs, total RNA was extracted from infected cells at the indicated time points using the RNeasy Minikit (Qiagen, Valencia, CA) as recommended by the manufacturer. Total RNA was treated with DNase I (Ambion, Austin, TX) at 37°C for 30 min. Equal amounts of total RNA from infected cells were used for cDNA synthesis using Superscript III Plus RNase H reverse transcriptase (RT) (Invitrogen, CA) and random primers. Real-time qPCR was done in triplicate using the Power SYBR green PCR Master Mix kit in a 20-μl reaction volume, as recommended by the manufacturer (Applied Biosystems, CA), using specific primers. The PCR conditions were 2 min at 94°C initially, followed by 10 s at 96°C, 20 s at 47°C, and 15 s at 72°C for 40 cycles. Changes in mRNA expression were determined by the comparative CT method (threshold cycle number at the cross point between amplification plot and threshold), and values were normalized to 16S RNA. Negative or positive values were considered downregulation or upregulation when there was a minimum of 2-fold difference of gene expression.
Preferential plasma membrane permeabilization and loading of the cytosol with antibodies.
Human monocyte-derived macrophages were isolated and maintained as described previously (20). Monocytes were seeded in 24-well plates at 1 × 106 cells/well. Cells were treated with cytochalasin D (5 μg/ml), an actin polymerization inhibitor, prior to infection and throughout the experiment. hMDM plasma membranes were selectively permeabilized for 5 min at room temperature with an RPMI 1640 solution containing digitonin (50 μg/ml) as well as anti-AnkB antiserum, as we described previously (41, 42). Following permeabilization, cells were extensively washed with medium and infected with wild-type L. pneumophila as well as the ankB and dotA mutant strains at an MOI of 25, 50, or 75 for 15 min. The hMDMs were extensively washed with medium and incubated an additional 30 min for antibody-antigen interaction. Cells were then fixed with 3.7% formaldehyde for 15 min at room temperature. To ensure that cytochalasin D inhibited phagocytosis and bacteria remained extracellular, antibody labeling of L. pneumophila with specific rabbit polyclonal antiserum was performed prior to permeabilization, followed by Alexa Fluor 488 donkey anti-rabbit secondary antibody (Invitrogen). Cells were then permeabilized with 0.1% Triton X-100 for 10 min at room temperature, followed by anti-AnkB antiserum detection by Alexa Fluor 555-conjugated donkey anti-rabbit IgG (Invitrogen).
TEM translocation assay.
The U937 cells were grown in RPMI 1640 containing 10% fetal bovine serum (FBS), seeded in black clear-bottom 96-well plates at 1 × 105 cells/well, and treated with phorbol myristate acetate (PMA) for 48 h prior to infection. L. pneumophila strains containing the TEM-1 fusion proteins were grown for 3 days on BCYE containing chloramphenicol (5 μg/ml) and then streaked onto BCYE containing chloramphenicol and 0.5 mM IPTG to induce expression of the fusion proteins. Cell monolayers were loaded with the β-lactamase substrate CCF4 by adding 20 μl of 6× CCF4-AM solution (LiveBLAzer-FRET B/G loading kit; Invitrogen) containing 0.1 M probenecid. Cells were incubated with the solution for 2 h at room temperature. U937 cells were treated with the actin polymerization inhibitor cytochalasin D (5 μg/ml) for 30 min prior to infection and maintained throughout the infection using different MOIs. Plates were centrifuged (1,000 rpm, 5 min) to initiate bacterium-cell contact and incubated for 10 min. Fluorescence was quantified on a BioTek Synergy HT microplate reader with excitation at 405 nm, and emission was detected at 460 nm and 530 nm. Bacterial effector translocation was determined by the emission ratio 460 nm/530 nm to normalize the β-lactamase activity to noninfected substrate loaded cells.
Recruitment of host farnesylation and ubiquitination machinery to sites of L. pneumophila attachment.
A total of 5 × 105 hMDMs on glass coverslips in 24-well plates were pretreated for 30 min with cytochalasin D (5 μg/ml) and then infected with wild-type L. pneumophila and the ankB and dotA mutants at an MOI of 10 for 15 min. Processing of infected cells for confocal microscopy was performed as described previously (21). Briefly, fixed and permeabilized cells were blocked for 1 h with 3% bovine serum albumin (BSA)–phosphate-buffered saline (PBS) and then mouse anti-L. pneumophila antiserum (dilution, 1/1,000), and anti-Skp1, anti-Cul1, anti-FTα, anti-RCE1, and anti-IcmT antibodies (dilution, 1/200) (Abcam, Cambridge, MA) were added to 3% BSA–PBS and incubated at room temperature for 1 h. Following extensive washing with 3% BSA–PBS, bound antibodies were detected with Alexa Fluor 488- or 555-conjugated donkey anti-rabbit or mouse IgG antibodies (Invitrogen, Carlsbad, CA) for 1 h. Following this, the glass coverslips were mounted on glass slides using ProLong Gold antifade reagent (Invitrogen, Carlsbad, CA). The fixed cells were examined with an Olympus FV1000 laser scanning confocal microscope as we described previously (21). On average, 8 to 15 0.2-μM serial Z sections of each image were captured and stored for further analyses, using Adobe Photoshop CS5.
GC-MS analyses of free amino acids.
The cellular levels of free amino acids were determined as part of the global metabolomics profile. The hMDMs cells were seeded in 6-well plates at 1 × 106 cells/well, and prior to infection, the cells were treated with 1 μM cytochalasin D for 30 min. The hMDMs were infected with WT or ankB mutant L. pneumophila at an MOI of 100 for 1 h, and the infected cells were lysed in aqueous 90% methanol. Lysates were stored at −20°C for 1 h and then centrifuged (21,000 × g at 4°C) for 10 min. The resulting supernatants were dried using a Speed-Vac and prepared for gas chromatography-mass spectrometry (GC-MS).
All GC-MS analyses were performed at the University of Utah Metabolomic core facility using a Waters GCT Premier mass spectrometer fitted with an Agilent 6890 gas chromatograph and a Gerstel MPS2 autosampler, as we described previously (32). The dried samples were suspended in 40 μl of 40-mg/ml O-methoxylamine hydrochloride in pyridine and incubated for 1 h at 30°C. A 25-μl sample of this solution was transferred to autosampler vials followed by the addition of N-methyl-N-trimethylsilyltrifluoracetamide and further incubated for 30 min at 37°C with shaking. A 1-μl sample was injected to the gas chromatograph inlet in the split mode set to a 10:1 ratio. Injector temperature was held at 250°C. The gas chromatograph had an initial temperature of 95°C for 1 min followed by a 40°C/min ramp to 110°C with a hold time of 2 min. This was followed by a second 5°C/min ramp to 250°C and then a third ramp to 350°C and a final hold time of 3 min. A 30-m Restek Rxi-5 MS column with a 5-m-long guard column was employed for analysis. Data were collected by MassLynx 4.1. Data analysis for free cellular amino acids was performed using QuanLynx, which quantified the area under the curve for each amino acid. All data were saved to an Excel spread sheet for further analysis. The GC-MS analysis gives relative results of the area under the curve for the same amino acid and is not quantitative relative to other amino acids within the same sample. Since the GC-MS analyses compare levels of the same metabolite/amino acid between different samples/treatments, the results are presented as ratios of infected to uninfected cells.
RESULTS
Triggering expression of ankB upon attachment of L. pneumophila to human macrophages.
Upon entry of the ankB mutant to macrophages or amoebas, the bacterium exhibits a dramatic starvation response and differentiation into the nonreplicative phase, but both phenotypes are circumvented by amino acid supplementation (32). Similar phenotypes are also exhibited by the wild-type strain upon entry into proteasome-inhibited cells, and in both cases the respective phenotypes are circumvented upon supplementation of amino acids (32). We tested the hypothesis that WT L. pneumophila likely employs AnkB during initial stages of interaction with the host cell to circumvent the amino acid starvation response and the associated phenotypic modulations. We determined whether attachment of L. pneumophila to host cells triggered expression of AnkB. To determine this, human monocyte-derived macrophages (hMDMs) were pretreated with cytochalasin D for 30 min to block phagocytosis and then infected with wild-type L. pneumophila at an MOI of 10 for 0, 7.5, and 15 min. This bacterial attachment protocol resulted in attachment of 1 or 2 bacteria/cell in ∼50% of the cells in the monolayers. Blocking of bacterial entry into hMDMs by cytochalasin D was confirmed by sterilization of the infected cytochalasin D-treated monolayers by gentamicin treatment, indicating that the bacteria were extracellular. Following RNA purification and cDNA synthesis, expression of the ankB gene was determined by real-time qPCR using expression of the 16S RNA as an internal control, as we described previously (11, 18, 20). Expression of the constitutively expressed mompS gene was used as a control. Compared to expression of ankB at 0 min postattachment, its expression was increased 9-fold and 26-fold at 7.5 and 15 min, respectively, following bacterial attachment (Fig. 1). During the course of the attachment experiment, no change was observed in expression of mompS (Fig. 1). These data show that transcription of ankB is triggered immediately upon attachment of L. pneumophila to hMDMs.
FIG 1.
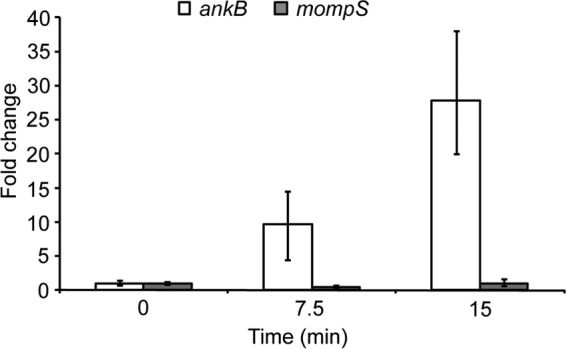
Expression of ankB upon intimate attachment to hMDMs. Quantitative real-time PCR of ankB gene expression by L. pneumophila at 0, 7.5, and 15 min after attachment to hMDMs. The hMDMs were pretreated with cytochalasin D and infected for the indicated time periods, followed by total isolation of RNA and RT-PCR. Expression of ankB or the control gene mompS was compared to 16S RNA levels. Fold changes in gene expression were compared to levels measured at time zero min. Error bars indicate standard errors of the means (SEM). The data are representative of three independent experiments.
Translocation of AnkB into macrophages upon bacterial attachment.
To determine temporal and spatial translocation of AnkB upon intimate attachment of L. pneumophila to macrophages, we generated an L. pneumophila strain expressing a β-lactamase-AnkB reporter fusion construct to monitor real-time translocation in cells preloaded with the CCF4 fluorometric β-lactamase substrate (43). The human macrophage U937 cell line preloaded with CCF4 was pretreated with cytochalasin D and then infected with L. pneumophila expressing β-lactamase fusions at various MOIs for 15 min (Fig. 2). Hydrolysis of CCF4 was measured by quantifying blue (460 nm) and green (530 nm) fluorescence emission and is reported as the 460 nm/530 nm ratio, where a ratio greater than 1 represents positive translocation. The data showed that AnkB was equally translocated into untreated and cytochalasin D-treated U937 macrophages. Translocation of AnkB by attached extracellular bacteria was totally dependent on a functional Dot/Icm T4SS, since the dotA translocation-defective mutant failed to translocate the effector (Fig. 2). As expected, increasing the MOI resulted in increased AnkB translocation in a dose-response manner, as evident by greater CCF4 hydrolysis (Fig. 2). No translocation was detected for the fatty acid biosynthetic enzyme enoyl-coenzyme A (enoyl-CoA) reductase (FabI), which was used as a negative control (Fig. 2) (43). These data show that attachment of L. pneumophila to macrophages triggers rapid translocation of the AnkB effector into the host cell.
FIG 2.
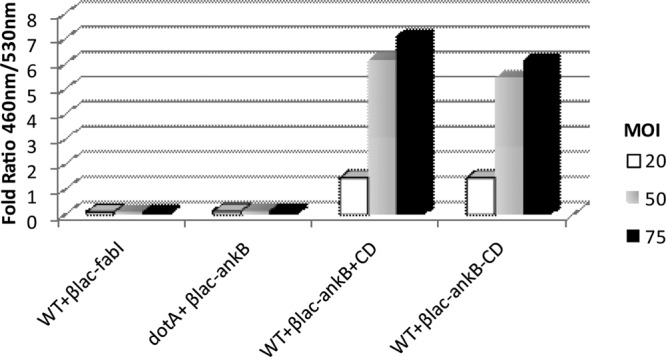
Translocation of BlaM-AnkB fusion protein by L. pneumophila upon intimate attachment to U937 cells. Cytochalasin D-treated U937 cells were infected with increasing MOIs of L. pneumophila strains expressing various BlaM fusion constructs, and translocation was measured by monitoring hydrolysis of the fluorogenic substrate CCF4. A 460 nm/530 nm ratio greater than 1 indicates translocation of the fusion construct. Data represent mean values of three independent experiments.
Host-mediated farnesylation and anchoring of AnkB to the cytosolic side of the plasma membrane beneath attachment sites of extracellular bacteria.
We decided to determine cellular location of the native AnkB effector injected by attached extracellular bacteria. Regular labeling of cytochalasin D-treated infected cells to detect cellular location of AnkB injected by attached extracellular bacteria was difficult to interpret by confocal microscopy. This was due to our findings that the injected AnkB was not detected in the cytosol but seemed to be localized beneath the attached extracellular WT bacteria, which also bound the anti-AnkB antibody. The resolution was not sufficient to differentiate injected AnkB localized intracellularly beneath bacterial attachment from AnkB contained in the attached extracellular bacteria. Since the β-lactamase-AnkB reporter fusion was clearly injected by attached extracellular bacteria (Fig. 2), our microscopy findings suggested that the injected AnkB was likely localized exclusively beneath bacterial attachment sites.
To overcome the above-mentioned caveat and to determine whether the injected native AnkB by attached extracellular bacteria was located beneath bacterial attachment sites, the cytosol of live hMDMs was preloaded with anti-AnkB antibodies prior to bacterial attachment. This would allow the antibody to bind AnkB upon its injection by attached extracellular bacteria (13, 41, 42). This strategy also provides a clear and more solid interpretation of the data, since the anti-AnkB antibody is loaded to the host cell cytosol prior to inoculation of the bacteria. To load the host cell cytosol with the anti-AnkB antibodies prior to infection, the plasma membrane of live hMDMs was preferentially permeabilized with a low concentration of digitonin (13, 41, 42). After loading the cells with the antibody and allowing the cells to heal the membrane damage for few minutes, the integrity of the plasma membrane was confirmed by impermeability to trypan blue. Without digitonin treatment, the plasma membrane of hMDMs was impermeable to anti-AnkB antibodies, as expected (Fig. 3). The antibody-loaded hMDMs were treated with cytochalasin D to prevent phagocytosis and then infected at an MOI of 10 with WT L. pneumophila or the isogenic mutant dotA or ankB. The cells were then fixed and processed for confocal microscopy. This bacterial attachment protocol resulted in an average attachment of 1 or 2 bacteria/cell in ∼50% of the cells in the monolayers. To allow differentiation between extracellular and intracellular bacteria, extracellular L. pneumophila cells were labeled with specific antibodies prior to permeabilization of the infected cells. When the hMDMs were permeabilized with digitonin and loaded with anti-AnkB antibodies, the loaded antibody was detectable as red patches throughout the cytosol of ∼98% of the cells, indicating successful loading of the cells with anti-AnkB antibodies to prior to infection (Fig. 3). When hMDMs were infected with wild-type L. pneumophila, 52% of attached extracellular bacteria colocalized with AnkB exclusively beneath the site of bacterial attachment (Fig. 3). As expected, the dotA translocation-defective mutant and the ankB mutant did not colocalize with AnkB (5% and 0%, respectively) (Student's t test, P < 0.007 and 0.003, respectively) (Fig. 3).
FIG 3.
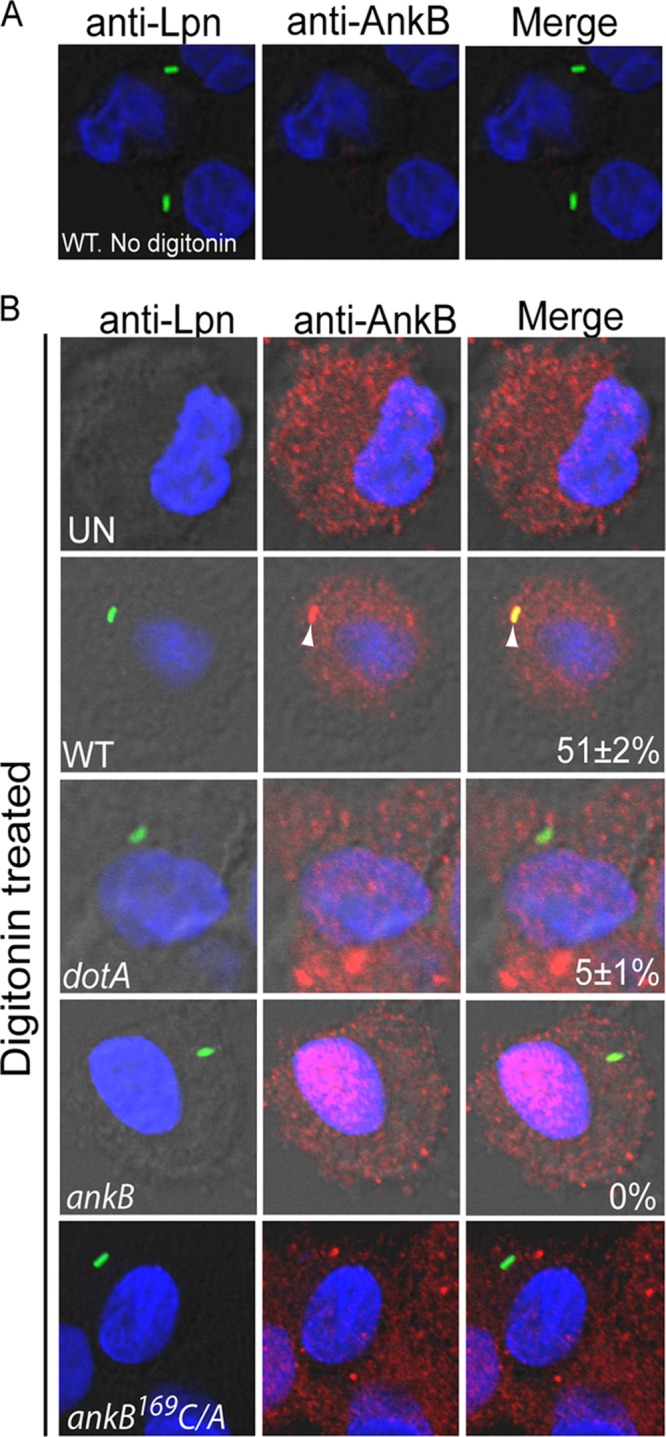
Anchoring of native AnkB injected by attached L. pneumophila to the cytosolic side of the plasma membrane of hMDMs beneath bacterial attachment sites. Representative confocal microscopy images of cytochalasin D-treated infected hMDMs that were preloaded with anti-AnkB antisera (red) in the absence (A) or presence (B) of digitonin. The hMDMs were infected by L. pneumophila strains (green) for 15 min. The numbers in the merged images of all panels are means and standard deviations of the frequency of anchoring AnkB to the plasma membrane beneath attached extracellular bacteria. The data represent analyses of 100 infected cells and are representative of three independent experiments.
Since AnkB is hydrophilic and its anchoring to the LCV membrane is mediated by host farnesylation within amoebas and macrophages (22, 25), we determined whether host farnesylation was required for the exclusive localization of AnkB to the cytosolic side of the plasma membrane beneath the sites of bacterial attachment. The cytosol of hMDMs was preloaded with anti-AnkB antiserum prior to infection, as described above. Cytochalasin-D treated cells were infected by the farnesylation-defection ankB-169C-A substitution mutant in the CaaX motif (22). The data showed that infection by the ankB-169C-A substitution mutant resulted in failure to anchor AnkB to the plasma membrane beneath bacterial attachment sites (Student's t test, P < 0.005) (Fig. 3). Therefore, host farnesylation anchors the injected AnkB by attached extracellular L. pneumophila to the cytosolic side of the plasma membrane directly and exclusively beneath bacterial attachment sites. This is the first demonstration of farnesylation-mediated anchoring of an injected bacterial effector to the inner leaflet of the plasma membrane beneath bacterial attachment sites.
Recruitment of the host farnesylation machinery to the plasma membrane beneath attached extracellular L. pneumophila.
Since during infection host-mediated farnesylation of AnkB anchors it to the LCV membrane and AnkB was exclusively localized beneath bacterial attachment sites, we tested the hypothesis that the host farnesylation enzymes FTase, IcmT, and RCE1 were recruited to the plasma membrane by attached extracellular bacteria to anchor AnkB to the plasma membrane. Cytochalasin D-treated hMDMs were infected with wild-type L. pneumophila and the isogenic mutants ankB and dotA at an MOI of 10 for 15 min. The cells were then immediately fixed and processed for confocal microscopy to determine if FTase, IcmT, and RCE1 were recruited beneath the sites of bacterial attachment. The data showed that FTase, IcmT, and RCE1 were all recruited beneath attachment sites of wild-type bacteria at a frequency of ∼85% (Fig. 4A, B, and C). In contrast, FTase, IcmT, and RCE1 were recruited at a frequency of only ∼10%, beneath attachment sites of the dotA mutant (Student's t test, P < 0.006, 0.007, and 0.006, respectively) (Fig. 4). FTase, IcmT, and RCE1 were recruited at a significantly reduced frequency of 43, 47, and 41%, respectively (Student's t test, P < 0.01), beneath attachment sites of the ankB mutant bacteria (Fig. 4). The moderate reduction in recruitment of the host enzymes by the ankB mutant is most likely due to the fact that the other ∼12 farnesylated effectors of Legionella injected by the ankB mutant (28, 29) interact with the host farnesylation enzymes, while the translocation-defective dotA mutant is severely defective in recruitment of the host enzymes (22). These data indicate that upon attachment of L. pneumophila the Dot/Icm apparatus is essential for recruitment of the host enzymes into the plasma membrane beneath the sites of bacterial attachment. This is the first example of recruitment of the host farnesylation machinery by attached extracellular bacteria to anchor an injected effector to the cytosolic side of the plasma membrane exclusively beneath bacterial attachment sites.
FIG 4.

The farnesylation machinery components FTα, RCE1, and IcmT are recruited beneath attachment sites of L. pneumophila to hMDMs. The hMDMs pretreated with cytochalasin D were infected by wild-type (WT) L. pneumophila and the isogenic dotA or ankB mutant for 15 min. Representative confocal microscopy images of infected hMDMs showing colocalization of FTα (A), RCE1 (B), and IcmT (C) proteins to attached WT, ankB mutant, or dotA mutant bacteria. Bacteria were labeled with anti-Lpn antibody (green), and FTα, RCE1, and IcmT were labeled with the respective specific antibodies (red) and then analyzed by confocal microscopy. The arrowheads indicate intense colocalization of FTα, RCE1, or IcmT with the WT strain. The numbers in the merged images of all panels are means and standard deviations of the frequency of recruitment of FTα, RCE1, or IcmT beneath attached extracellular bacteria. The data represent analyses of 100 infected cells and are representative of three independent experiments.
Recruitment of the host SCF1 ubiquitin ligase complex beneath attached extracellular L. pneumophila.
During ectopic expression, the AnkB effector interacts with the host Skp1 component of the SCF1 E3-ubiquitin ligase complex, but the location of this interaction during infection is not known (21, 23). Since AnkB is exclusively localized to the LCV membrane during infection and to the plasma membrane beneath attached extracellular L. pneumophila, we tested the hypothesis that the SCF1 ubiquitin ligase was recruited to the LCV, where it interacts with AnkB, and that this recruitment was rapidly initiated at the plasma membrane beneath bacterial attachment sites. To determine recruitment of SCF1 to the LCV, hMDMs were infected at an MOI of 10 for 1 h with wild-type L. pneumophila or the ankB or dotA translocation-defective isogenic mutant. The data showed that both Skp1 and Cul1 components of the SCF1 were recruited to the LCV of the WT strain at a frequency of 82 to 84% (Fig. 5). Recruitment of both host cell components was dependent on a functional Dot/Icm translocation system, since the dotA translocation-defective mutant was severely defective in this recruitment (Fig. 5). To determine whether recruitment of the SCF1 components was initiated by attached extracellular bacteria, cytochalasin D-treated hMDMs were preloaded with anti-Skp1 or -Cul1 antibodies and then infected at an MOI of 10 for 15 min. Gentamicin treatment of cytochalasin D-treated control cells sterilized the monolayer, which confirms effectiveness of blocking bacterial entry by cytochalasin D. The cells were fixed immediately and processed for confocal microscopy. The data showed that Skp1 and Cul1 were recruited beneath bacterial attachment sites at a frequency of ∼80%. This recruitment was dependent on a functional Dot/Icm type IV secretion system, since Skp1 and Cul1 were recruited at a frequency of only ∼10% by attached translocation-defective dotA mutant bacteria (Student's t test, P < 0.008) (Fig. 5). Only 34 and 30% of attached ankB mutant bacteria recruited Skp1 and Cul1, respectively (Fig. 5), which was significantly less than wild-type bacteria (Student's t test, P < 0.01). These data show that the Dot/Icm translocation system of L. pneumophila is essential for recruitment of the SCF1 ubiquitin ligase to the LCV and that this recruitment is initiated at the plasma membrane beneath sites of bacterial attachment. This is the first example of recruitment of the host SCF1 to a pathogen-containing vacuole and the initiation of this process at the cytosolic side of the plasma membrane beneath bacterial attachment sites.
FIG 5.

Recruitment of the SCF ubiquitin ligase components Skp1 and Cul1 beneath attachment sites of L. pneumophila to hMDMs and their subsequent compartmentalization to the LCV. Untreated (A) or cytochalasin D-pretreated (B) hMDMs preloaded with anti-Skp1 or anti-Cul1 antibodies were infected by wild-type (WT) L. pneumophila or the isogenic dotA or ankB mutant for 15 min, followed immediately by fixation and processing for microscopy. Representative confocal microscopy images of infected hMDMs cells show colocalization of Skp1 and Cul1 proteins (red) with the LCV (green) (A) and with attached extracellular L. pneumophila (B). The arrowheads indicate intense colocalization of Skp1 or Cul1 with the WT bacteria. The numbers in the merged images of all panels are quantifications of the frequency of recruitment of Skp1 or Cul1 beneath attached extracellular bacteria. The data represent analyses of 100 infected cells and are representative of three independent experiments.
Elevated levels of cellular amino acids triggered by attached extracellular L. pneumophila.
The ultimate function of the LCV membrane-anchored AnkB effector is to generate high levels of cellular amino acids through host proteasomal degradation of K48-linked polyubiquitinated proteins (32). Therefore, we determined whether the injected AnkB by attached extracellular L. pneumophila resulted in elevated levels of cellular amino acids through degradation of the polyubiquitinated proteins assembled beneath bacterial attachment sites (21). To achieve this, cytochalasin D-treated hMDMs were infected by the wild-type strain or the isogenic ankB mutant L. pneumophila. Trypan blue straining of the cells showed that there was no detectable effect of cytochalasin D on permeability of the plasma membrane. The hMDMs were lysed, and the relative levels of free amino acids were determined by GC-MS. The data showed that attached wild-type L. pneumophila triggered a rapid rise in the levels of amino acids, relative to uninfected cells (Student's t test, P < 0.001) (Fig. 6). In contrast, attachment of the ankB mutant bacteria to hMDMs did not alter cellular levels of amino acids relative to uninfected cells (Fig. 6). Thus, translocation of AnkB by attached extracellular L. pneumophila results in increased levels of cellular amino acids, which are needed to block a potential starvation response and differentiation of L. pneumophila into the nonreplicative phase (see model in Fig. 7). This is the first example of a strategy by an intracellular pathogen to trigger rapid nutritional remodeling of the host cell upon attachment to the plasma membrane, and as a result, a gratuitous surplus of cellular amino acids is generated to support proliferation of the incoming pathogen.
FIG 6.
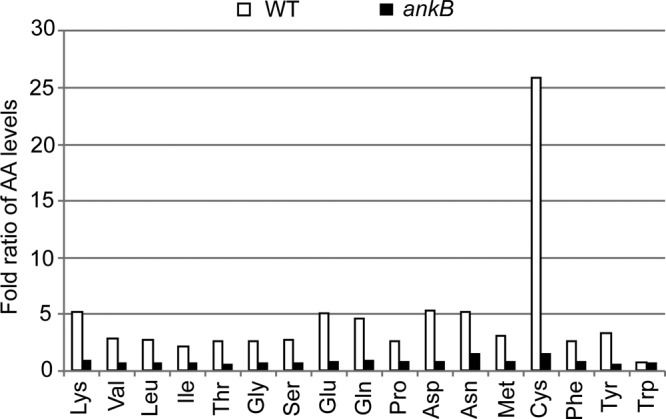
Intimate attachment of L. pneumophila to hMDMs triggers an increase in cellular levels of free amino acids. The hMDMs pretreated with cytochalasin D were infected for 15 min, followed by preparation of cellular lysates and determination of the relative levels of cellular amino acids by GC-MS. Amino acid levels are expressed as the fold ratio of infected/uninfected hMDMs. The analyses were performed in triplicate, and the data shown are from one of three representative experiments. The data represent analyses of 100 infected cells and are representative of three independent experiments.
FIG 7.
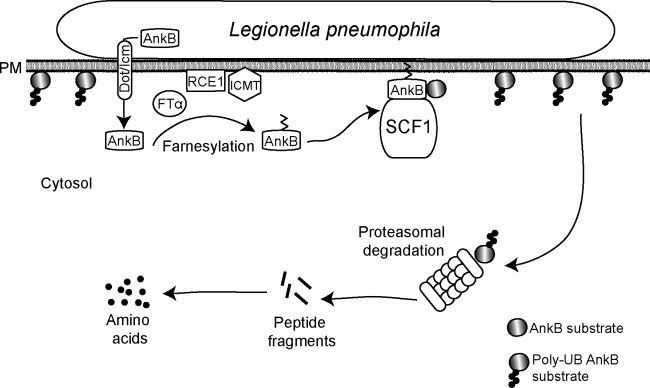
Working model of AnkB-mediated nutritional preparation of the host cell by attached extracellular L. pneumophila. Upon bacterial attachment, the Dot/Icm translocation system is triggered to inject bacterial effectors, one of which is AnkB. The host farnesylation enzymes are recruited beneath bacterial attachment sites and are essential for farnesylation of AnkB, which enables anchoring of this effector to the cytosolic side of the plasma membrane beneath bacterial attachment sites. The host SCF1 ubiquitin ligase complex is recruited to the plasma membrane, where it interacts with the F-box domain of AnkB to trigger assembly of K48-linked polyubiquitinated proteins beneath bacterial attachment sites. Proteasomal degradation of the K48-linked polyubiquitinated proteins generates higher levels of cellular amino acids, particularly the limiting ones, such as cysteine, which is a metabolically preferable source of carbon and energy for L. pneumophila. The availability of higher levels of cell amino acids upon entry of L. pneumophila circumvents a potential starvation response and differentiation into the motile nonreplicative phase for the entering bacterium. The elevated cellular levels of amino acids trigger differentiation of L. pneumophila into the replicative phase and are main sources of carbon and energy that feed the TCA cycle to power intracellular bacterial proliferation.
DISCUSSION
Upon entry of WT L. pneumophila into proteasome-inhibited cells or entry of the ankB mutant into untreated cells, both populations of bacteria exhibit a dramatic starvation response (32), which leads to bacterial differentiation into the nonreplicative phase (34, 35), and these differentiations are totally overcome upon supplementation of amino acids (32). Based on these observations, we tested the hypothesis that the function of AnkB is likely to be rapidly triggered upon attachment of L. pneumophila to the host cell to ensure that levels of amino acids in the host cell are sufficient to prevent a starvation response and a counterproductive differentiation into the nonreplication motile phase. Our data show that both expression and translocation of AnkB are rapidly triggered upon bacterial attachment to the host cell. Since translocation of 10 other Ank effectors of L. pneumophila requires bacterial entry (19, 20), our findings show that AnkB is among a small group of L. pneumophila effectors that are translocated by attached extracellular bacteria (44, 45). Upon its injection by attached bacteria, AnkB is the first L. pneumophila effector known to be exclusively anchored to the cytosolic side of the plasma membrane beneath the sites of bacterial attachment (see model in Fig. 7). Upon phagocytosis and initial formation of the phagosomal membrane from the plasma membrane, AnkB remains exclusively anchored to the outer leaflet of the LCV membrane (22), which is derived from the inner leaflet of the plasma membrane. Specific anchoring of AnkB to the inner leaflet of the plasma membrane upon bacterial attachment is dependent on eukaryotic farnesylation of the cysteine residue within the C-terminal CaaX motif of AnkB (22). Anchoring of AnkB to the plasma membrane by attached extracellular bacteria is essential for the biological function of AnkB (see model in Fig. 7). It would be interesting to determine the subcellular location of the AnkB homologue of the Paris strain of L. pneumophila (23), which lacks the farnesylation motif, upon bacterial attachment as well as after formation of the LCV. It is most likely that the structural differences in AnkB between various strains of L. pneumophila account for functional differences and role of the effector in intracellular replication (26), similar to what has been recently shown for the AnkI effector (19) (LegAS4/RomA) of two different strains of L. pneumophila (26, 46, 47). However, genomic redundancy may also play a factor (1, 9). Nevertheless, AnkB is the first example of an injected effector by attached extracellular L. pneumophila in which host-mediated farnesylation anchors it to the cytosolic side of the plasma membrane exclusively beneath bacterial attachment sites.
We have previously demonstrated that the farnesylation enzymes cytosolic FTase and ER-bound RCE1 and IcmT localize to the LCV in a Dot/Icm-dependent manner (22). Our data show that all three farnesylation enzymes are rapidly recruited to the plasma membrane beneath bacterial attachment sites (Fig. 7). Recruitment of these enzymes is Dot/Icm dependent but is only partially dependent on AnkB, since other farnesylated Dot/Icm-translocated effectors are likely to be involved in recruitment of the farnesylation machinery at the plasma membrane (28, 29).
During infection, the host SCF1 ubiquitin ligase is recruited to the LCV, indicating that AnkB-SCF1 interaction occurs on the LCV membrane (21, 27). Importantly, recruitment of SCF1 is initiated at the inner leaflet of the plasma membrane beneath bacterial attachment sites, similar to the host farnesylation machinery (Fig. 7). This recruitment is only partially dependent on AnkB, indicating that other L. pneumophila F-box proteins (23, 31, 48, 49) are likely to be involved in recruitment of the SCF1 complex to the plasma membrane beneath sites of bacterial attachment. The ability of L. pneumophila to rapidly recruit both the farnesylation machinery and the E3 SCF1 ubiquitin ligase machinery to the cytosolic side of the plasma membrane beneath sites of bacterial attachment allows AnkB to assemble K48-linked polyubiquitinated proteins on the plasma membrane beneath bacterial attachment sites (Fig. 7) (21, 32). Proteasomal degradation of the AnkB-assembled K48-linked polyubiquitinated proteins triggered by injection of AnkB by attached extracellular bacteria generates higher levels of cellular amino acids upon entry of L. pneumophila into the host cell (Fig. 7). As amino acids are the main sources of carbon and energy production by L. pneumophila through metabolism in the TCA cycle (40), our findings are the first example of rapid microbial trigger of nutritional preparation of the host cell upon entry of the microbe.
Rapid nutritional remodeling of the host cell upon contact and invasion of the host cell by L. pneumophila (Fig. 7) is most likely linked to the biphasic life cycle of L. pneumophila, since nutritional availability seems to be the main known trigger of this differentiation (34). L. pneumophila switches from a motile nonreplicative transmissive form to a nonmotile replicative form following invasion of a host cell and availability of sufficient nutrients to circumvent a starvation response (34, 50, 51). To differentiate into the replicative form and suppress a potential starvation response upon entry into host cells, L. pneumophila requires higher levels of cellular amino acids to feed the TCA cycle (40). Therefore, what is the biological relevance of rapidly injecting AnkB by attached extracellular bacteria, since phagocytosis and bacterial entry are a rapid process? The AnkB-dependent generation of higher levels of cellular amino acids upon bacterial attachment and entry is clearly a bacterial strategy to circumvent a counterproductive starvation response and differentiation into the nonreplicative motile phase in response to the basal levels of cellular amino acids. This will ensure nutritional preparation of the host cell through a rapid trigger to raise the levels of cellular amino acids needed for nutrition of the incoming pathogen. Importantly, intracellular replication of L. pneumophila is not possible without elevated cellular levels of amino acids, particularly cysteine, which is the most limiting amino acid in eukaryotes (40) but ironically is a metabolically favorable amino acid for L. pneumophila to feed the TCA cycle upon conversion of cysteine to pyruvate (2, 32). As host nutrient retrieval strategies are potential targets for antimicrobial therapy (52), interference with AnkB function is a promising antibacterial therapy.
Our data highlight the complexity of the temporal and spatial dynamics of host-pathogen interaction and a novel microbial strategy to ensure nutritional preparation of the host cell upon bacterial occupancy. To achieve nutritional preparation of the host cell, attached extracellular L. pneumophila rapidly translocates AnkB, which exploits the host farnesylation and ubiquitin-proteasome systems at the plasma membrane to elevate cellular levels of amino acids upon bacterial occupancy. This is the first example of rapid host nutritional remodeling by an intracellular pathogen upon its attachment and entry into the host cell. It will be intriguing to examine whether other intracellular pathogens trigger nutritional remodeling of their host cell from the onset of infection to ensure an adequate supply of host nutrients to power intracellular bacterial proliferation and its high demands for gratuitous sources of carbon and energy.
ACKNOWLEDGMENTS
We thank J. Cox at the University of Utah Metabolomic Core Facility for the amino acid analyses.
The Y.A.K. lab is supported by Public Health Service Awards R01AI069321 and R21AI107978 from NIAID and by the commonwealth of Kentucky Research Challenge Trust Fund.
Footnotes
Published ahead of print 14 October 2013
REFERENCES
- 1.Isberg RR, O'Connor TJ, Heidtman M. 2009. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7:13–24. 10.1038/nrmicro1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Price CTD, Richards AM, Von Dwingelo JE, Samara HA. 2013. Legionella pneumophila synchronization of amino acid auxotrophy and its role in adaptation and pathogenic evolution to the amoeba host. Environ. Microbiol.. 10.1111/1462-2920.12290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Quadan T, Price CT, Abu Kwaik Y. 2012. Exploitation of evolutionarily conserved amoeba and mammalian processes by Legionella. Trends Microbiol. 20:299–306. 10.1016/j.tim.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richards AM, Von Dwingelo JE, Price CT, Abu Kwaik Y. 2013. Cellular microbiology and molecular ecology of Legionella-amoeba interaction. Virulence 4:307–314. 10.4161/viru.24290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Price C, Abu Kwaik Y. 2012. Amoebae and mammals deliver protein-rich Atkins diet meals to Legionella. Microbe 7:506–513 [Google Scholar]
- 6.Franco IS, Shuman HA, Charpentier X. 2009. The perplexing functions and surprising origins of Legionella pneumophila type IV secretion effectors. Cell. Microbiol. 11:1435–1443. 10.1111/j.1462-5822.2009.01351.x [DOI] [PubMed] [Google Scholar]
- 7.Luo ZQ. 2012. Legionella secreted effectors and innate immune responses. Cell. Microbiol. 14:19–27. 10.1111/j.1462-5822.2011.01713.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo Z-Q. 2011. Striking a balance: modulation of host cell death pathways by Legionella pneumophila. Front. Microbiol.. 10.3389/fmicb.2011.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo Z-Q. 2011. Targeting one of its own: expanding roles of substrates of the Legionella pneumophila Dot/Icm type IV secretion system. Front. Microbiol.. 10.3389/fmicb.2011.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molmeret M, Bitar D, Han L, Abu Kwaik Y. 2004. Disruption of the phagosomal membrane and egress of Legionella pneumophila into the cytoplasm during late stages of the intracellular infection of macrophages and Acanthamoeba polyphaga. Infect. Immun. 72:4040–4051. 10.1128/IAI.72.7.4040-4051.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Khodor S, Al-Quadan T, Abu Kwaik Y. 2010. Temporal and differential regulation of expression of the eukaryotic-like ankyrin effectors of L. pneumophila. Environ. Microbiol. Rep. 2:677–684. 10.1111/j.1758-2229.2010.00159.x [DOI] [PubMed] [Google Scholar]
- 12.Al-Khodor S, Kalachikov S, Morozova I, Price CT, Abu Kwaik Y. 2009. The PmrA/PmrB two-component system of Legionella pneumophila is a global regulator required for intracellular replication within macrophages and protozoa. Infect. Immun. 77:374–386. 10.1128/IAI.01081-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molmeret M, Jones S, Santic M, Habyarimana F, Esteban MT, Kwaik YA. 2010. Temporal and spatial trigger of post-exponential virulence-associated regulatory cascades by Legionella pneumophila after bacterial escape into the host cell cytosol. Environ. Microbiol. 12:704–715. 10.1111/j.1462-2920.2009.02114.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segal G, Purcell M, Shuman HA. 1998. Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc. Natl. Acad. Sci. U. S. A. 95:1669–1674. 10.1073/pnas.95.4.1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogel JP, Andrews HL, Wong SK, Isberg RR. 1998. Conjugative transfer by the virulence system of Legionella pneumophila. Science 279:873–876. 10.1126/science.279.5352.873 [DOI] [PubMed] [Google Scholar]
- 16.Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo ZQ. 2011. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6:e17638. 10.1371/journal.pone.0017638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cazalet C, Jarraud S, Ghavi-Helm Y, Kunst F, Glaser P, Etienne J, Buchrieser C. 2008. Multigenome analysis identifies a worldwide distributed epidemic Legionella pneumophila clone that emerged within a highly diverse species. Genome Res. 18:431–441. 10.1101/gr.7229808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Habyarimana F, Price CT, Santic M, Al-Khodor S, Kwaik YA. 2010. Molecular characterization of the Dot/Icm-translocated AnkH and AnkJ eukaryotic-like effectors of Legionella pneumophila. Infect. Immun. 78:1123–1134. 10.1128/IAI.00913-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Habyarimana F, Al-Khodor S, Kalia A, Graham JE, Price CT, Garcia MT, Kwaik YA. 2008. Role for the Ankyrin eukaryotic-like genes of Legionella pneumophila in parasitism of protozoan hosts and human macrophages. Environ. Microbiol. 10:1460–1474. 10.1111/j.1462-2920.2007.01560.x [DOI] [PubMed] [Google Scholar]
- 20.Al-Khodor S, Price CT, Habyarimana F, Kalia A, Abu Kwaik Y. 2008. A Dot/Icm-translocated ankyrin protein of Legionella pneumophila is required for intracellular proliferation within human macrophages and protozoa. Mol. Microbiol. 70:908–923. 10.1111/j.1365-2958.2008.06453.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price CT, Al-Khodor S, Al-Quadan T, Santic M, Habyarimana F, Kalia A, Kwaik YA. 2009. Molecular mimicry by an F-box effector of Legionella pneumophila hijacks a conserved polyubiquitination machinery within macrophages and protozoa. PLoS Pathog. 5:e1000704. 10.1371/journal.ppat.1000704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price CT, Al-Quadan T, Santic M, Jones SC, Abu Kwaik Y. 2010. Exploitation of conserved eukaryotic host cell farnesylation machinery by an F-box effector of Legionella pneumophila. J. Exp. Med. 207:1713–1726. 10.1084/jem.20100771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lomma M, Dervins-Ravault D, Rolando M, Nora T, Newton HJ, Sansom FM, Sahr T, Gomez-Valero L, Jules M, Hartland EL, Buchrieser C. 2010. The Legionella pneumophila F-box protein Lpp2082 (AnkB) modulates ubiquitination of the host protein parvin B and promotes intracellular replication. Cell. Microbiol. 12:1272–1291. 10.1111/j.1462-5822.2010.01467.x [DOI] [PubMed] [Google Scholar]
- 24.Al-Khodor S, Price CTD, Kalia A, Abu Kwaik Y. 2010. Functional diversity of ankyrin repeats in microbial proteins. Trends Microbiol. 18:132–139. 10.1016/j.tim.2009.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Quadan TP, Price CT, London N, Schueler-Furman O, Abu Kwaik Y. 2011. Anchoring bacterial effectors to host membranes through host-mediated prenylation: A common paradigm. Trends Microbiol. 19:573–579. 10.1016/j.tim.2011.08.003 [DOI] [PubMed] [Google Scholar]
- 26.Price CT, Abu Kwaik Y. 2013. One bacterial effector with two distinct catalytic activities by different strains. EMBO Rep. 14:753–754. 10.1038/embor.2013.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Quadan T, Abu Kwaik Y. 2011. Molecular characterization of exploitation of the polyubiquitination and farnesylation machineries of Dictyostelium discoideum by the AnkB F-box effector of Legionella pneumophila. Front. Microbiol. 10.3389/fmicb.2011.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price CT, Jones SC, Amundson KE, Abu Kwaik Y. 2010. Host-mediated post-translational prenylation of novel Dot/Icm-translocated effectors of Legionella pneumophila. Front. Microbiol. 10.3389/fmicb.2010.00131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanov SS, Charron G, Hang HC, Roy CR. 2010. Lipidation by the host prenyltransferase machinery facilitates membrane localization of Legionella pneumophila effector proteins. J. Biol. Chem. 285:34686–34698. 10.1074/jbc.M110.170746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulman BA, Carrano AC, Jeffrey PD, Bowen Z, Kinnucan ER, Finnin MS, Elledge SJ, Harper JW, Pagano M, Pavletich NP. 2000. Insights into SCF ubiquitin ligases from the structure of the Skp1-Skp2 complex. Nature 408:381–386. 10.1038/35042620 [DOI] [PubMed] [Google Scholar]
- 31.Price CT, Abu Kwaik Y. 2010. Exploitation of host polyubiquitination machinery through molecular mimicry by eukaryotic-like bacterial F-box effectors. Front. Microbiol.. 10.3389/fmicb.2010.00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price CT, Al-Quadan T, Santic M, Rosenshine I, Abu Kwaik Y. 2011. Host proteasomal degradation generates amino acids essential for intracellular bacterial growth. Science 334:1553–1557. 10.1126/science.1212868 [DOI] [PubMed] [Google Scholar]
- 33.Hammer BK, Swanson MS. 1999. Co-ordination of Legionella pneumophila virulence with entry into stationary phase by ppGpp. Mol. Microbiol. 33:721–731. 10.1046/j.1365-2958.1999.01519.x [DOI] [PubMed] [Google Scholar]
- 34.Dalebroux ZD, Svensson SL, Gaynor EC, Swanson MS. 2010. ppGpp conjures bacterial virulence. Microbiol. Mol. Biol. Rev. 74:171–199. 10.1128/MMBR.00046-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Molofsky AB, Swanson MS. 2004. Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol. Microbiol. 53:29–40. 10.1111/j.1365-2958.2004.04129.x [DOI] [PubMed] [Google Scholar]
- 36.Rasis M, Segal G. 2009. The LetA-RsmYZ-CsrA regulatory cascade, together with RpoS and PmrA, post-transcriptionally regulates stationary phase activation of Legionella pneumophila Icm/Dot effectors. Mol. Microbiol. 72:995–1010. 10.1111/j.1365-2958.2009.06705.x [DOI] [PubMed] [Google Scholar]
- 37.Altman E, Segal G. 2008. The response regulator CpxR directly regulates expression of several Legionella pneumophila icm/dot components as well as new translocated substrates. J. Bacteriol. 190:1985–1996. 10.1128/JB.01493-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feldman M, Segal G. 2007. A pair of highly conserved two-component systems participates in the regulation of the hypervariable FIR proteins in different Legionella species. J. Bacteriol. 189:3382–3391. 10.1128/JB.01742-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dorer MS, Kirton D, Bader JS, Isberg RR. 2006. RNA interference analysis of Legionella in Drosophila cells: exploitation of early secretory apparatus dynamics. PLoS Pathog. 2:e34. 10.1371/journal.ppat.0020034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abu Kwaik Y, Bumann D. 2013. Microbial quest for food in vivo: “nutritional virulence” as an emerging paradigm. Cell. Microbiol. 15:882–890. 10.1111/cmi.12138 [DOI] [PubMed] [Google Scholar]
- 41.Santic M, Asare R, Skrobonja I, Jones S, Abu Kwaik Y. 2008. Acquisition of the vacuolar ATPase proton pump and phagosome acidification are essential for escape of Francisella tularensis into the macrophage cytosol. Infect. Immun. 76:2671–2677. 10.1128/IAI.00185-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santic M, Molmeret M, Barker JR, Klose KE, Dekanic A, Doric M, Abu Kwaik Y. 2007. A Francisella tularensis pathogenicity island protein essential for bacterial proliferation within the host cell cytosol. Cell. Microbiol. 9:2391–2403. 10.1111/j.1462-5822.2007.00968.x [DOI] [PubMed] [Google Scholar]
- 43.Charpentier X, Gabay JE, Reyes M, Zhu JW, Weiss A, Shuman HA. 2009. Chemical genetics reveals bacterial and host cell functions critical for type IV effector translocation by Legionella pneumophila. PLoS Pathog. 5:e1000501. 10.1371/journal.ppat.1000501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagai H, Cambronne ED, Kagan JC, Amor JC, Kahn RA, Roy CR. 2005. A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc. Natl. Acad. Sci. U. S. A. 102:826–831. 10.1073/pnas.0406239101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cambronne ED, Roy CR. 2007. The Legionella pneumophila IcmSW complex interacts with multiple Dot/Icm effectors to facilitate type IV translocation. PLoS Pathog. 3:e188. 10.1371/journal.ppat.0030188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rolando M, Sanulli S, Rusniok C, Gomez-Valero L, Bertholet C, Sahr T, Margueron R, Buchrieser C. 2013. Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe 13:395–405. 10.1016/j.chom.2013.03.004 [DOI] [PubMed] [Google Scholar]
- 47.Li T, Lu Q, Wang G, Xu H, Huang H, Cai T, Kan B, Ge J, Shao F. 2013. SET-domain bacterial effectors target heterochromatin protein 1 to activate host rDNA transcription. EMBO Rep. 14:733–740. 10.1038/embor.2013.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cazalet C, Rusniok C, Bruggemann H, Zidane N, Magnier A, Ma L, Tichit M, Jarraud S, Bouchier C, Vandenesch F, Kunst F, Etienne J, Glaser P, Buchrieser C. 2004. Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat. Genet. 36:1165–1173. 10.1038/ng1447 [DOI] [PubMed] [Google Scholar]
- 49.Ensminger AW, Isberg RR. 2010. E3 ubiquitin ligase activity and targeting of BAT3 by multiple Legionella pneumophila translocated substrates. Infect. Immun. 78:3905–3919. 10.1128/IAI.00344-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dalebroux ZD, Edwards RL, Swanson MS. 2009. SpoT governs Legionella pneumophila differentiation in host macrophages. Mol. Microbiol. 71:640–658. 10.1111/j.1365-2958.2008.06555.x [DOI] [PubMed] [Google Scholar]
- 51.Faucher SP, Mueller CA, Shuman HA. 2011. Legionella pneumophila transcriptome during intracellular multiplication in human macrophages. Front. Microbiol.. 10.3389/fmicb.2011.00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abu Kwaik Y. 14 October 2013. Targeting nutrient retrieval by Francisella tularensis. Front. Cell. Infect. Microbiol. 10.3389/fcimb.2013.00064 [DOI] [PMC free article] [PubMed] [Google Scholar]