

Abstract
N6-methyladenosine (m6A) is the most prevalent and reversible internal modification in mammalian messenger and non-coding RNAs. We report here that human METTL14 catalyzes m6A RNA methylation. Together with METTL3, the only previously known m6A methyltransferase, these two proteins form a stable heterodimer core complex of METTL3-14 that functions in cellular m6A deposition on mammalian nuclear RNAs. WTAP, a mammalian splicing factor, can interact with this complex and affect this methylation.
N6-methyladenosine (m6A) is a conserved internal modification found in almost all eukaryotic nuclear RNAs1–3, as well as in the viral RNA that replicates inside host nuclei4. The discoveries of functionally significant demethylases that reverse this methylation5,6, together with the recently revealed m6A distributions in mammalian transcriptomes7,8, strongly indicate regulatory functions of this dynamic modification.
The m6A modification is post-transcriptionally installed by a multi-component N6-adenosine methyltransferase (MT) complex yet to be fully identified and characterized. Of a ~200 kD MT complex isolated from mammalian cell nuclear extract that exhibits methyltransferase activity, only a 70 kD protein was identified and named MT-A70 or METTL3 (methyltransferase like 3)9. The knockdown of METTL3 led to apoptosis of human HeLa cells1, while the deficiency of its homologues in other species resulted in developmental arrest or defects in gametogenesis10–12.
In mammals, the mRNA methylation occurs within a consensus sequence of Pu[G>A]m6AC[A/C/U] (Pu=purine) though only a portion of these putative methylation sites contain m6A (ref. 7). How the methylation pattern and methylation level on mRNA are regulated in vivo is unclear largely because the methyltransferase complex itself has not yet been revealed. A phylogenetic analysis of the MT-A70 (METTL3) family of methyltransferases suggested that METTL14 (methyltransferase like 14), which shares 43% identity with METTL3 (Supplementary Results, Supplementary Note 1), is a homologue of METTL313. METTL3 and METTL14 are highly conserved in mammals (Supplementary Notes 2, 3), a feature that prompted us to ask if METTL14 contributes to mRNA methylation in mammalian cells. Meanwhile, recent studies in Arabidopsis and yeast suggested that homologues of a mammalian pre-mRNA splicing regulator WTAP (Wilms’ Tumor1-Associating Protein)14 are involved in RNA methylation12,15. We therefore also included WTAP in our investigation.
We knocked down METTL3, METTL14, and WTAP, respectively, to check the m6A levels in HeLa and 293FT cells using siRNAs (over 80% knockdown after 48 h; Supplementary Fig. 1 and Table 1). The LC-MS/MS results indicated that knockdown of cellular METTL3, METTL14, and WTAP decreased the m6A level in polyadenylated RNA by ~30%, ~40%, and ~50% in HeLa cells, respectively, and ~20%, ~35%, and ~42% in 293FT cells, respectively (Fig. 1a, Supplementary Fig. 2). Both METTL14 and WTAP affect them6A level more significantly than METTL3. In contrast, when we knocked down METTL4 (close to 80% knockdown efficiency), a close mammalian homologue of METTL3 and METTL14, we did not observe any noticeable change of the m6A level in the isolated polyadenylated RNA (Supplementary Fig. 1a).
Figure 1. METTL3, METTL14, and WTAP affect the cellular m6A level in polyadenylated RNA with METTL3 and METTL14 forming a stable complex.
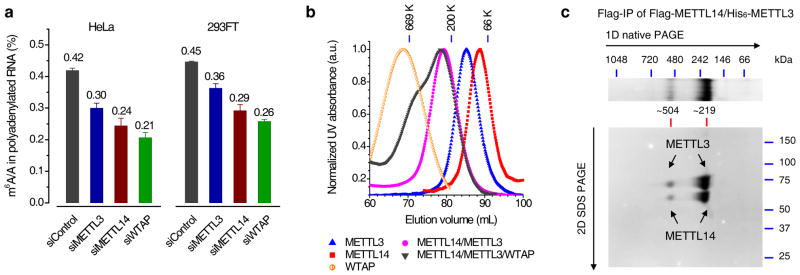
(a) LC-MS/MS quantification of the m6A/A ratio in polyadenylated RNA isolated from HeLa and 293FT with the control and single knockdown of METTL3, METTL14, or WTAP. Both groups of data were assessed using student’s t-test with P value < 1e-6 (calculated between control and specific knockdown sample). Error bars indicate mean ± s.d. (n = 10 for HeLa, five biological replicates × two technical replicates, and n = 8 for 293FT, four biological replicates × two technical replicates). (b) Gel filtration traces of individual Flag-tagged METTL3, METTL14, and WTAP, co-expressed Flag-METTL14/His6-METTL3 as well as mixed Flag-METTL14/Flag-METTL3/Flag-WTAP with equal molar amount. All proteins were expressed in insect cells and purified by Flag-IP. Markers: 669 kDa (thyroglobulin, bovine), 200 kDa (β-amylase from sweet potato), and 66 kDa (bovine serum albumin). (c) Coomassie staining of two-dimensional native/SDS PAGE of the Flag-IP product from insect cells co-expressing Flag-METTL14/His6-METTL3. The band of ~219 kDa corresponds to the METTL3-14 heterodimer, while the band of ~504 kDa represents dimer of dimer. Full images of gels are presented in Supplementary Fig. 15.
We expressed the recombinant proteins of METTL3, METTL14, and WTAP from insect cells (with different tags including Flag, GST, and His6) for biochemical characterizations (Supplementary Fig. 3a). Each Flag-tagged protein was purified by the anti-Flag resins and subjected to gel filtration analysis. METTL3 and METTL14 form a stable METTL3-14 complex in the gel filtration experiment (Fig. 1b and Supplementary Fig. 3b, c). Subsequent two-dimensional native/SDS PAGE analysis of the co-expressed METTL3 and METTL14 further confirmed formation of a complex between these two proteins with a stoichiometry of 1/1 (Fig. 1c and Supplementary Fig. 3d). WTAP appears to form aggregates as revealed by its much larger apparent molecular weight calculated from the gel filtration trace (Fig. 1b). WTAP can bind to the METTL3-14 complex; however, a much lower stoichiometry of WTAP to METTL3 or METTL14 was observed in the co-immunoprecipitations (co-IP) experiment (Supplementary Fig. 3a), indicating a relatively weaker interaction between WTAP and these two methyltransferases.
To study the cellular interactions among these proteins Flag-tagged METTL3, METTL14, or WTAP were expressed in HeLa cells and pulled down by the anti-Flag beads. Western blotting, silver staining, and mass spectrometry protein identification were employed in order to characterize the protein components in each IP fraction (Supplementary Fig. 4a and Supplementary Tables 4–9). Indeed, the pull-down product in each IP experiment contained the other two proteins. Close examination of the cell extract input, IP, and flow-through (FT) fractions by western blotting led us to conclude that METTL3 and METTL14 exist as a stable complex inside cells (Supplementary Fig. 4b). Consistent with the in vitro observation, the interactions between WTAP and the two methyltransferases are weaker. As a control, none of the IP products contained the homologous methytransferase METTL4 (Supplementary Fig. 4c).
Next, we tested N6-adenosine methylation activity for each recombinant protein and the combinations of METTL3/METTL14 toward various synthetic RNA probes with or without the consensus sequence GGACU and the stem-loop secondary structure (Fig. 2 and Supplementary Fig. 5). S-(5′-Adenosyl)-L-methionine-d3 (d3-SAM, 1) with a deuterium-substituted methyl group was utilized as the cofactor for accurate mass spectrometry quantification. We calculated the molar ratio of the formed d3-m6A (2) to each RNA probe in order to quantify the methylation efficiency. Since RNA probe 1 has two consensus motifs located in both stem and loop, the theoretical maximum value for d3-m6A/probe is 2. All other probes have the d3-m6A/probe maxima of 1.
Figure 2. In vitro methylation activity of METTL3, METTL14, and WTAP.

The in vitro RNA N6-adenosine methylation activities of Flag-tagged METTL3, METTL14, or WTAP as well as the combination of METTL3 and METTL14 were tested using different RNA probes (numbered 1–6) with/without the consensus sequence of GGACU in the stem and/or loop, and with/without a stem-loop secondary structure in the presence of isotope-labeled cofactor d3-SAM (S-(5′-adenosyl)-L-methionine-d3). The methylation yields were calculated based on the molar ratio of d3-m6A to specific probe, measured by LC-MS/MS. Error bars indicate mean ± s.d., n = 4 (two biological replicates × two technical replicates).
WTAP itself showed no methyltransferase activity with all probes tested, while both METTL3 and METTL14 exhibited methyltransferase activity with METTL14 showing much higher activity (close to 10-fold with several probes) than METTL3 (Fig. 2). For instance, when RNA probe 1 was tested, Flag-tagged METTL14 afforded a d3-m6A/probe value of 0.24 compared to 0.02 of that observed for METTL3 under the same conditions. Furthermore, the combination of METTL3/METTL14 (~1.04, d3-m6A/probe) dramatically enhances methyltransferase activity compared to each individual protein, demonstrating a synergistic effect. The same reactivity trend was also observed for other probes containing the consensus sequence. In addition, when we combined the cellular IP products of METTL3 and METTL14 and compared the methylation activity with that of the each individual IP fraction, the same synergistic effect was observed (Supplementary Fig. 4d).
Both METTL3 and METTL14 preferentially methylate RNA substrates containing the previously revealed consensus sequence. When RNA probes 1 and 5 (contain a GGACU motif) were used, ~11- to 64-fold higher methylation efficiency was observed when compared to the same experiments conducted using RNA probes 4 and 6, which possess a change of C to U (GGAUU) in the consensus sequence of GGACU (Fig. 2). We further synthesized RNA probes 2 and 3 with only one GGACU sequence in the loop or stem, respectively. The combination of METTL3/METTL14 appears to exhibit slightly higher activity towards probe 3 (GGACU in the stem) than probe 2 (GGACU in the loop). Since very little activity was observed for probe 4, our results confirm that the GGACU sequence is required for optimal activity either in the loop or stem. Additionally, the METTL3/METTL14 combination showed very high activity to probe 5 with a random structure, indicating no obvious preference of these methyltransferases to the RNA secondary structure for m6A deposition. These results indicate that these methyltransferases have sequence specificity but show less structural preference to RNA substrates.
We next isolated the native HeLa cell nuclear extract (NE) according to the published procedure (Supplementary Fig. 6a)16, and tested the methylation activity of each NE fraction at each separation step (Fig. 3a). As a result, the NE fractions A2-(19–21), which exhibited the highest activity, were found to be enriched with METTL3 and METTL14 (Fig. 3a). These three fractions were combined. Western blotting of native PAGE gel of the combined fractions showed that both METTL3 and METTL14 migrate to the same front marked with an apparent molecular weight of ~240 kDa, indicating their presence in a heterodimer complex (Supplementary Fig. 6b). Indirect immunofluorescence analysis was employed with the imaging results in HeLa cells clearly demonstrating that METTL3, METTL14, and WTAP co-localize well with each other (Pearson correlation coefficient, PCC = 0.83–0.91) in well-defined, irregular dot-like structures inside the nucleus, with no fluorescence signal observed in the cytoplasm (Supplementary Fig. 7). We also found that these three proteins all co-localize well with various pre-mRNA processing factors residing in the nuclear speckles (Supplementary Fig. 8). Taken together, we concluded that METTL3 and METTL14 can form a stable complex to perform the m6A methylation function inside mammalian cells. WTAP can interact with this complex to affect cellular m6A deposition.
Figure 3. RNA N6-adenosine methylation by the METTL3-14 complex isolated from HeLa cell nuclear extract and identification of their RNA-binding sites.

(a) Left panel: Methylation activity tests of HeLa cell nuclear extracts, which were separated step-by-step according to the scheme shown in Supplementary Fig. 6a. Right panel: Methylation activity of gel filtration products of NE-A2. The corresponding western blotting of each fraction was shown at the bottom of each panel. Error bars indicate mean ± s.d., n = 2 (two biological replicates). Full images of gels are presented in Supplementary Fig. 16. (b) Consensus motifs identified within 4SU-PAR-CLIP binding sites of METTL3 (P = 1e-93), METTL14 (P = 1e-47), and METTL3/METTL14 overlay (P = 1e-79). (c) A schematic illustration for the reversible methylation of N6-adenosine in RNA. METTL3 and METTL14 form a heterodimeric methyltransferase complex within the cell nuclei that performs the RNA methylation function. WTAP interacts with the METTL3-14 complex to affect the m6A deposition. The demethylase removes the m6A mark, demonstrating a reversible process.
We further performed PAR-CLIP (photoactivatable ribonucleoside enhanced crosslinking and immunoprecipitation)17 with 4SU (4-thiouridine)-treated HeLa cells to identify the direct binding sites of these proteins. The crosslinked RNA segments were isolated, converted into cDNA libraries, and subjected to high-throughput sequencing. Analyses of sequenced clusters yielded the enriched binding motifs of GGAC for METTL14 and METTL3, and GACU for WTAP (Fig. 3b and Supplementary Fig. 9a), all of which are consistent with the previously identified consensus sequence of Pu[G>A]m6AC[A/C/U] for m6A. METTL14 and METTL3 share an average of ~56% binding sites in common while all three proteins have ~36% common binding sites (Supplementary Fig. 9b).
Our subsequent analyses revealed that a large fraction of the binding sites for all three proteins fall into intergenic regions (~43–49%) and introns (~29–34%) (Supplementary Fig. 9c and Supplementary Data Sets 1–3). Given that METTL3-14 is thought to work on pre-mRNA and is co-localized with splicing factors in the nuclear speckles, it may not be surprising that many intron sites were identified. The binding sites that could be assigned to genes are mainly located in CDS, and then 3′UTR (Supplementary Fig. 9d), consistent with the peak distribution of m6A in mammalian nuclear RNA7,8. METTL3 (2,110 genes) and METTL14 (1,147 genes) share an average of ~57% common target genes (Supplementary Fig. 9e). Notably, 261 of the METTL3 target genes are enriched in the functional pathways related to “cell death and cellular response to stress” (Supplementary Fig. 10). Consistent with the previous report1, the viability of HeLa cells was dramatically reduced after 72 h knockdown of METTL3. The knockdown of the other two proteins also led to cell death (Supplementary Fig. 11).
The PAR-CLIP sites were compared to the transcriptome-wide m6A distribution obtained in the same cell line. The genes identified by PAR-CLIP of the three proteins have ~50% overlap with the m6A-containing genes (Supplementary Fig. 12). Meanwhile, we found that silencing of METTL3, METTL14, or WTAP led to noticeable increases (~15–26%) of the abundance of their m6A target transcripts compared to the average abundance of all transcripts (Supplementary Fig. 13 and Supplementary Data Sets 4–6). We further revealed that the reduced global m6A methylation increases the lifetimes of nascent RNAs (Supplementary Fig. 14). Together, these results indicate an overall negative impact on gene expression by installing m6A on mRNA, consistent with an m6A-dependent mRNA degradation process as one main function of the m6A methylation18.
In summary, we discovered a new methyltransferse METTL14 that forms a stable heterodimer with METTL3. This METTL3-14 dimer mediates m6A deposition on nuclear RNA inside mammalian cells. WTAP does not possess methylation activity, but it interacts with the METTL3-14 complex to significantly affect cellular m6A deposition. The METTL3-14 complex and the recently discovered m6A RNA demethylases could dynamically regulate m6A in mRNA and other nuclear RNA through opposing enzymatic functions (Fig. 3c). Contributions from each component to the methylation and their impacts on different biological pathways need to be carefully investigated. By characterizing the methyltransferase core complex, this study provides the basis for these future mechanistic investigations of biological functions associated with m6A deposition1,2,5,6,7,8,19.
ONLINE METHODS
Cloning and expression of METTL3, METTL14, and WTAP
The recombinant proteins METTL3 (GenBank Accession No. NP_062826.2), METTL14 (GenBank Accession No. NP_066012.1), and WTAP (GenBank Accession No. NP_001257460.1) were expressed in insect cells with different N-terminal tags (Flag, His6, and GST) by using Bac-to-Bac baculovirus expression system according to a previously published procedure20. The METTL3, METTL14, and WTAP plasmids used for over-expression in mammalian cells were constructed by cloning the corresponding cDNAs (Open Biosystems) into a mammalian vector pcDNA3 (Invitrogen) with N-terminal Flag tag.
Cell lines, antibodies, chemicals, and others
Human HeLa and 293FT cell lines were used in this study. The primary antibodies were purchased from commercial sources: rabbit anti-METTL3 for western blot (15073-1-AP; Proteintech Group); mouse anti-METTL3 for immunostaining (H00056339-B01P; Novus); rabbit anti-METTL14 (HPA038002; Sigma); mouse anti-WTAP (60188-1-lg, Proteintech Group); rabbit anti-METTL4 (ab107540; abcam); goat HRP-anti-GAPDH (A00192; GenScript); mouse HRP-anti-Flag for western blot (A5892; Sigma); rat anti-Flag for immunostaining (637304; Biolegency); rabbit anti-m6A (202003; Synaptic Systems); mouse anti-SC35 (S4045; Sigma); mouse anti-SM (ab3138; Abcam); mouse anti-ASF/SF2 (32-4600; Invitrogen); and mouse anti-SRPK1 (611072; BD Transduction Laboratories). The secondary antibodies used for immunostaining: Alexa 488-conjugated donkey anti-rat IgG (H+L) (A21208; Molecular Probes); Alexa 488-conjugated goat anti-rabbit IgG (H+L) (A11034; Molecular Probes); Alexa 594-conjugated goat anti-mouse IgG (H+L) (A11032; Molecular Probes), and Alexa 647-conjugated goat anti-mouse IgG (H+L) (A21236; Molecular Probes).
d3-SAM (S-(5′-adenosyl)-L-methionine-d3) was purchased from C/D/N ISOTOPES INC (D-4093). N6-CD3-Adenosine free nucleoside was synthesized according to the Supplementary Note 4. The SDS PAGE (NP0335BOX and NP0336BOX), native PAGE (BN1003BOX and BN1001BOX), and two-dimensional native/SDS PAGE (NP0326BOX) gels as well as the corresponding gel staining kits were purchased from Invitrogen.
Mammalian cell culture, siRNA knockdown, and plasmid transfection
Human HeLa cell line was grown in DMEM (Gibco, 11965) media supplemented with 10% FBS and 1% 100× Pen Strep (Gibco, 15140). Human 293FT cell line was grown in DMEM (Gibco, 11995) media supplemented with 10% FBS and 1% 100× Pen Strep. METTL3, METTL14, METTL4, and WTAP siRNAs were purchased from QIAGEN with sequences shown in Supplementary Table 1. Transfection was achieved by using Lipofectamine RNAiMAX (Invitrogen) for siRNA, or Lipofectamine 2000 (Invitrogen) for the plasmid following the manufacturer’s protocols.
RNA isolation for LC-MS/MS
Total RNA was isolated from transiently transfected cells with TRIZOL reagent (Invitrogen). Polyadenylated RNA was extracted using PolyATtract mRNA isolation systems IV (Promega), followed by removal of contaminated rRNA with RiboMinus transcriptome isolation kit (Invitrogen).
RNA m6A quantification by LC-MS/MS
The polyadenylated RNA from transfected cells was isolated using a biotinylated poly(dT) probe followed by a rRNA depletion step to ensure depletion of rRNA (Supplementary Fig. 1e). The isolated RNA was subsequently digested into nucleosides and the amount of m6A was measured by LC-MS/MS following the published procedure5; the total contents of m6A and A were quantified based on the corresponding standard curves generated using pure standards (Supplementary Fig. 2), from which the m6A/A ratio was calculated. Typically, 200–300 ng of polyadenylated RNA was digested by nuclease P1 (2 U) in 25 μL of buffer containing 25 mM NaCl, and 2.5 mM ZnCl2 at 37 °C for 2 h, followed by additions of NH4HCO3 (1 M, 3 μL) and alkaline phosphatase (0.5 U) and incubation at 37 °C for 2 h. The sample was then filtered (0.22 μm pore size, 4 mm diameter, Millipore), and 5 μL of the solution was injected into LC-MS/MS. The nucleosides were separated by reverse phase ultra-performance liquid chromatography on a C18 column with online mass spectrometry detection using Agilent 6410 QQQ triple-quadrupole LC mass spectrometer in positive electrospray ionization mode. The nucleosides were quantified by using the nucleoside-to-base ion mass transitions of 285 to 153 (d3-m6A), 282 to 150 (m6A), and 268 to 136 (A). Quantification was performed in comparison with the standard curve obtained from pure nucleoside standards running on the same batch of samples. The ratio of m6A to A was calculated based on the calibrated concentrations.
Biochemistry assay for m6A methyltransferase activity in vitro
In vitro methyltransferase activity assay was performed in a standard 50 μL of reaction mixture containing the following components: 0.15 nmol RNA probe, 0.15 nmol each recombinant protein (single METTL3, METTL14, WTAP, or their combinations with a molar ratio of 0.15 nmol/0.15 nmol for two components, 0.8 mM d3-SAM, 80 mM KCl, 1.5 mM MgCl2, 0.2 U μL−1 RNasin, 10 mM DTT, 4% glycerol, and 15 mM HEPES (pH 7.9). Prior to the reaction, the RNA probes were annealed with a program of (i) 90 °C for 3 min, and (ii) −2 °C/cycle for 40 cycles within 30 min.
The reaction was incubated at 16 °C for 12 h. The resultant RNA was recovered by phenol/chloroform (low pH) extraction followed by ethanol precipitation, and digested by nuclease P1 and alkaline phosphatase for QQQ LC-MS/MS analysis. The nucleosides were quantified by using the nucleoside to base ion mass transitions of 285 to 153 (d3-m6A) and 284 to 152 (G). G served as an internal control to calculate the amount of RNA probe in each reaction mixture.
RNA probes
The sequence of RNA probe 1 was derived from Rous sarcoma virus RNA21. RNA probes 2–6 were designed in our lab. All of the probes were purchased from Integrated DNA Technologies.
In vitro and in vivo co-IP assays
For the in vitro co-IP assay to investigate the interactions between METTL3 and METTL14, METTL3 and WTAP, or METTL3 and METTL14, various combinations of proteins with different tags were co-expressed in insect cells. The driver protein was designed with Flag tag. The cell lysates were incubated with anti-Flag beads (Sigma), and the pull-down products were then analyzed by SDS or native PAGE and western blotting.
A typical example for in vitro IP is described as follows. The insect cells harvested from a 15-cm plate were lysed in 1.6 mL buffer A (20 mM Tris-HCl, pH 7.4, 300 mM NaCl, 10% glycerol, 1 mM DTT, and 1/100 protease inhibitor cocktail (Nacalai USA, Inc., Cat. no. 25955-11)) and sonicated for 2 min at a moderate amplitude. After removal of the cell debris by centrifuge at 14 krpm for 40 min, the supernatant of cell lysate was collected and incubated with 60 μL of magnetic anti-Flag beads for 2 h. The Flag-tagged protein and their interacting partner from the lysate were then immobilized on the beads, whereas the unbound proteins were washed away with 5 × 300 μL portions of buffer A. Subsequently, the protein-protein complex was eluted with 60 μL buffer A containing 0.3 mg/mL of 3× Flag peptides (Sigma). The eluted product was then analyzed by SDS PAGE and western blotting.
For in vivo co-IP, HeLa cells individually expressing Flag-tagged METTL3, METTL14, or WTAP were collected by cell lifter, and pelleted by centrifuge at 1 kpm for 5 min. The cell pellet was re-suspended with 2 volumes of lysis buffer (150 mM KCl, 10 mM HEPES (pH 7.6), 2 mM EDTA, 0.5% NP-40, 0.5 mM DTT, 1/100 protease inhibitor cocktail), and incubated on ice for 10 min. To remove the cell debris, the lysate solution was centrifuged at 15 krpm for 15 min at 4 °C, and the resultant supernatant was passed through a 0.22-μm membrane syringe filter. While 50 μL of cell lysate was saved as Input, the rest was incubated with the anti-Flag M2 magnetic beads in ice-cold NT2 buffer (200 mM NaCl, 50 mM HEPES (pH 7.6), 2 mM EDTA, 0.05% NP-40, 0.5 mM DTT, 1/100 protease inhibitor cocktail) for 4 h at 4 °C. The beads were collected by magnetic stand, and the supernatant was saved as Flow-through (FT). The beads were subsequently subjected to extensive wash with 8 × 1 mL portions of ice-cold NT2 buffer, followed by incubation with the elution solution containing 3× Flag peptide (0.5 mg/mL in NT2 buffer) at 4 °C for another 2 h. Finally, 3× Flag peptide was removed from the eluted solution by using a 5-kD cutoff column, and the resultant eluted products were saved as IP. All the Input, FT, and IP products were analyzed by SDS/native PAGE for silver staining and western blotting. Alternatively, the in vivo RNA N6-adenosine methylation activity of each IP product and their various combinations were tested as described above.
Protein identification
The in-gel samples were trypsin digested, resuspended in 12 μL and run by LC-MS/MS using LTQ-Orbitrap Velos with 1 h gradient, which was performed by the Proteomics & Mass Spectrometry Facility, at Donald Danforth Plant Science Center. The data was searched against the NCBInr_Human database. The recommended criteria for a significant protein identification based on publication requisitions are: (1) at least 2 unique peptides per protein identified; (2) each peptide must show a probability higher than 90% (about a Mascot Ion Score higher than 30). Filters were locked using these criteria and data were manually inspected in order to ensure that only significant proteins were identified.
Nuclear extract preparation
The nuclear extract preparation was performed according to the published procedure with a slight modification as shown in Supplementary Fig. 616. To each buffer used 0.5 mM DTT and 0.5 mM PMSF were added. The dialysis time was shortened to 6–8 h in order to avoid protein precipitation. Gel filtration was run on a Superdex-200 column (Pharmacia).
Immunofluorescence microscopy and cell imaging
HeLa cells were seeded on an 8-well chamber (Lab-Tek) at a density of 3 × 104 cells/well. After treatment indicated in each experiment, cells were fixed with 100% methanol for 15 min at −20 °C, and then permeabilized with 0.1% Triton X-100 and 0.5% NP-40 in PBS for 15 min on ice. After 30-min of blocking in 10% FBS in PBST (PBS with 0.05% Tween-20), cells were incubated sequentially with an appropriate amount of primary antibodies for 12 h, and then secondary antibodies for 30 min. Finally, cells were mounted and counterstained with DAPI by using an anti-fade reagent with DAPI (S36939, Invitrogen).
Fluorescence images were captured by Leica SP5 II STED-CW super-resolution laser scanning confocal microscopy equipped with an oil immersion objective (63×, NA = 1.40). For double staining, control scans confirmed that no bleed-through was detectable under the conditions used. The co-localization was quantified by JAcoP (ImageJ plug-in), and the Pearson coefficients were acquired under Costes’ automatic threshold22.
PAR-CLIP
HeLa cells were transfected with the plasmid containing Flag-tagged METTL3, METTL14, or WTAP by Lipofectamine2000. At 6 h post-transfection, 4SU (4-thiouridine) was added to the cell culture medium with a final concentration of 150 μM. At 24 h post-transfection, PAR-CLIP was performed according to the previous protocol23 with the following modifications. For UV-crosslinking, plates of cells were irradiated on ice using Stratalinker 2400 (Stratagene) twice at 0.15 J/cm2 with a 1 min break. METTL3, METTL14, and WTAP were immunoprecipitated with the anti-Flag M2 magnetic beads, respectively. For RNase T1 digestion, we used 0.2 U/μL for 15 min (instead of 1 U/μL in the original protocol) for the initial cleavage in the lysates, and 30 U/μL for 15 min (instead of 100 U/μL) for the second digestion on the beads. The library was prepared by using the TruSeq small RNA sample preparation kit (Illumina), and 15 PCR cycles were used for the final library amplification step. Each experiment was conducted with two biological replicates; sequencing quality is shown in Supplementary Table 10.
m6A-seq
Scrambled control or siRNAs against METTL3, METTL14, and WTAP were transfected into HeLa cells, respectively, by using Lipofectamine RNAiMAX reagent. After 48 h, total RNA was isolated using TRIZOL reagent. Polyadenylated RNA was further enriched from total RNA using FastTrack MAG Maxi mRNA isolation kit (Invitrogen). In particularly, an additional DNase-I digestion step was applied to all the samples to avoid DNA contamination. RNA fragmentation, m6A-seq, and library preparation were performed according to the previously protocol24. Each experiment was conducted in two biological replicates, with their sequencing quality shown in Supplementary Table 11.
Pre-processing reads
The raw reads of PAR-CLIP were trimmed of 3′ adaptors by using the HOMER software. The reads shorter than 13 bp were discarded. Processed reads were aligned to the hg18 version of the human genome assembly, which was downloaded from the University of California at Santa Cruz (UCSC, http://genome.cse.ucsc.edu), by using the Bowtie software. First 36 bp of the sequence reads were used as seed sequence. Two mismatches at most were allowed.
The raw reads of m6A-seq were aligned to the hg18 version of the human genome assembly by using the TopHat software with default parameters. The RefSeq gene annotations were downloaded from UCSC Table Browser and used as the transcriptome indexes.
PAR-CLIP data analysis
PARalyzer was used to detect the reads groups and the binding sites of each specific protein. The T-to-C mutation was used for the identification of 4SU-PAR-CLIP binding sites. The parameters used in PARalyzer were referred to a previously published paper25.
m6A-seq data analysis
The m6A-enriched regions in each m6A-IP sample were extracted by using the model-based analysis of ChIP-seq (MACS) peak-calling algorithm26, with the corresponding m6A-Input sample serving as the input control. For each library, the enriched peaks with P < 1e-5 were used for further analysis. We used edgeR (R package) to find the peaks with differential m6A level (defined as the m6A-IP/Input ratio) upon the knockdown of METTL3, METTL14, or WTAP. P values were calculated by comparing the coefficients of the m6A-IP/Input ratios between the control and specific knockdown samples by using the glmLRT function. Peaks with a statistically significant decrease in m6A level (P < 0.05) upon knockdown were thus defined as the putative target peaks of the specific protein studied.
The gene differential expression analyses upon the knockdown of METTL3, METTL14, or WTAP were performed by using DESeq (R package). For each RefSeq gene, RPKM (reads per kilobase per million reads) value was calculated to represent m6A or RNA expression level.
Peak merge, annotation, and motif analysis
The PAR-CLIP binding sites or m6A peaks from two replicates were merged by using the command of mergePeaks in HOMER software. This command was also used to analyze the overlap of the PAR-CLIP binding sites and m6A peaks under different experiment conditions. Genome or exon annotation analysis of the PAR-CLIP sites was performed by using the command of annotatePeaks in HOMER software. Motifs with 5–7 bp length were detected within 50 bp around PAR-CLIP reads group by using Homer software. The sequences located in repeat regions were masked.
Cell proliferation assay
HeLa cells were seeded in a 96-well plate at an initial density of 5,000 cells per well. After 24 h, METTL3, METTL14, WTAP, or control siRNAs were transfected into cells using Lipofectamine RNAiMAX. The treated cells were incubated in a humidified environment with 5% CO2 at 37 °C for 48 h, 72 h, or 96 h. 20 μL of 5 mg/mL MTT reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Invitrogen) was then added to each well. The cells were further incubated for 4 h at 37 °C. The medium was then removed and replaced by 100 μL of DMSO. The plate was gently agitated for 15 min before the absorbance (A) at 490 nm was recorded by a microplate reader. The relative cell viability (x) was calculated by , where AsiRNA and Asicontrol represent the absorbance of cells treated with specific siRNA and control siRNA, respectively, while Ablank represents the absorbance of cells cultured with fresh culture medium.
RNA metabolism assay
Both nascent RNA synthesis and RNA stability were determined by using EU (5-ethynyl uridine) nascent RNA detection kit named Click-iT® RNA Alexa Fluro® 488 HCS Assay (Invitrogen, C10327) according to the manufacturer’s instructions. Briefly, HeLa cells were grown in black 96-well plate and used for nascent RNA synthesis. METTL3, METTL14, WTAP, and control siRNAs were transfected into cells, respectively, by using Lipofectamine RNAiMAX as described above. At 48 h post-transfection, EU was added to cell culture medium with a final concentration of 1 mM. After incubation at 37 °C for different time intervals as indicated, EU-labeled cells were stained with Alexa 488 fluorescent dye via click chemistry and then subjected to fluorescence intensity measurement by using Tecan Safire2 Multi-detection Microplate Reader. The amount of newly transcribed RNA was quantified by the normalization of relative fluorescent intensity units (RFU) to the protein mass of the cells, which is proportional to the cell number. Nascent RNA stability assay was performed by using HeLa cells in a similar way except that actinomycin D (SIGMA, A9415) was added 1 h after EU incorporation with a final concentration of 5 μg/mL and incubated until the indicated time. The degradation rate of RNA (k) was estimated by the following equation
where t is the transcription inhibition time (h), Nt and N0 stand for the RNA quantities at time t and 0. Through exponential decay fitting of Nt/N0 versus time, the k can be derived and thus the lifetime (t1/2) can be calculated.
Real-time PCR (RT-PCR)
RT-PCR was performed to assess the relative abundance and lifetimes of the selected target mRNAs in different siRNA-treated HeLa samples. To measure the mRNA lifetime, actinomycin D (5 μg/mL) was added to the cell culture medium at 48 h post-transfection, and incubated with cells for 0 h, 3 h, and 6 h, respectively. Total RNA was then isolated by RNeasy kit (QIAGEN) with an additional DNase-I digestion step on column. RT-PCR was performed by using the Platinum SYBR Green One-Step qRT-PCR kit (Invitrogen) with 200–400 ng of total RNA templates. The lifetime (t1/2) of each target mRNA was calculated by using the equations described above.
Statistical analysis
For experiments except PAR-CLIP and m6A-seq, the statistical analyses were determined by Student’s unpaired t-test (two-tailed). For PAR-CLIP and m6A-seq, the statistical analyses were performed as described in the corresponding data analysis part.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health (GM071440 and GM088599). We thank Drs. P. Faber and L. Dore for helping with high-throughput sequencing experiments and S. F. Reichard, MA for editing the manuscript.
Footnotes
Author Contributions
C. H. conceived the project. J. L. and Y. Y. designed and performed most experiments. D. H. and Z. L. performed high-throughput sequencing data analyses. X.W. and Y. F. helped perform PAR-CLIP experiment, biochemistry assay, and data analysis. L. Z. and M. Y. assisted in expressing recombinant proteins in insect cells. G. J. and W. C. participated in the subcloning. X. D. participated in nuclear extract separation. Q. D. synthesized the d3-m6A standard for LC-MS/MS analysis. J. L., Y. Y., and C. H. wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
Supplementary information is available in the online version of the paper. The PAR-CLIP and m6A-seq data were deposited in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession number GEO46705.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Bokar JA. The biosynthesis and functional roles of methylated nucleosides in eukaryotic mRNA. In: Grosjean H, editor. Fine-Tuning of RNA Functions by Modification and Editing. Vol. 12. Springer-Verlag; Berlin Heidelberg: 2005. pp. 141–177. [Google Scholar]
- 2.Jia G, Fu Y, He C. Trends in Genetics. 2013;29:108–115. doi: 10.1016/j.tig.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Motorin Y, Helm M. Wiley Interdiscip Rev RNA. 2011;2:611–631. doi: 10.1002/wrna.79. [DOI] [PubMed] [Google Scholar]
- 4.Beemon K, Keith J. J Mol Biol. 1977;113:165–179. doi: 10.1016/0022-2836(77)90047-x. [DOI] [PubMed] [Google Scholar]
- 5.Jia G, et al. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng G, et al. Mol cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dominissini D, et al. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 8.Meyer KD, et al. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. RNA. 1997;3:1233–1247. [PMC free article] [PubMed] [Google Scholar]
- 10.Clancy MJ, Shambaugh ME, Timpte CS, Bokar JA. Nucleic Acids Res. 2002;30:4509–4518. doi: 10.1093/nar/gkf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hongay CF, Orr-Weaver TL. Proc Natl Acad Sci. 2011;108:14855–14860. doi: 10.1073/pnas.1111577108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhong S, et al. Plant Cell. 2008;20:1278–1288. doi: 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bujnicki JM, Feder M, Radlinska M, Blumenthal RM. J Mol Evol. 2002;55:431–444. doi: 10.1007/s00239-002-2339-8. [DOI] [PubMed] [Google Scholar]
- 14.Horiuchi K, et al. Proc Natl Acad Sci. 2006;103:17278–17283. doi: 10.1073/pnas.0608357103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agarwala SD, Blitzblau HG, Hochwagen A, Fink GR. Plos Genetics. 2012;8:e1002732. doi: 10.1371/journal.pgen.1002732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F. J Biol Chem. 1994;269:17697–704. [PubMed] [Google Scholar]
- 17.Hafner M, et al. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, et al. Nature. doi: 10.1038/nature12730. advance online publication, 27 November 2013. [DOI] [Google Scholar]
- 19.Fustin JM, et al. Cell. 2013;155:793–806. doi: 10.1016/j.cell.2013.10.026. [DOI] [PubMed] [Google Scholar]
- 20.Yu M, et al. Nat Protocols. 2012;7:2159–2170. doi: 10.1038/nprot.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kane SE, Beemon K. Mol Cell Biol. 1985;5:2298–2306. doi: 10.1128/mcb.5.9.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolte S, CordeliÈRes FP. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 23.Hafner M, et al. J Vis Exp. 2010;41:e2034. [Google Scholar]
- 24.Dominissini D, et al. Nat Protocols. 2013;8:176–189. doi: 10.1038/nprot.2012.148. [DOI] [PubMed] [Google Scholar]
- 25.Ascano M, et al. Nature. 2012;492:382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, et al. Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.