

Abstract
The cancer stem cell (CSC) theory depicts such cells as having the capacity to produce both identical CSCs (symmetrical division) and tumor-amplifying daughter cells (asymmetric division). CSCs are thought to reside in niches similar to those of normal stem cells as described for neural, intestinal, and epidermal tissue, are resistant to chemotherapy, and are responsible for tumor recurrence. We recently described the niche-like nature of mouse renal adenocarcinoma (RENCA) cells following encapsulation in agarose macrobeads. In this paper we tested the hypothesis that encapsulated RENCA colonies function as an in vitro model of a CSC niche and that the majority of cells would undergo chemotherapy-induced death, followed by tumor recurrence. After exposure to docetaxel (5 µg/ml), 50% of cells were lost one week post-treatment while only one or two cells remained in each colony by 6 weeks. Surviving cells expressed OCT4 and reformed tumors at 16 weeks post-treatment. Docetaxel-resistant cells also grew as monolayers in cell culture (16–17 weeks post-exposure) or as primary tumors following transplantation to Balb/c mice (6 of 10 mice) or NOD.CB17-Prkdcscid/J mice (9 of 9 mice; 10 weeks post-transplantation or 28 weeks post-exposure). These data support the hypothesis that a rare subpopulation of OCT4+ cells are resistant to docetaxel and these cells are sufficient for tumor recurrence. The reported methodology can be used to obtain purified populations of tumor-initiating cells, to screen for anti-tumor-initiating cell agents, and to investigate the in vitro correlate of a CSC niche, especially as it relates to chemo-resistance and tumor recurrence.
Keywords: RENCA, macrobeads, encapsulation, tumor-initiating cells, OCT4
Introduction
The cancer stem cell theory puts forth the hypothesis that tumor growth originates from a rare population of stem-cell like cells that undergo asymmetrical division to give rise to either replacement cancer stem cells or to a more lineage-restricted population of tumor-amplifying cells1 as first described for leukemia2 and later for numerous solid organ cancers (reviewed in refs. 3 and 4). Tumor-amplifying cells make up the bulk of a given tumor and because these cells rapidly proliferate, they are the target of many common chemotherapeutic drugs. The rare cancer stem cells, in contrast, are thought to be slow-cycling quiescent cells that are resistant to those forms of chemotherapy that target rapidly dividing cells.5 While the bulk of a tumor may therefore be susceptible to various forms of chemotherapy, the resistant cancer stem cells are thought to be responsible for cancer recurrence, although this remains an active area of investigation.6
Recently, three independent groups have employed lineage tracing techniques in mouse models of various cancers to uncover the cells of origin in gut, skin, and brain tumors. Schepers et al., using the colorful Cre-reporter Rosa-26Confetti, demonstrated that Lgr5+ intestinal crypt stem cells were the source of developing adenomas.7 A mouse model of skin cancer was used by Driessens et al. to illustrate that, in benign papillomas, only a small proportion of cells act as tumor-initiating cells, while the more aggressive squamous cell carcinomas are primarily composed of cells that display the stem cell characteristics of indefinite proliferation and reduced differentiation.8 In a third study, a green fluorescent protein (GFP) transgene capable of only labeling neural stem cells was bred into glioma-prone mice by Chen et al. to show that developing gliobastomas were primarily composed of GFP− cells.9 Further, treatment with the DNA alkylating agent temozolomide, which is frequently used to treat glioblastoma multiforme patients, resulted in the selective depletion of the non-stem and GFP− cells with tumor recurrence originating from GFP+ stem cells. Importantly, elimination of the GFP+ stem cells significantly improved survival in these mice.
In people, mortality from cancer recurrence, following chemotherapy and a period of “remission”, is estimated to account for the majority of cancer deaths. Thus, a better understanding of cancer stem cell chemo-resistance and tumor recurrence may provide significant opportunities to reduce mortality.
We have recently reported the development of agarose–agarose macrobeads containing encapsulated cells from a mouse renal cortical adenocarcinoma cell line (RENCA) in the inner layer of agarose.10 The macrobeads are approximately 8 mm in diameter with a 4–6 mm diameter core and a 1–2 mm outer coating of agarose. The core of the macrobead is initially seeded with 150 000 RENCA cells although over the first couple of weeks in culture approximately 99% of these cells die. During the next several weeks in culture, small elliptical tumor colonies begin to form, presumably originating from the few surviving cells. Over the next few months these tumor colonies enlarge in size and ultimately are composed of around 100 cells each with a one or two cell layer on the edges of the ellipsoid and cellular debris in the center of the tumor colonies. The size of the tumor colonies is stable over at least 3 y in culture and is maintained by active growth (proliferating cell nuclear antigen-positive cells, Ki67-positive cells, and BrdU-positive cells) and death (activated caspase-3-positive cells and TUNEL-positive cells) mechanisms.10 Encapsulated tumor cells do not escape the confines of the inner layer of the agarose as the cells are unable to penetrate the more concentrated outer layer of agarose. The outer layer of agarose also serves to provide strength to the macrobead as well as immuno-protection to the encapsulated cells as recipient immune cells cannot infiltrate the macrobead.
The encapsulated tumor colonies are nourished by culture media in vitro and peritoneal fluid in vivo following implantation into the abdominal cavity of cancer patients. Implanted macrobeads remain free-floating in the peritoneal cavity and have been shown to secrete numerous tumor-inhibitory proteins which suppress freely-growing cancer cells (human, murine, feline, and canine cancers to date), as demonstrated through both macrobead conditioned media and by tumor cell lines cultured together with RENCA macrobeads.11 Whether or not these same tumor inhibitory proteins also act in an autocrine or paracrine manner on the encapsulated RENCA cells is not known although this is likely given that the colonies only reach a given size and do not become confluent within the inner agarose layer. Furthermore, the ability to inhibit tumor growth increases with the age of the macrobeads until the colonies within individual macrobeads reach their maximal size; at which point the inhibition of external freely-growing tumors is greatest. Although RENCA cells are routinely used for the production of macrobeads as is currently being investigated in clinical trials for the treatment of patients with treatment-resistant metastatic cancer, similar properties have been demonstrated when various human tumor cell lines are used in place of the RENCA cells.10
In the current study, we hypothesized that the tumor colonies within the RENCA macrobeads are composed of tumor-initiating cells and tumor-amplifying daughter cells, and could therefore serve as an in vitro model of cancer stem cell niches. We set out to determine if the tumor colonies within the RENCA macrobeads were consistent with the concept of cancer stem cell niches containing both chemo-sensitive cells and chemo-resistant cells capable of re-forming tumor colonies.
Results
Cell recovery following treatment with paclitaxel or docetaxel in the macrobead
Microscopic examination of RENCA macrobeads, as early as 1 week post-exposure to either paclitaxel or docetaxel, revealed a partial, but not complete, loss of cellular viability (Fig. 1). RENCA macrobeads treated with paclitaxel (intermediate dose of 3.5 µg/ml) demonstrated a loss of cells through week 6 post-treatment, but then gradually returned to pre-treatment cell numbers by week 18. RENCA macrobeads treated with docetaxel (intermediate dose of 5 µg/ml) consistently displayed one or two cells remaining in the encapsulated colonies by week 6 post-treatment. By week 18 post-docetaxel treatment, approximately 10% of treated macrobeads developed one or two large colonies composed of numerous cells while the majority of colonies were devoid of cells. Control macrobeads exposed to the vehicle DMSO, maintained normal morphology throughout the observation period of 18 weeks as evidenced by elliptical tumor colonies composed of a rim of cells 1–2 cells thick with a central area of cell debris (Fig. 1).

Figure 1. Cell recovery following treatment with paclitaxel or docetaxel in the RENCA macrobead. Representative H&E staining of control and treated RENCA macrobeads. Normal RENCA macrobead tumor morphology was maintained post- vehicle treatment (0.07% DMSO, top panel). Progressive loss of cell viability following paclitaxel treatment (3.5 µg/ml, center panel) restored to pre-treatment levels after 18 weeks. Extensive cell loss by week 6 post-docetaxel treatment (5.0 µg/ml, bottom panel) and viable cells in a minority of colonies at week 18 post-treatment. For all panels, original magnification = 400×, scale bar = 20 µm.
To quantify cellular loss after treatment with paclitaxel or docetaxel, representative macrobeads were sectioned for histological analysis. Individual cell nuclei were counted to determine cell number and the average numbers of cells per colony were graphed (Fig. 2). Macrobeads exposed to the vehicle (DMSO) exhibited typical macrobead cell number and 95–100% of the colonies remained viable throughout the study (Fig. 2A). Following paclitaxel treatment, macrobeads had an initial loss (weeks 1–3 post-treatment) of approximately 25% of cells per colony, although the majority of colonies contained viable cells (Fig. 2B). By 18 weeks post-paclitaxel treatment, encapsulated colonies contained an equivalent number of viable cells per colony as control macrobeads. Docetaxel-treated macrobeads rapidly lost viable cell numbers such that by 6 weeks post-treatment only about one or two cells per colony remained (Fig. 2C). By 18 weeks post-docetaxel treatment, approximately 10% of treated macrobeads developed one or two large colonies ranging in size from about 200–1000 microns (approximately 2–5 times the size of a normal RENCA colony) and composed of numerous cells. This experiment demonstrates that rare docetaxel-resistant RENCA cells can reform colonies within the macrobead.
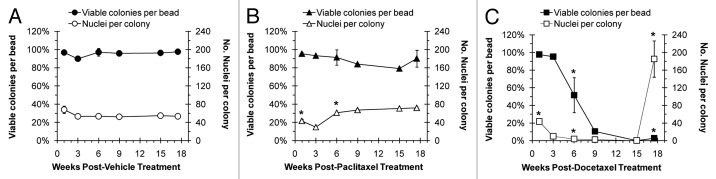
Figure 2. Viability of RENCA macrobeads following paclitaxel or docetaxel treatment. A quantitative assessment of the percent of colonies that remained viable and the average number of viable nuclei per colony in treated macrobeads. Typical levels of colony viability and the number of viable nuclei per colony were maintained in vehicle-treated macrobeads throughout the course of the study (A). When possible, an average of 3 replicate experiments was considered. Statistically significant outcome differences for paclitaxel-treated macrobeads (B) or docetaxel-treated macrobeads (C), as compared with vehicle-treatment, are denoted with an asterisk (*), P < 0.05.
Paclitaxel and docetaxel exposure reduces metabolic activity and tumor inhibition
The metabolic activity of RENCA macrobeads exposed to paclitaxel was not affected at the low and intermediate dosages. The high dose of paclitaxel, however, produced an approximate 50% reduction in metabolic activity by week 6 and through week 9 (Fig. 3A). All dosages of docetaxel resulted in significant dose-dependent reductions of metabolic activity throughout the nine-week observation period (Fig. 3B).

Figure 3. Metabolic activity and tumor inhibitory capacity of RENCA macrobeads exposed to paclitaxel and docetaxel. Metabolic activity (as assessed by MTT assay, see Methods) of RENCA macrobeads exposed to the intermediate dose of paclitaxel (A) or docetaxel (B) as compared with untreated or vehicle treated RENCA macrobeads. Tumor inhibitory capacity of RENCA macrobeads, on exogenous tumor cells, following exposure to paclitaxel (C) or docetaxel (D) as compared with untreated or vehicle treated RENCA macrobeads.
Exposure to paclitaxel or docetaxel also affected the tumor inhibitory capacity of RENCA macrobeads. Paclitaxel exposure transiently reduced the tumor inhibitory capacity at the intermediate and high dosages, although the reduction at the highest dose required the longest time to return to within 83% of control levels (Fig. 3C). All doses of docetaxel treatment resulted in a suppression of the tumor inhibitory effect of the RENCA macrobeads, which only returned to 30–50% of control levels by 17 weeks post-exposure (Fig. 3D).
Docetaxel-resistant RENCA cells proliferate in vitro and form tumors in vivo
To determine if docetaxel-resistant RENCA cells were capable of forming tumors, RENCA colonies were dislodged from the core of macrobeads and primary cultures of surviving cells were transplanted to syngeneic mice. In vitro, the recovered cells were approximately 10 times larger than normal RENCA cells grown in monolayer and were not observed to undergo cell growth for approximately 16 weeks (Fig. 4A). Between 16–17 weeks post-culture, the cells formed plaques (Fig. 4B) and upon routine passage began to proliferate and grow as a monolayer over the surface of the culture dish. Within 2 weeks, these cells were observed to have comparable growth rates and morphology (Fig. 4C) of typical RENCA monolayer cells (Fig. 4D).

Figure 4. Docetaxel-resistant cells cultured in vitro and after transplantation. Morphological appearance of docetaxel-resistant cells one week post-isolation from docetaxel treated macrobeads (A). Maintained in vitro for 16–17 weeks docetaxel-resistant cells developed plaques (B). These cells were passaged at week 16 post-treatment, and within 2 weeks the harvested cells assumed normal RENCA morphology (C). As a reference, RENCA monolayers demonstrating normal RENCA cell morphology (D). Representative H&E stained tissue of a Balb/cJ mouse at 56 d post-transplantation shows tumor formation inthe left kidney (E) and lung metastasis (F). Representative H&E stained tissue from tumor formation in the left kidney (G) and lung metastasis (H) of a NOD.CB17-Prkdcscid/J mouse at 66 d post-transplantation. Original magnification for all panels = 200×, scale bar: 40 µm.
To determine the ability of docetaxel-resistant cells to form tumors in vivo, Balb/cJ mice and NOD.CB17-Prkdcscid/J mice received 200 cells under the left kidney capsule. Between 42 and 67 d post-transplantation, 6 of 10 Balb/cJ mice and 9 of 9 NOD.scid mice developed tumors under the kidney capsule (Fig. 4E and G respectively). At necropsy, 4 of the 6 Balb/cJ mice and 6 of the 9 NOD.CB17-Prkdcscid/J mice also presented with lung metastases (Fig. 4F and H respectively).
Docetaxel-resistant RENCA cells express the stem cell marker OCT4
To determine whether the surviving cells from docetaxel-treated RENCA macrobeads expressed markers of embryonic stem cells, macrobead sections were stained for the presence of OCT4. The majority of surviving cells at 6 weeks post-docetaxel treatment demonstrated positive staining for the presence of OCT4 while only an occasional OCT4+ cell was observed in vehicle-treated control colonies (Fig. 5). By 16–18 weeks post-docetaxel exposure, when colonies reformed in some macrobeads as discussed above, only a minority of cells within the newly formed colonies expressed OCT4.

Figure 5. Docetaxel-resistant RENCA cells express OCT4. OCT4 expression (green fluorescence) and nuclear staining (DAPI, blue fluorescence) is limited to a minority of the cell population composing tumor colonies in control RENCA macrobeads (A). Cells surviving in RENCA macrobeads at 6 weeks post-docetaxel treatment were positive for OCT4 expression (B–D). Original magnification of panels (A–D) = 200×, insets in panels (A and B) = 400×; scale bar = 20 µm.
Discussion
In this study we have demonstrated that exposure of mature RENCA macrobeads to the chemotherapeutic agent docetaxel results in the elimination of the majority of the encapsulated cells that form individual cancer colonies. Only rare cells within the colonies survive to 6 weeks post-treatment and express the stem cell transcription factor OCT4. Follow-up to 18 weeks post-exposure results in the re-formation of tumor colonies within some macrobeads. The chemo-resistant OCT4+ cells, when harvested from docetaxel-treated macrobeads at 6 weeks post-treatment, are able to proliferate in vitro and form tumors in vivo. The fact that the harvested cells required a greatly extended time to form monolayers in vitro or to form tumor colonies in vivo, as compared with normal RENCA cells, is in line with the stem cell-like nature of these cells given the well-known slow cycling and quiescent properties of normal stem cells. These results support the hypothesis that the tumor colonies within mature RENCA macrobeads provide an in vitro model of the hypothesized cancer stem cell or tumor-initiating cell niche.
We believe it is likely that other agents could produce similar results to those of the docetaxel-treated RENCA macrobeads. In fact, paclitaxel produced similar macrobead morphology as the docetaxel-treated macrobeads, albeit at different post-exposure times and dosages. These results suggest that the survival of only one or two cells per colony may be a matter of optimizing exposure and dose for different agents. Furthermore, other cell lines can be encapsulated in agarose–agarose macrobeads, including MMT (mouse mammary tumor), K12 (feline mammary cancer), and the human tumor cell lines DU145 (prostate carcinoma), HCT 116 (colorectal carcinoma), J82 (urinary bladder transitional cell carcinoma), and MCF7 (mammary gland adenocarcinoma). These cell lines mature to form tumor colonies similar to those of the RENCA macrobeads as we have previously reported.10 This suggests that a comparable system could be developed with other cell lines, and possibly primary tumor cells, to study the biology of tumor-initiating cells and their niches, as well as to screen existing or novel anti-tumor-initiating cell agents.
The methodology reported in the current paper allows for the selection and isolation of a pure population of cells with tumor-initiating properties. Recently, Zhou et al. reported a technique similar to that reported herein to isolate cancer stem cells.12 Zhou et al. focused on primary human sarcoma tumor cells microencapsulated originally as single cells in alginate while we worked with the macro-encapsulation of 150 000 cells from an established murine renal carcinoma cell line. Following exposure to epirubicin, Zhou et al. noted that resistant cells formed tumor spheres within the alginate capsules which were then dissolved to isolate the spheres for analysis. This study reported the presence of OCT3/4 and Nanog staining cells in the core of the tumor spheres.12 These cells were shown to be capable of inducing tumors in at least some mice. The group of Ma et al. also recently used chemoresistance to purify cancer stem cells.13 In this work, the human ovarian cancer cell line SKOV3 was not encapsulated, but simply cultured in the presence of cisplatin and paclitaxel.13 Resistant cells, then transferred to serum-free stem cell culture conditions, formed non-adherent spheres, similar in appearance to those of Zhou et al. with a majority of cells appearing to be positive for both OCT4 and Nanog staining. As with Zhou et al., at least some mice transplanted with 500 of these tumor sphere cells, went on to develop tumors. We found that at 6 weeks post-docetaxel treatment the only surviving cells were one or occasionally two cells per colony that were OCT4+ and were harvested as a pure population and shown to proliferate in vitro and form tumors in vivo. Previous work from our laboratories demonstrated that only a minority of cells within individual tumor colonies expressed OCT4 and that these cells were predominantly located at the tips of the ovoid colonies.10 In the current study, the docetaxel-resistant cells within a tumor colony were primarily located at the tips of the colonies and were OCT4+.
OCT4, a transcription factor within the POU-domain family, is expressed in normal pluripotent embryonic stem cells.14 This transcription factor has been shown to be essential for self-renewal of embryonic stem cells as OCT4 gene knock out is lethal to developing embryos15 and is a critical factor in the generation of pluripotency from adult murine somatic cells16 and adult human somatic cells.17,18 OCT4 has also been shown to be upregulated in various tumor-initiating cell populations, as assessed in xenotransplantation assays from human primary tumors including breast cancer,19 non-small cell lung cancer,20 and Ewing sarcoma.21 Dysplasia22 and tumor formation23 following transduction of OCT4 in normal epithelial tissues have also been reported. Under the conditions of the present paper, we have shown OCT4-expressing cells to be a defining characteristic of a rare population of cells that survive docetaxel treatment. This finding is consistent with the work of others that have shown cancer stem cell chemo-resistance (reviewed in refs. 24–27) including the specific descriptions of OCT4+ cells that exhibit chemo-resistant properties via several mechanisms including an overexpression of drug efflux pumps such as ABCG2/MDR1 and BCRP/Pgp in the human prostate cell line CWR-R1,28 the activation of the AKT pathway in human hepatocellular cell lines29 and the induction of the Stat3/Survivin pathway.30 Recently, OCT4 has been shown to be capable of being transported from one cell to another as a possible mechanism to expand the number of stem cells during the annual regrowth of deer antlers.31 In agreement with our nuclear and cytoplasmic OCT4 staining of docetaxel-resistant cells, this paper describes the varied cellular localization of OCT4, including nuclear, cytoplasmic, and transporting nanotubular structures.31 Our data are consistent with these previous works and supports the hypothesis that the OCT4+ cells we have isolated are indeed tumor-initiating cells.
Further, the data reported in these studies support the concept that the tumor colonies within mature RENCA macrobeads function as cancer stem cell niches, notwithstanding the absence of a vasculature network. Just as normal stem cells reside in distinct niches that provide regulatory support for both the maintenance of stemness and for lineage differentiation,32 it is thought that cancer stem cells must also rely on a similarly distinct environment.33-35 Indeed, the influence of the microenvironment on normal stem cells is so great that male testis stem cells have been shown to differentiate into lactating mammary glands when transplanted to the mammary fat pad.36 To date, a thorough description and localization of cancer stem cell niches has been elusive owing to the complex anatomy and heterogeneous nature of tumors,37 the uncertainty as to whether or not cancer stem cells commandeer normal stem cell niches or create new niches,38 and even the definitive identification of the cancer stem cells themselves.6 However, Xie et al., after first describing the normal germline stem cell niche in a structure known as the germarium within the ovary of Drosophila39 went on to more thoroughly characterize the biology within this niche.40 In this later study, the Xie laboratory used neoplastic germline stem cells to define the role of E-cadherin-dependent adhesion on stem cell niche occupancy.40 Although we have not investigated similar characteristics of the RENCA macrobead cancer stem cell niches, we find an astonishing similarity between mature RENCA macrobead colonies and the niche descriptions of Xie, including the morphology and the significantly upregulated expression of E-cadherin.10
Additional studies would be required to better understand why tumors reformed in only a small proportion of macrobeads. It is likely that the immediate environment of surviving cells changed significantly within the macrobeads after exposure to chemotherapy and that this change inhibited long-term cell survival and colony reformation. The importance of mechanical forces on normal stem cell development has been reported and represents an exciting and rapidly emerging field of study.41-43 In the same way, manipulation of the extracellular matrix in the cancer microenvironment has been shown to change the proliferative behavior of cancer cells,44 as we have also observed when varying the concentration, and thus the rigidity, of the agarose used to encapsulate different cell lines (unpublished observations). Nonetheless, the isolation of a pure population of tumor-initiating cells using the method reported here will allow a thorough genetic, phenotypic, and importantly, sensitization characterization of such cells and the environmental conditions conducive for or non-favorable to tumor-initiating cell maintenance.
The data reported herein can also be used to develop therapeutic strategies involving combination treatments employing RENCA macrobeads and various anti-neoplastic drugs or immunotherapy. Additional studies, however, would be required to determine the long-term effects of such drugs on the inhibitory capacity of the macrobeads. Similar studies could also be performed in order to determine the feasibility and timing of alternating macrobead and chemotherapy as a treatment option, especially the requirements of any potential washout periods. To date, the RENCA macrobeads have only been used independently in Phase I and Phase II clinical trials for treatment-resistant epithelial cancers (ClinicalTrials.gov identifier NCT00283075 and NCT01053013, respectively).
We believe the implications of this paper to the study of cancer biology are highly significant, and will allow for the accurate and thorough characterization of tumor-initiating cells and their microenvironment. These data are in line with the hypothesis that OCT4+ cells are resistant to docetaxel and that these cells are sufficient for tumor recurrence. The methodology reported in this paper also provides a pure population of tumor-initiating cells that can be studied within the three-dimensional environment of the macrobeads. Alternatively, the tumor-initiating cells can be harvested and studied under numerous in vitro and in vivo conditions. The macrobeads can also be used to encapsulate and study various human tumor cell lines and possibly primary human tumor cells, including as a screening method for anti-cancer stem cell agents. It is our hope that the data reported in this manuscript will lead to a greater understanding of tumor-initiating cells and the application of this knowledge to improve patients’ lives and survival.
Materials and Methods
Cell lines
The RENCA cell line used for these experiments is a renal cortical adenocarcinoma that arose spontaneously in Balb/c mice, originally obtained from the National Cancer Institute. RENCA cells were maintained in vitro (5% CO2 + air at 37 °C) in tissue culture flasks (BD Biosciences, 353136) containing RPMI 1640 supplemented with 25 mM HEPES + l-Glutamine (HyClone, SH3A2475) and 10% newborn calf serum (NCS; Invitrogen, 04-4002). Cell passages were limited to no more than 20 from a frozen stock of these cells which were routinely screened and found to be negative for the presence of Mycoplasma.
RENCA macrobeads
RENCA agarose-agarose macrobeads were prepared as previously described.10 Briefly, 100 µl of 0.8% agarose (HSB-LV; Lonza Copenhagen ApS) in MEM (Sigma-Aldrich, M5775) was mixed with 1.5 × 105 RENCA cells. The agarose/cell suspension was expelled from a transfer pipette into mineral oil to form the core of the macrobead. Following washing with RPMI 1640 and overnight culture at 37 °C in 5% CO2 and air, the core was rolled in approximately 1 ml of 4.5% agarose to apply an outer coat and then transferred to mineral oil. Macrobeads were cultured in 90-mm petri dishes (Nunc, 4031) at 10 macrobeads/40 ml of RPMI 1640 with 10% NCS.
Metabolic activity assay
A colorimetric assay which utilizes the ability of mitochondrial succinate dehydrogenase to reduce yellow 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetraozlium bromide (MTT) to purple formazan was employed to assess metabolic activity. Following a 3-h incubation in 1 mg/ml MTT (Sigma-Aldrich, M5655) prepared in RPMI + 10% NCS, at 37 °C, the resulting formazan crystals were solubilized by crushing each macrobead in 2 ml isopropanol (Sigma-Aldrich I9516) and allowing dissolution overnight. The samples were centrifuged for 3 min at 10 000 RPM in order to pellet cell and agarose debris, and 850 µl of supernatant was transferred to a 48-well plate. Absorbance was read at 570 nm on a Bio-Tek Synergy 2 Plate Reader.
Tumor inhibitory capacity
RENCA cells (15 000/well) were seeded in 6-well plates (BD Falcon, 353046) in 4 ml fresh culture media or 5-d macrobead-conditioned media. Following 5 d of culture, the cells were methanol-fixed, and stained with 0.33% (w/v) neutral red (Sigma-Aldrich, N2889). The stain was extracted in 1 ml/well 1.25% (w/v) sodium dodecyl sulfate (Life Technologies, 15525-017) and absorbance read at 540 nm, 630 nm reference wavelength. Tumor inhibitory capacity (reported as percent inhibition) was defined as the percent difference in Abs540 nm–630 nm between conditioned and fresh media.
RENCA macrobead exposure to chemotherapeutic agents
Mature RENCA macrobeads (>12 weeks of age) were exposed to paclitaxel (Sigma-Aldrich, T7402) and docetaxel (Sigma-Aldrich, 01885) at concentrations of each drug based on target blood levels in human patients (considered the intermediate dose), one log-dose below (low dose) and one log-dose above (high dose). The lyophilized chemotherapeutic agents were reconstituted with 0.07% dimethyl sulfoxide (DMSO; Sigma-Aldrich, D2650) and were diluted in macrobead culture media to their final treatment concentration. Macrobeads were incubated in the presence of paclitaxel or docetaxel for the in vivo half-life of that drug (Table 1; refs. 45 and 46). Prior to transferring macrobeads to fresh culture media, macrobeads were washed twice with RPMI 1640 to remove traces of residual drug. Control conditions included untreated macrobeads and macrobeads exposed to the drug vehicle DMSO. After drug exposure, macrobeads were followed for up to 18 weeks in vitro and assessed at various times for metabolic activity (MTT assay), tumor inhibitory capacity, and histology.
Table 1. Description of chemotherapeutic agents and dosages.
| Agent | Class | Mechanism of action | Typical cancers | Clinical dose range | Half-life (in vivo) | Macrobead treatment | |
|---|---|---|---|---|---|---|---|
| Time | Dose | ||||||
| Paclitaxel (Taxol®) | Microtubule inhibitor | Blocks microtubule disassembly | Breast, lung, ovarian | 135–175 mg/m2 | 13–20 h | 1 d | 0.35 µg/ml |
| 3.5 µg/ml | |||||||
| 35 µg/ml | |||||||
| Docetaxel (Taxotere®) | Microtubule inhibitor | Blocks microtubule disassembly | Breast, lung, prostate | 60–100 mg/m2 over 1 h | 12 h | 1 d | 0.5 µg/ml |
| 5.0 µg/ml | |||||||
| 50 µg/ml | |||||||
Harvesting of RENCA macrobead cells
Following drug exposure, surviving cells were harvested by slicing macrobeads in half and removing the inner core of agarose. The inner core of agarose was placed under a dissecting microscope and individual tumor colonies were removed with the aid of surgical forceps. Using a pipette, the colonies were transferred to a 60 mm culture dish (BD Biosciences, 353002) containing fresh culture media, and maintained in vitro (5% CO2 + air at 37 °C) for up to 17 weeks.
Animals
Experiments were reviewed and approved by the IACUC of The Rogosin Institute. Balb/cJ mice (n = 10) and NOD.CB17-Prkdcscid/J mice (n = 9) were obtained from The Jackson Laboratory. After a 7-d acclimatization period, 200 RENCA cells were embedded in a clot of the recipient’s blood and transplanted under the capsule of the left kidney.
Histology
Macrobeads and tumor tissues were fixed in 10% neutral-buffered formalin overnight and transferred to phosphate-buffered saline (PBS, Amresco, E703–1L) until processing, embedded in paraffin and 5 µm sections were stained with hematoxylin and eosin (H&E; Vector Laboratories, H3404; Polysciences, 09859, respectively). Formalin-fixed, paraffin-embedded macrobead sections were labeled with rabbit polyclonal antibody to OCT4 (Abcam, ab19857) followed by an Alexa Fluor 488-conjugated goat polyclonal antibody to rabbit IgG (Invitrogen, A11034). Nuclei were stained with 4’, 6’ diamidino-2-phenylindole dihydrochloride (DAPI) and sections were coverslipped with ProLong Gold anti-fade reagent (Invitrogen, P36935).
Image acquisition
Formalin-fixed macrobead sections stained with H&E were viewed with an inverted microscope (Zeiss Axiovert s100; Carl Zeiss Vision GmbH) using brightfield illumination while fluorescent-stained sections used mercury vapor illumination (AttoArc 2 HBO 100 w). Images were photographed (Axiocam MRc) using Axiovs40 v4.8.1.0 software.
Disclosure of Potential Conflicts of Interest
The work described in this manuscript is part of the long-term agarose–agarose cancer macrobead project developed and conducted by The Rogosin Institute, a not-for-profit clinical care and research institute affiliated with Weill-Cornell Medical College and The New York Presbyterian Hospital and its Healthcare System in New York City, New York. The project has received substantial financial support from Metromedia Bio-Sciences LLC (MMBS) under an agreement providing exclusive licensing rights to the technology to MMBS. In addition, a royalty fee is to be paid to The Institute, if the product ultimately becomes commercially successful. Patents covering the technology are owned by The Rogosin Institute and the ongoing maintenance fees for these patents are paid for by The Rogosin Institute. None of the authors receives compensation of any kind directly from MMBS, and the work itself is performed entirely under the direction of The Rogosin Institute in a manner consistent with its scientific review and other policies, procedures, and governance structure. However, some costs associated with salaries and benefits of employees are included in the annual budgets under which support for the project is provided by MMBS.
Acknowledgments
The authors acknowledge the invaluable assistance of Bob Evans Farms, Inc., in enabling this work as well as the support of Brian Doll, Deborah Hoffer, Ashley Ewing, and colleagues at The Rogosin Institute. This work was financially supported by Metromedia Bio-Sciences LLC.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26455
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 3.Bomken S, Fiser K, Heidenreich O, Vormoor J. Understanding the cancer stem cell. Br J Cancer. 2010;103:439–45. doi: 10.1038/sj.bjc.6605821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–22. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 5.Kvinlaug BT, Huntly BJ. Targeting cancer stem cells. Expert Opin Ther Targets. 2007;11:915–27. doi: 10.1517/14728222.11.7.915. [DOI] [PubMed] [Google Scholar]
- 6.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 7.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337:730–5. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 8.Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488:527–30. doi: 10.1038/nature11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–6. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith BH, Gazda LS, Conn BL, Jain K, Asina S, Levine DM, Parker TS, Laramore MA, Martis PC, Vinerean HV, et al. Three-dimensional culture of mouse renal carcinoma cells in agarose macrobeads selects for a subpopulation of cells with cancer stem cell or cancer progenitor properties. Cancer Res. 2011;71:716–24. doi: 10.1158/0008-5472.CAN-10-2254. [DOI] [PubMed] [Google Scholar]
- 11.Smith BH, Gazda LS, Conn BL, Jain K, Asina S, Levine DM, Parker TS, Laramore MA, Martis PC, Vinerean HV, et al. Hydrophilic agarose macrobead cultures select for outgrowth of carcinoma cell populations that can restrict tumor growth. Cancer Res. 2011;71:725–35. doi: 10.1158/0008-5472.CAN-10-2258. [DOI] [PubMed] [Google Scholar]
- 12.Zhou S, Li F, Xiao J, Xiong W, Fang Z, Chen W, Niu P. Isolation and identification of cancer stem cells from human osteosarcom by serum-free three-dimensional culture combined with anticancer drugs. J Huazhong Univ Sci Technolog Med Sci. 2010;30:81–4. doi: 10.1007/s11596-010-0114-4. [DOI] [PubMed] [Google Scholar]
- 13.Ma L, Lai D, Liu T, Cheng W, Guo L. Cancer stem-like cells can be isolated with drug selection in human ovarian cancer cell line SKOV3. Acta Biochim Biophys Sin (Shanghai) 2010;42:593–602. doi: 10.1093/abbs/gmq067. [DOI] [PubMed] [Google Scholar]
- 14.Rosner MH, Vigano MA, Ozato K, Timmons PM, Poirier F, Rigby PW, Staudt LM. A POU-domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature. 1990;345:686–92. doi: 10.1038/345686a0. [DOI] [PubMed] [Google Scholar]
- 15.Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Schöler H, Smith A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–91. doi: 10.1016/S0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 16.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 17.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 18.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–6. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 19.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–11. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 20.Chen YC, Hsu HS, Chen YW, Tsai TH, How CK, Wang CY, Hung SC, Chang YL, Tsai ML, Lee YY, et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS One. 2008;3:e2637. doi: 10.1371/journal.pone.0002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suvà ML, Riggi N, Stehle JC, Baumer K, Tercier S, Joseph JM, Suvà D, Clément V, Provero P, Cironi L, et al. Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res. 2009;69:1776–81. doi: 10.1158/0008-5472.CAN-08-2242. [DOI] [PubMed] [Google Scholar]
- 22.Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–77. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 23.Beltran AS, Rivenbark AG, Richardson BT, Yuan X, Quian H, Hunt JP, Zimmerman E, Graves LM, Blancafort P. Generation of tumor-initiating cells by exogenous delivery of OCT4 transcription factor. Breast Cancer Res. 2011;13:R94. doi: 10.1186/bcr3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26:2839–45. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schatton T, Frank NY, Frank MH. Identification and targeting of cancer stem cells. Bioessays. 2009;31:1038–49. doi: 10.1002/bies.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore N, Lyle S. Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J Oncol. 2011;2011 doi: 10.1155/2011/396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrison R, Schleicher SM, Sun Y, Niermann KJ, Kim S, Spratt DE, Chung CH, Lu B. Targeting the mechanisms of resistance to chemotherapy and radiotherapy with the cancer stem cell hypothesis. J Oncol. 2011;2011:941876. doi: 10.1155/2011/941876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linn DE, Yang X, Sun F, Xie Y, Chen H, Jiang R, Chen H, Chumsri S, Burger AM, Qiu Y. A Role for OCT4 in Tumor Initiation of Drug-Resistant Prostate Cancer Cells. Genes Cancer. 2010;1:908–16. doi: 10.1177/1947601910388271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang XQ, Ongkeko WM, Chen L, Yang ZF, Lu P, Chen KK, Lopez JP, Poon RT, Fan ST. Octamer 4 (Oct4) mediates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology. 2010;52:528–39. doi: 10.1002/hep.23692. [DOI] [PubMed] [Google Scholar]
- 30.Guo Y, Mantel C, Hromas RA, Broxmeyer HE. Oct-4 is critical for survival/antiapoptosis of murine embryonic stem cells subjected to stress: effects associated with Stat3/survivin. Stem Cells. 2008;26:30–4. doi: 10.1634/stemcells.2007-0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rolf HJ, Niebert S, Niebert M, Gaus L, Schliephake H, Wiese KG. Intercellular transport of Oct4 in mammalian cells: a basic principle to expand a stem cell niche? PLoS One. 2012;7:e32287. doi: 10.1371/journal.pone.0032287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9:11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- 33.Borovski T, De Sousa E Melo F, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. 2011;71:634–9. doi: 10.1158/0008-5472.CAN-10-3220. [DOI] [PubMed] [Google Scholar]
- 34.Cabarcas SM, Mathews LA, Farrar WL. The cancer stem cell niche--there goes the neighborhood? Int J Cancer. 2011;129:2315–27. doi: 10.1002/ijc.26312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121:3804–9. doi: 10.1172/JCI57099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boulanger CA, Mack DL, Booth BW, Smith GH. Interaction with the mammary microenvironment redirects spermatogenic cell fate in vivo. Proc Natl Acad Sci U S A. 2007;104:3871–6. doi: 10.1073/pnas.0611637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805:105–17. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LaBarge MA. The difficulty of targeting cancer stem cell niches. Clin Cancer Res. 2010;16:3121–9. doi: 10.1158/1078-0432.CCR-09-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie T, Spradling AC. A niche maintaining germ line stem cells in the Drosophila ovary. Science. 2000;290:328–30. doi: 10.1126/science.290.5490.328. [DOI] [PubMed] [Google Scholar]
- 40.Jin Z, Kirilly D, Weng C, Kawase E, Song X, Smith S, Schwartz J, Xie T. Differentiation-defective stem cells outcompete normal stem cells for niche occupancy in the Drosophila ovary. Cell Stem Cell. 2008;2:39–49. doi: 10.1016/j.stem.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 42.Reilly GC, Engler AJ. Intrinsic extracellular matrix properties regulate stem cell differentiation. J Biomech. 2010;43:55–62. doi: 10.1016/j.jbiomech.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 43.Lee DA, Knight MM, Campbell JJ, Bader DL. Stem cell mechanobiology. J Cell Biochem. 2011;112:1–9. doi: 10.1002/jcb.22758. [DOI] [PubMed] [Google Scholar]
- 44.Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–47. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 45.TAXOL® product insert [Internet]. Bristol-Myers Squibb: c2011 [cited 2013 June 26]. Available at: http://packageinserts.bms.com/pi/pi_taxol.pdf
- 46.TAXOTERE® product insert [Internet]. Sanofi-Aventis US LLC; c2010 April [cited 2013 June 26]. Available at: http://products.sanofi.us/taxotere/taxotere.html