

Abstract
ETV1 (ETS variant 1) is a transcription factor from the ETS family and an oncogene in several types of human malignancies. Paradoxically, a predicted inactivating mutation in ETV1 was previously found in a clone of HT1080 cells with reduced activity of p53. We report that elevated expression of ETV1 makes p53-null tumor cells hypersensitive to restoration of said tumor suppressor. Furthermore, elevated levels of either wild-type ETV1 or its truncated derivative, dETV1, which mimics the product of an oncogenic rearrangement in certain tumors, results in increased expression of mRNA for p14ARF, a known activator of p53. Accordingly, expression of a luciferase reporter, which is driven by a putative ARF promoter, was elevated by concomitant expression of either ETV1 or dETV1. Our observations point to yet another example of a tumor suppressor gene being activated by a potentially oncogenic signal. A better understanding of the mechanisms that allow a cell to bypass such safeguards is needed in order to predict and prevent the development of an oncogene-tolerant state during cancer evolution.
Keywords: oncogenic transformation, tumor suppression, p14ARF, p53, ETV1
Introduction
Altered transcriptional regulation is a common characteristic, and often a driving force of oncogenesis. Deregulation of transcription factors may result from aberrations in upstream elements of cell signaling cascades. Alternatively, abnormal activity of a transcription factor may result from direct mutation of the corresponding gene. Both types of events are known to contribute to hyperactivation of the ETS family of transcriptional regulators, the founding member of which is an oncogene in a transforming retrovirus.1 Its closest human homologs are key mediators of transforming functions of common human oncogenes2 and can transform cells upon overexpression in experimental models.3,4 These and other ETS proteins have been shown to control genes whose products accelerate cell growth, motility, and survival.5
Not surprisingly, multiple ETS family members are frequently affected by mutations leading to cancer. For example, ETV1 is commonly involved in chromosomal translocations that result in multiple fusion proteins including EWS-ETV1 in Ewing sarcoma and at least 10 ETV1 partners in prostate cancer.6 While some gene fusion events change amino acids at the N-terminus of ETV1, others results in N-terminal truncation or overexpression of the full-length protein. In addition, to the aforementioned chromosomal rearrangements, ETS genes, such as ETV1, are also known to be genetically deregulated by other means, leading to prostate cancer,7 melanoma,8 and gastrointestinal stromal tumors (GISTs)9. For instance, 50–70% of prostate cancers overexpress of full-length or truncated forms of ETS genes,10,11 while greater than 40% of melanomas and greater than 50% of GISTs display increased levels of ETV1 in particular.7,8
The exact mechanism by which elevated ETV1 activity affects cancer progression is not fully understood. Previously, it was shown that various ETS family members, including ETV1,12 can be phosphorylated in response to the Ras/MAPK signaling pathway, resulting in increased stimulation of target genes that are often associated with the promotion of cancer cell phenotypes, such as proliferation, migration, and invasion.13,14 Furthermore, Hollenhorst et al. recently reported that oncogenic ETS proteins, and ETV1 in particular, can directly initiate elements of a transcriptional program characteristic of cells transformed by oncogenic Ras/MAPK signaling.15 Interestingly, some ETS family members function as bona fide tumor and metastasis suppressors, while others evoke a pro-mitogenic transcriptional program concomitantly with activation of tumor suppressor genes or their products.16-19 A possible outcome is that a mitogenically-stimulated cell becomes hypervigilant to potentially mutagenic impacts,20 and, consequently, the incidence of cancer is reduced due to induction of cell death or growth arrest in dangerously affected cells.
Intriguingly, an insertional event creating an anti-sense transcript from the ETV1 gene has been found in a genetic screen for the mutants with reduced transcriptional activity of p53 (ref. 21 and unpublished). This prompted us to investigate a possible mechanism of ETV1-p53 connection. In this study we report that ETV1 reduces tolerance of cancer cells to wild-type p53 and increases transcription of the key regulator of p53, p14ARF. This phenomenon may serve as one of the blocks on the way of cancer progression.
Results
ETV1 reduces tolerance of p53-null cancer cells to re-introduction of wild-type p53
To investigate whether increasing ETV1 expression can stimulate p53 activity we employed p53-null Saos-2 osteosarcoma cells, which are generally suppressed by re-expression of p53. However, there is some background survival upon reintroduction of functional p53, allowing for the establishment of clones that stably express p53,22 perhaps, because the level or activity of the protein is not high enough to induce death. We speculated that if ETV1 potentiates the tumor-suppressive function of p53, then expression of ETV1 would reduce the tolerance of Saos-2 cells to p53. This could be detected as a reduction in the number of viable clones upon reintroduction of this tumor suppressor. For this purpose, we generated Saos-2 derivatives retrovirally transduced with wild type ETV1, anti-sense ETV1, or the corresponding empty vector (pBabePuro). Stably engineered cultures were then infected with either a p53-expressing construct or the respective empty vector (pMV12), and were selected for the presence of the vector-encoded hygromycin resistance marker. The general susceptibility to retroviral infection was not affected by the ETV1 status, as is evidence by a similar yield of hygromycin-resistant cells in all three cultures infected with pMV12 (Fig. 1). In contrast, we noticed a dramatic reduction in the yield of colonies among p53-transduced cells that were pre-engineered with ETV1, as compared with the cells that were pre-engineered with anti-sense ETV1 or the empty vector (Fig. 1A). We concluded that overexpression of ETV1 strongly reduces the tolerance of p53-null cells to re-introduction of this tumor suppressor.
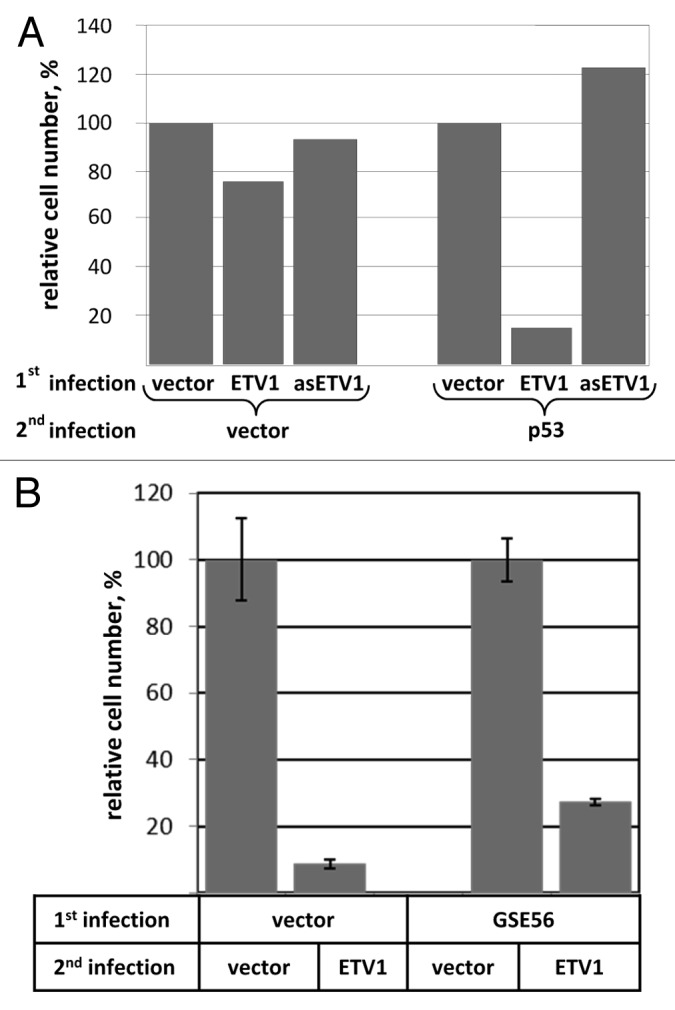
Figure 1. (A) ETV1 reduces tolerance of Saos2 to re-expression of p53. p53-deficient Saos-2 cells harboring an expression construct for ETV1, anti-sense ETV1 (asETV1) or the corresponding empty vector (pBabePuro) were superinfected with a p53-expressing construct or the corresponding empty vector (pMX12). The cells were selected for the presence of the pMX12-encoded hygromycin resistance marker. The numbers of remaining cells were compared using methylene blue staining and extraction method. Results for each arm of the experiment were normalized for the number of cells in pBabePuro-transduced populations. (B) Tolerance to ETV1 overexpression is enhanced by interference with the function of p53. hTERT-HME1 harboring either dominant-negative p53 fragment (GSE56) or the respective empty vector control (pLXSN) were infected with an ETV1-expressing retrovirus, or the respective empty vector control (pBabePuro). The yield of puromycin-resistant cells was measured using methylene blue staining and extraction method. The results were adjusted for virus titer and are scaled relative to the numbers of cells in pBabePuro-transduced populations.
The response of non-transformed human cells to ETV1 is partially dependent on p53
We hypothesized that if an increase in ETV1 activity enhances suppressive functions of p53, then the cells that still harbor wild-type p53 may be sensitive to overexpression of ETV1. Furthermore, in such a system, a reduction in p53 activity would be expected to improve tolerance to ETV1. In order to test this hypothesis, we examined tolerance to ectopic expression of ETV1 among immortalized non-transformed human mammary epithelial cells (hTERT-HME1) that were pre-engineered with an expression construct for dominant-negative p53 fragment (GSE 5623) or the respective empty vector (pLXSN). These cells were infected with a retrovirus encoding ETV1 or the respective empty vector (pBabePuro), and the number of viable cells was compared upon completion of puromycin selection. When adjusted for the titer of each retrovirus, there was a dramatic decrease in the yield of ETV1-transduced cells, indicating that hTERT-HME1 are very sensitive to this transgene (Fig. 1B). By this criterion, the tolerance to ETV1 was approximately three times higher among the cells that express dominant-negative p53 (Fig. 1B), confirming involvement of this tumor suppressor in the response of hTERT-HME1 to ETV1.
Overexpression of ETV1 elevates the levels of ARF mRNA
One of the mechanisms by which activity of p53 is elevated is by relieving this protein from the inhibitory and destabilizing effects of MDM2. The ability to sequester MDM2 away from p53 has been attributed to the product of an alternative reading frame of INK4A locus (a.k.a. ARF),24 and expression of the latter is known to be upregulated by oncogenic signals.25 Furthermore, elevated ARF expression is known to sensitize p53-resistant tumor cells to p53.26 We set forth to investigate whether elevated expression of ETV1 could upregulate ARF. Importantly, ARF may also have some p53-independent functions,27 and this phenomenon (along with limited efficiency of GSE56) might explain why interference with the function of p53 only partially averted sensitivity to ETV1 in hTERT-HME1 cells (Fig. 1B).
We have generated Saos-2 cells stably transduced with either wild-type human ETV1 or its truncated form, dETV1, which mimics the variant overexpressed in some cancers (Fig. 2A). Interestingly, while dETV1 may be missing parts of the N-terminal transactivation domain, it retains the putative C-terminal transactivation domain,12 as well as the ability to increase expression of, at least, some of the ETV1-responsive genes, thereby initiating ETV1-dependent biological processes.7
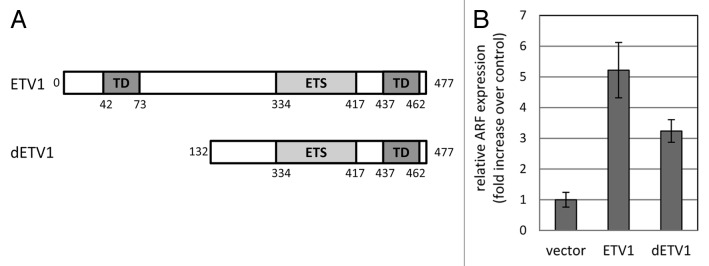
Figure 2. Effect of ETV1 and dETV1 expression on p14ARF mRNA expression. (A) Schematic representation of ETV1 deletion mutant, dETV1. The dETV1 variant lacks 131 N-terminal amino acids, which results in the loss of a transactivation domain position from amino acids 42 to 73. The DNA-binding ETS domain, as well as another putative transactivation domain spanning amino acids 437–462, remains intact at the C-terminal end. The scheme is not drawn to scale. (B) p14ARF mRNA expression in Saos-2 cells constitutively expressing ETV1 or dETV1 was measured by quantitative RT-PCR, normalized to mean GAPDH transcript levels and reported relative to that in cells harboring the respective empty vector control (pBabePuro). Results are shown as means and standard deviations of three independent replicas.
Quantitative RT-PCR revealed that the cells expressing either transgene had elevated levels of ARF, as compared with the cells transduced with the respective control vector (Fig. 2B).
ETV1 upregulates ARF promoter
We hypothesized that ETV1 exhibits its effect on ARF mRNA primarily by enhancing the function of the corresponding promoter. In order to test this prediction, we created a luciferase construct driven by a fragment of approximately 1.2 kbps from human ING4A locus. This sequence corresponds to the translation start site and 5′-untranslated region of ARF mRNA, as well as the immediate upstream genomic fragment (Fig. 3A). Co-transfection of this plasmid with an ETV1-expressing construct into 293T cells resulted in elevated luciferase expression (Fig. 3B). On the other hand, co-transfection with ETV1 did not affect a luciferase reporter driven by the immediate-early promoter of human cytomegalovirus (Fig. 3B), suggesting that the effect on the ARF-driven reporter was not due to a non-specific increase in luciferase abundance or function.

Figure 3. The effect of ETV1 and dETV1 on ARF promoter activity. (A) The structure of the ARF-luciferase reporter pARF-luc. A fragment of human genome that corresponds to a portion of INK4A exon 1b and the putative ARF promoter was introduced into pGL3Basic plasmid upstream of the firefly luciferase coding region. Translation start sites corresponding to p14ARF and luciferase (show in bold) are maintained in the same open reading. Nucleotide positions are numbered relative to the transcription start site of ARF (“TSS”). The scheme is not drawn to scale. (B) ETV1 increases ARF promoter activity. pARF-luc was co-transfected with a constitutive β-galactosidase reporter pRSV-βgal and either an ETV1-expressing construct or the corresponding empty vector control (pBabePuro). The luciferase activity was normalized for that of β-galactosidase and shown relative to that in vector-transfected cells. (C) ETV1 does not increase the activity of CMV immediate early promoter. CMV-driven luciferase construct pLNCLuc was co-transfected with pRSV-βgal and either an ETV1-expressing construct or the corresponding empty vector control (pBabePuro). (D) ETV1 and dETV1 promote ARF gene expression in Saos-2 cells. p14ARF promoter activity in Saos-2 cells was measured following co-transfection of pARF-luc and pRSV-βgal into Saos-2 cells pre-engineered with the indicated transgenes (see Fig. 2). Luciferase activity was normalized to that of β-galactosidase and is presented relative to the levels in cells expressing empty vector control (pBabePuro). Results are shown as the means and standard deviations of three independent replicas.
We further explored the function of the ARF promoter reporter in the Saos-2 cells engineered with ETV1- or dETV1-expressing constructs or the corresponding empty vector. Luciferase activity was significantly elevated upon transfection into the cells that expressed either one of the ETV1 variants, as compared with the activity of the reporter in cells harboring the corresponding empty vector (Fig. 3C). We conclude that ETV1 and its tumor-derived variant can specifically enhance the function of ARF promoter.
Discussion
Our results indicate that elevated levels of ETV1 increase the sensitivity of cancer cells to the inhibitory effects of the tumor suppressor, p53. This phenomenon coincides with increased expression of the gene for the p53 activator, ARF. Importantly, both the wild-type ETV1 and a truncated ETV1 variant, which mimics the forms of the protein found in some cancers, are able to increase the activity of ARF promoter. The quantitative differences in the activities of the ETV1 variants may represent bona fide biological distinctions, or merely differences in the expression levels achieved in specific assays. Importantly, when acting as oncogenes, these forms are found overexpressed to various degrees relative to the wild type protein in normal cells. Hence, both would be expected to elevate ARF transcription, but the exact effect would largely depend on the level of overexpression of a particular form in a given tumor.
The phenomenon described here follows suit with the many documented cases of growth-promoting factors also increasing the likelihood of growth-suppressive or apoptotic responses in a cell (e.g., refs. 20 and 28–31). The prevalence of this theme in intracellular signaling networks could be easily explained. Persistent proliferative signaling may indicate an oncogenic aberration. Moreover, even a normal proliferating cell is at a higher risk of converting incidental DNA damage into a dangerous mutation. This warrants extra-vigilant surveillance for the proliferative signals that appear out of the proper cell cycle context or coincidentally with genotoxic stress. Such surveillance is naturally achieved through coupling such signals with expression of tumor suppressors. For example, various potentially oncogenic transcription factors stimulate transcription of ARF32-35 or change properties in the presence of this protein.36 Consequently, many growth-promoting oncogenes are poor initiators of transformation in normal cells; rather than causing sustained tumor growth, they tend to cause death or growth arrest of the affected cell. Therefore, in the most efficient scenario of tumor progression, activation of classical oncogenes has to be accompanied by changes that would make it more tolerable to a cell. In some cases, this is further complicated by the engagement of multiple parallel tumor-suppressive mechanisms by the same oncogene. In this case, the individual role of a particular tumor suppressor varies depending on the genetic and environmental circumstances, and inactivation of just one of the tumor suppressors may be insufficient to fully induce the state of oncogene tolerance (discussed by Gil and Peters37). Interestingly, akin to other ETS family members,38 ETV1 has been identified recently as a possible regulator of the p16-specific promoter in the INK4A locus.39 Together with the data presented here, this suggests that the activity of ETV1 is also under intense scrutiny of tumor suppressors. The need to establish a state of tolerance toward ETS-family members before a cancer may benefit from these oncogenes could explain why upregulation of these proteins represents a progression, rather than initiation event,40 and why overexpression of ETV1 is found predominantly in clinical samples from advanced tumors.41 Furthermore, the relatively late activation of ETS family members, including ETV1, is supported by the fact that such activation is sometimes achieved via different genetic events in different tumor cells from the same patient.42,43
The fragment of ARF promoter used in our studies contains sequences, which conform to the ETV1-binding motif.44 However, binding specificity of ETS-family proteins is known to change depending on posttranslational modifications and interactions with other proteins.13,45 Thus, we cannot rule out that the phenomenon observed here is mediated by modified ETV1 or an ETV1-containing complex, which binds to a non-canonical site. Furthermore, it remains possible that induction of ARF transcription is mediated by ETV1 indirectly, through activation of additional transcription factors.
The role of ETS family proteins as drivers of tumor progression positions them as desirable therapeutic targets, and prospective drugs targeting these proteins are being actively developed.46 However, activation of tumor suppressors by ETV1 also brings up the paradoxical possibility that therapeutic targeting of ETV1 in certain situations may be undesirable. In particular, ARF is an activator of p53, which is known to sensitize some cancers to killing by chemotherapy and divert others into the senescence program.47,48 ARF may also sensitize cells to certain treatments in a p53-independent manner.49 Consequently, ETV1 activity in the presence of intact ARF and p53 in certain context might have a net positive effect on the therapeutic responsiveness of the disease. It remains to be seen whether the ARF-p53 pathway is functionally retained in any of the tumors with elevated ETV1 activity.
The observation that ETV1 upregulates tumor suppressors poses a critical question: what mechanisms are employed by cancer cells to circumvent those safeguards? Although ARF, p16, and p53 are sometimes mutated in prostate cancer,50 the frequency of these events appears lower than the frequency at which ETS family members are activated in this cancer.11 It is also important to note that ARF is able to induce both p53-dependent and -independent tumor-suppressive responses,27 which makes it even harder to circumvent its activity. In this regard, attention must be given to mechanisms of epigenetic control of the entire INK4 region, which are expected to govern production of 3 tumor suppressors at once (p15, p16, and ARF),37 as well as to the proposed simultaneous and synergistic suppression of the products of this locus at both epigenetic and posttranslational levels.51,52 A better understanding of the state of oncogene tolerance is needed in order to predict the likelihood of tumor origination and progression, as well as to devise future interventions, which would reverse such tolerance and turn an activating oncogene into an engine of tumor suppression.
Materials and Methods
hTERT-HME1 (Clontech, Inc., C4002-1) were propagated as previously reported.53 The culture of other cell lines and retroviral transduction were performed as described elsewhere.54 Virus titers were determined on p53-deficient H1299 cells. Cell numbers were compared using methylene blue staining and extraction method.55
ETV1 coding region was amplified from a pool of total cDNA of human fibrosarcoma HT1080 cells and cloned into pBabePuro56 to create pBabePuroETV1 construct. The sequences coding for the first 59 amino acids following the translational start codon were removed from pBabePuroETV1 to make pBabePurodETV1.
In order to construct ARF luciferase reporter (pARF-luc), a fragment of human genomic DNA was amplified from BAC clone 478M20 (acquired from a library at Roswell Park Cancer Institute) using GCCGCTCCTT CCTTTCCTTG and GATGACTCCT CGGTCGCAGA primers. The PCR fragment was cut FatI (New England Biolabs, R0650) and RsaI (New England Biolabs, R0167) and inserted between EcoIRCI (Promega, R6951) and NcoI (New England Biolabs, R0193) sites of pGL3 Basic immediately upstream of the Luciferase gene.
ARF transcripts were detected and quantified using quantitative real-time RT-PCR. GAPDH was the endogenous control used for normalization of the data. Each primer set was positioned in different exons of the gene in order to avoid amplification of contaminating genomic DNA. The nucleotide sequences were: ARF primers, CTACTGAGGA GCCAGCGTCT and CACGGGTCAG GTGAGAGTG, GAPDH primers, GTCTCCTCTG ACTTCAACGC G and ACCACCCTGT TGCTGTAGCA A. RNA from the Saos-2 cells was isolated using the RNeasy, RT kit (Qiagen, 74104). cDNA was synthesized using SuperScript® III Reverse Transcriptase (Invitrogen, 18080044). PCR reactions were performed using an ABI Prism 7900 Sequence Detection System and IQ SYBR Green SuperMix (BioRad, 1708880). The thermal cycling conditions comprised 2 min at 50 °C, 10 min at 90 °C, and 1 min at 60 °C for 40 cycles. The data was analyzed using Applied Biosystems RQ Manager 1.2.1.
All transient transfections were performed using Lipofectamine® LTX and Plus Reagent (Invitrogen, 15338030) according to the manufacturer’s recommendations. For transient transfection of 293T cells, pBabePuro or pBabePuroETV1 was mixed with pARF-luc or pLNCLuc57 and a constitutively active β-galactosidase reporter pRSV-βgal58 in a 2:1:2 ratio, respectively. For transient transfection of Saos-2-derived cells, pARF-luc and pRSV-βgal were mixed in 1:2 ratio. Forty-eight hours post transfection, the cells were lysed, the activity of the reporters was measured using a β-Galactosidase Enzyme Assay System (Promega, E2000) and a Luciferase Assay System (Promega, E1500) kits, and luciferase activity was normalized for that β-galactosidase.
Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors would like to thank Dr George Stark for helpful discussions and comments. The work was supported in part by funds from the Roswell Park Alliance Foundation.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26883
References
- 1.Leprince D, Gegonne A, Coll J, de Taisne C, Schneeberger A, Lagrou C, Stehelin D. A putative second cell-derived oncogene of the avian leukaemia retrovirus E26. Nature. 1983;306:395–7. doi: 10.1038/306395a0. [DOI] [PubMed] [Google Scholar]
- 2.Galang CK, García-Ramírez J, Solski PA, Westwick JK, Der CJ, Neznanov NN, Oshima RG, Hauser CA. Oncogenic Neu/ErbB-2 increases ets, AP-1, and NF-kappaB-dependent gene expression, and inhibiting ets activation blocks Neu-mediated cellular transformation. J Biol Chem. 1996;271:7992–8. doi: 10.1074/jbc.271.14.7992. [DOI] [PubMed] [Google Scholar]
- 3.Seth A, Papas TS. The c-ets-1 proto-oncogene has oncogenic activity and is positively autoregulated. Oncogene. 1990;5:1761–7. [PubMed] [Google Scholar]
- 4.Seth A, Watson DK, Blair DG, Papas TS. c-ets-2 protooncogene has mitogenic and oncogenic activity. Proc Natl Acad Sci U S A. 1989;86:7833–7. doi: 10.1073/pnas.86.20.7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. doi: 10.1016/S0378-1119(02)01156-3. [DOI] [PubMed] [Google Scholar]
- 6.Clark JP, Cooper CS. ETS gene fusions in prostate cancer. Nat Rev Urol. 2009;6:429–39. doi: 10.1038/nrurol.2009.127. [DOI] [PubMed] [Google Scholar]
- 7.Hermans KG, van der Korput HA, van Marion R, van de Wijngaart DJ, Ziel-van der Made A, Dits NF, Boormans JL, van der Kwast TH, van Dekken H, Bangma CH, et al. Truncated ETV1, fused to novel tissue-specific genes, and full-length ETV1 in prostate cancer. Cancer Res. 2008;68:7541–9. doi: 10.1158/0008-5472.CAN-07-5930. [DOI] [PubMed] [Google Scholar]
- 8.Jané-Valbuena J, Widlund HR, Perner S, Johnson LA, Dibner AC, Lin WM, Baker AC, Nazarian RM, Vijayendran KG, Sellers WR, et al. An oncogenic role for ETV1 in melanoma. Cancer Res. 2010;70:2075–84. doi: 10.1158/0008-5472.CAN-09-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, Fletcher JA, Dewell S, Maki RG, Zheng D, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010;467:849–53. doi: 10.1038/nature09409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, Cao X, Wei JT, Rubin MA, Shah RB, et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 11.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 12.Janknecht R. Analysis of the ERK-stimulated ETS transcription factor ER81. Mol Cell Biol. 1996;16:1550–6. doi: 10.1128/mcb.16.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh S, Shin S, Janknecht R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta. 2012;1826:1–12. doi: 10.1016/j.bbcan.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu T, Trojanowska M, Watson DK. Ets proteins in biological control and cancer. J Cell Biochem. 2004;91:896–903. doi: 10.1002/jcb.20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, Graves BJ. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011;25:2147–57. doi: 10.1101/gad.17546311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, Sharrocks AD, Peters G, Hara E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–70. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- 17.Funaoka K, Shindoh M, Yoshida K, Hanzawa M, Hida K, Nishikata S, Totsuka Y, Fujinaga K. Activation of the p21(Waf1/Cip1) promoter by the ets oncogene family transcription factor E1AF. Biochem Biophys Res Commun. 1997;236:79–82. doi: 10.1006/bbrc.1997.6909. [DOI] [PubMed] [Google Scholar]
- 18.Venanzoni MC, Robinson LR, Hodge DR, Kola I, Seth A. ETS1 and ETS2 in p53 regulation: spatial separation of ETS binding sites (EBS) modulate protein: DNA interaction. Oncogene. 1996;12:1199–204. [PubMed] [Google Scholar]
- 19.Xu D, Wilson TJ, Chan D, De Luca E, Zhou J, Hertzog PJ, Kola I. Ets1 is required for p53 transcriptional activity in UV-induced apoptosis in embryonic stem cells. EMBO J. 2002;21:4081–93. doi: 10.1093/emboj/cdf413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satyanarayana A, Greenberg RA, Schaetzlein S, Buer J, Masutomi K, Hahn WC, Zimmermann S, Martens U, Manns MP, Rudolph KL. Mitogen stimulation cooperates with telomere shortening to activate DNA damage responses and senescence signaling. Mol Cell Biol. 2004;24:5459–74. doi: 10.1128/MCB.24.12.5459-5474.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kandel ES, Lu T, Wan Y, Agarwal MK, Jackson MW, Stark GR. Mutagenesis by reversible promoter insertion to study the activation of NF-kappaB. Proc Natl Acad Sci U S A. 2005;102:6425–30. doi: 10.1073/pnas.0502463102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radinsky R, Fidler IJ, Price JE, Esumi N, Tsan R, Petty CM, Bucana CD, Bar-Eli M. Terminal differentiation and apoptosis in experimental lung metastases of human osteogenic sarcoma cells by wild type p53. Oncogene. 1994;9:1877–83. [PubMed] [Google Scholar]
- 23.Ossovskaya VS, Mazo IA, Chernov MV, Chernova OB, Strezoska Z, Kondratov R, Stark GR, Chumakov PM, Gudkov AV. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc Natl Acad Sci U S A. 1996;93:10309–14. doi: 10.1073/pnas.93.19.10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60:3689–95. [PubMed] [Google Scholar]
- 25.Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731–7. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 26.Lu W, Lin J, Chen J. Expression of p14ARF overcomes tumor resistance to p53. Cancer Res. 2002;62:1305–10. [PubMed] [Google Scholar]
- 27.Kuo ML, Duncavage EJ, Mathew R, den Besten W, Pei D, Naeve D, Yamamoto T, Cheng C, Sherr CJ, Roussel MF. Arf induces p53-dependent and -independent antiproliferative genes. Cancer Res. 2003;63:1046–53. [PubMed] [Google Scholar]
- 28.Conzen SD, Gottlob K, Kandel ES, Khanduri P, Wagner AJ, O’Leary M, Hay N. Induction of cell cycle progression and acceleration of apoptosis are two separable functions of c-Myc: transrepression correlates with acceleration of apoptosis. Mol Cell Biol. 2000;20:6008–18. doi: 10.1128/MCB.20.16.6008-6018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Radhakrishnan SK, Feliciano CS, Najmabadi F, Haegebarth A, Kandel ES, Tyner AL, Gartel AL. Constitutive expression of E2F-1 leads to p21-dependent cell cycle arrest in S phase of the cell cycle. Oncogene. 2004;23:4173–6. doi: 10.1038/sj.onc.1207571. [DOI] [PubMed] [Google Scholar]
- 30.Sreeramaneni R, Chaudhry A, McMahon M, Sherr CJ, Inoue K. Ras-Raf-Arf signaling critically depends on the Dmp1 transcription factor. Mol Cell Biol. 2005;25:220–32. doi: 10.1128/MCB.25.1.220-232.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kollmann K, Heller G, Sexl V. c-JUN prevents methylation of p16(INK4a) (and Cdk6): the villain turned bodyguard. Oncotarget. 2011;2:422–7. doi: 10.18632/oncotarget.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ameyar-Zazoua M, Wisniewska MB, Bakiri L, Wagner EF, Yaniv M, Weitzman JB. AP-1 dimers regulate transcription of the p14/p19ARF tumor suppressor gene. Oncogene. 2005;24:2298–306. doi: 10.1038/sj.onc.1208424. [DOI] [PubMed] [Google Scholar]
- 33.Berkovich E, Lamed Y, Ginsberg D. E2F and Ras synergize in transcriptionally activating p14ARF expression. Cell Cycle. 2003;2:127–33. doi: 10.4161/cc.2.2.293. [DOI] [PubMed] [Google Scholar]
- 34.Palmero I, Pantoja C, Serrano M. p19ARF links the tumour suppressor p53 to Ras. Nature. 1998;395:125–6. doi: 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- 35.Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–33. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boone DN, Hann SR. The Myc-ARF-Egr1 pathway: unleashing the apoptotic power of c-Myc. Cell Cycle. 2011;10:2043–4. doi: 10.4161/cc.10.13.15711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–77. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 38.Simboeck E, Di Croce L. p16INK4a in cellular senescence. Aging (Albany NY) 2013;5:590–1. doi: 10.18632/aging.100592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irelan JT, Gutierrez Del Arroyo A, Gutierrez A, Peters G, Quon KC, Miraglia L, Chanda SK. A functional screen for regulators of CKDN2A reveals MEOX2 as a transcriptional activator of INK4a. PLoS One. 2009;4:e5067. doi: 10.1371/journal.pone.0005067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carver BS, Tran J, Chen Z, Carracedo-Perez A, Alimonti A, Nardella C, Gopalan A, Scardino PT, Cordon-Cardo C, Gerald W, et al. ETS rearrangements and prostate cancer initiation. Nature. 2009;457:E1–, discussion E2-3. doi: 10.1038/nature07738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shin S, Kim TD, Jin F, van Deursen JM, Dehm SM, Tindall DJ, Grande JP, Munz JM, Vasmatzis G, Janknecht R. Induction of prostatic intraepithelial neoplasia and modulation of androgen receptor by ETS variant 1/ETS-related protein 81. Cancer Res. 2009;69:8102–10. doi: 10.1158/0008-5472.CAN-09-0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shaikhibrahim Z, Braun M, Nikolov P, Boehm D, Scheble V, Menon R, Fend F, Kristiansen G, Perner S, Wernert N. Rearrangement of the ETS genes ETV-1, ETV-4, ETV-5, and ELK-4 is a clonal event during prostate cancer progression. Hum Pathol. 2012;43:1910–6. doi: 10.1016/j.humpath.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 43.Svensson MA, LaFargue CJ, MacDonald TY, Pflueger D, Kitabayashi N, Santa-Cruz AM, Garsha KE, Sathyanarayana UG, Riley JP, Yun CS, et al. Testing mutual exclusivity of ETS rearranged prostate cancer. Lab Invest. 2011;91:404–12. doi: 10.1038/labinvest.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010;29:2147–60. doi: 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verger A, Duterque-Coquillaud M. When Ets transcription factors meet their partners. Bioessays. 2002;24:362–70. doi: 10.1002/bies.10068. [DOI] [PubMed] [Google Scholar]
- 46.Barber-Rotenberg JS, Selvanathan SP, Kong Y, Erkizan HV, Snyder TM, Hong SP, Kobs CL, South NL, Summer S, Monroe PJ, et al. Single enantiomer of YK-4-279 demonstrates specificity in targeting the oncogene EWS-FLI1. Oncotarget. 2012;3:172–82. doi: 10.18632/oncotarget.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blagosklonny MV. Wt p53 impairs response to chemotherapy: make lemonade to spare normal cells. Oncotarget. 2012;3:601–7. doi: 10.18632/oncotarget.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertheau P, Espié M, Turpin E, Lehmann J, Plassa LF, Varna M, Janin A, de Thé H. TP53 status and response to chemotherapy in breast cancer. Pathobiology. 2008;75:132–9. doi: 10.1159/000123851. [DOI] [PubMed] [Google Scholar]
- 49.Pimkina J, Murphy ME. Interaction of the ARF tumor suppressor with cytosolic HSP70 contributes to its autophagy function. Cancer Biol Ther. 2011;12:503–9. doi: 10.4161/cbt.12.6.15976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konishi N, Nakamura M, Kishi M, Nishimine M, Ishida E, Shimada K. Heterogeneous methylation and deletion patterns of the INK4a/ARF locus within prostate carcinomas. Am J Pathol. 2002;160:1207–14. doi: 10.1016/S0002-9440(10)62547-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qie S, Sang N. Release the ink4a/arf growth suppression by “u” and “me”? Cell Cycle. 2011;10:185–6. doi: 10.4161/cc.10.2.14471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberti A, Rizzolio F, Lucchetti C, de Leval L, Giordano A. Ubiquitin-mediated protein degradation and methylation-induced gene silencing cooperate in the inactivation of the INK4/ARF locus in Burkitt lymphoma cell lines. Cell Cycle. 2011;10:127–34. doi: 10.4161/cc.10.1.14446. [DOI] [PubMed] [Google Scholar]
- 53.Kan CE, Patton JT, Stark GR, Jackson MW. p53-mediated growth suppression in response to Nutlin-3 in cyclin D1 transformed cells occurs independently of p21. Cancer Res. 2007;67:9862–8. doi: 10.1158/0008-5472.CAN-07-0259. [DOI] [PubMed] [Google Scholar]
- 54.Singhal R, Bard JE, Nowak NJ, Buck MJ, Kandel ES. FOXO1 regulates expression of a microRNA cluster on X chromosome. Aging (Albany NY) 2013;5:347–56. doi: 10.18632/aging.100558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singhal R, Kandel ES. The response to PAK1 inhibitor IPA3 distinguishes between cancer cells with mutations in BRAF and Ras oncogenes. Oncotarget. 2012;3:700–8. doi: 10.18632/oncotarget.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–96. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schott B, Iraj ES, Roninson IB. Effects of infection rate and selection pressure on gene expression from an internal promoter of a double gene retroviral vector. Somat Cell Mol Genet. 1996;22:291–309. doi: 10.1007/BF02369568. [DOI] [PubMed] [Google Scholar]
- 58.Edlund T, Walker MD, Barr PJ, Rutter WJ. Cell-specific expression of the rat insulin gene: evidence for role of two distinct 5′ flanking elements. Science. 1985;230:912–6. doi: 10.1126/science.3904002. [DOI] [PubMed] [Google Scholar]