

Abstract
Chlamydia pneumoniae is a common respiratory pathogen that is considered a highly likely risk factor for atherosclerosis. C. pneumoniae is disseminated from the lung into systemic circulation via infected monocytes and lodges at the atherosclerotic sites. During transit, C. pneumoniae-infected monocytes in circulation are subjected to shear stress due to blood flow. The effect of mechanical stimuli on infected monocytes is largely understudied in the context of C. pneumoniae infection and inflammation. We hypothesized that fluid shear stress alters the inflammatory response of C. pneumoniae-infected monocytes and contributes to immune cell recruitment to the site of tissue damage. Using an in vitro model of blood flow, we determined that a physiological shear stress of 7.5 dyn/cm2 for 1 h on C. pneumoniae-infected monocytes enhances the production of several chemokines, which in turn is correlated with the recruitment of significantly large number of monocytes. Taken together, these results suggest synergistic interaction between mechanical and chemical factors in C. pneumoniae infection and associated inflammation.
Keywords: Chlamydia pneumoniae, Monocytes, Shear stress, Chemokines, Chemotaxis
Introduction
Chlamydia pneumoniae is a respiratory pathogen implicated in chronic inflammatory diseases like atherosclerosis, arthritis, and Alzheimer's disease.3,7,10 Of interest, there is compelling evidence from numerous studies including seroepidemiological,21,45 histopathological,15 animal models of disease development and treatment,4,41 short-term, and limited clinical intervention trials,9 and in vitro cell culture50 that suggest a major role of C. pneumoniae infection in atherosclerosis.1,7 In vivo studies have shown that C. pneumoniae infects neutrophils, alveolar macrophages, and is disseminated from the lungs to the vasculature through peripheral blood mononuclear cells to atherosclerotic foci.19,34,40 Since C. pneumoniae is ubiquitous, and chronic and reinfections of the lung are common, the infection often draws a chronic inflammatory response and immune cells to the site of infection, which perhaps in the process of clearing the infection, may lead to bystander tissue damage and disease sequelae.11,13,25
C. pneumoniae is an obligate intracellular bacterium and hence needs a host cell to survive and propagate. Following an initial infection, the infectious elementary bodies (EB) enter the host cell wherein they differentiate into non-infectious, replicating reticulate bodies (RB). The RB subsequently differentiates back to EB, and when the host cells die, the mature EBs are released and infect other susceptible host cells.28 C. pneumoniae infection cycle in monocytes/macrophages may last from 3–7 days during which the cells secrete a plethora of inflammatory cytokines, matrix metalloproteases, procoagulants, and upregulate adhesion and LDL-up-take receptor expression levels.31,36,48 C. pneumoniae is believed to be transmitted from the lung to other tissues including arterial wall and the brain by infected monocytes through circulation.19,30 While most of these in vitro studies focus on monocytes in static culture, in reality, during transit from the lungs by blood flow, the monocytes experience biophysical forces such as shear stress. Though it is established that hemodynamic forces indeed tightly regulate responses of cells in the vasculature including endothelial cells,51 platelets,27 and neutrophils,33,44 the effects of mechanical forces on infection and inflammation is grossly under-addressed. We have recently shown that shear stress exacerbates the release of cytokine IL-1 β in monocytes infected with the mouse respiratory pathogen, C. muridarum, and also increases endothelial adhesion.16
In this work, we evaluated the role of Chlamydia pneumoniae infection and shear stress on chemokine release from monocytes. Our results show that C. pneumoniae infection triggers the release of chemokines and monocyte migration, which are enhanced by shear stress, suggesting that infection and shear stress together may play a critical role in vascular inflammation and atherosclerosis.
Materials and Methods
Cells
Human monocyte cell line, THP-1 cells (ATCC, Manassas, VA) were cultured in RPMI 1640 (ATCC) supplemented with 10% FBS and .05 mM mercaptoethanol (Sigma-Aldrich, St. Louis, MO), at 37 °C and 5% CO2. The cells were passaged into fresh media when the cells reached a density of 106/mL. The cell viability was measured by trypan blue exclusion assay (Countess automated cell counter, Life Technologies, Grand Island, NY).
C. pneumoniae Propagation
Chlamydia pneumoniae TW183 (U Washington, Seattle, WA) in SPG buffer was added to a confluent monolayer of Hep2 cells (ATCC) in EMEM (ATCC) supplemented with 10% FBS (Life Technologies), 1 μg/mL gentamicin (Life Technologies) and 0.6 μg/mL cycloheximide (Sigma-Aldrich, St. Louis, MO). Genatmicin and cycloheximide were added to inactivate any extracellular C. pneumoniae, and to prevent Hep2 cell proliferation during the infection period, respectively. 72 h post infection, the Hep2 cells were harvested and lysed by vortexing using glass beads for 3 min. The elementary bodies (EB) were spun down at 30,000g using a high speed centrifuge, and aliquoted in sucrose-phosphate-glutamine buffer and stored at −80 °C. C. pneumoniae specific murine monoclonal antibody TT401 (U Washington, Seattle, WA) along with a FITC-conjugated secondary antibody (Abcam, Cambridge, MA) was used to establish the bacterial counts in stocks using fluorescence microscope (Leica DMI6000, Buffalo Grove, IL), following the published protocols for C. pneumoniae propagation.8
C. pneumoniae Infection of THP1 Cells
THP-1 monocytic cells were infected with C. pneumoniae EB at multiplicity of infection (MOI) of 2 by intermittent rocking at 35°C for 2.5 h. The inoculum was removed, the cells were resuspended at 1 × 106 cells/mL in RPMI-1640 supplemented with 10% FBS and 1 μg/mL gentamicin, and incubated for additional 72 h. To quantify infectivity, the adherent cells at 6, 18, 36 and 72 h post infection were fixed in freshly prepared 2% paraformaldehyde, permeabilized with 1× Permwash (BD Biosciences, San Jose, CA), and labeled with C. pneumoniae-specific murine primary antibody TT401 and FITC-conjugated anti-mouse secondary antibody and/or Alexa-Fluor 660 phalloidin antibody (Life Technologies). The nuclei were counter-stained with DAPI. 100 μL of 1 × 106 uninfected cells/mL were cytospun (CytoSpin 4, Thermo Scientific, Asheville, NC) at 1000 rpm for 5 min, and stained as described above. The cells were analyzed using a fluorescence microscope (Leica). The infectivity was also assayed using flow cytometry (LSR II, BD Biosciences, San Jose, CA). To quantify viability, the flask was incubated on ice for 5 min, the cells were isolated by gentle scraping, and counted after trypan blue staining using an automated cell counter (Countess, Life Technologies, Grand Island, NY).
Chemokine Assays
THP-1 monocytes were infected with mock PBS or Chlamydia pneumoniae EB (MOI 2) for 2.5 h and cultured for 72 h as described above. At 2, 6, 18, 36 and 72 h post infection, the cells were incubated on ice for 5 min, gently scraped, separated by centrifugation (5 min, 160g), and the supernatants were supplemented with recommended 1 × concentration of Halt protease inhibitor cocktail (Thermo Scientific, Rockford, IL) and stored at −80 °C for the analysis of chemokines IL-8, RANTES, MIP-1α, MIP-1β, MCP-1, and IP-10 using Bio-plex protein array system (Bio-Rad Laboratories, Hercules, CA). Briefly, 60 μL of supernatants were mixed with antibody-coupled, color-coded bead cocktail, and analyzed by luminex-based technology according to manufacturer's instructions. The chemokine concentrations in the suspension are automatically calculated using the standard curves derived from recombinant chemokine standards.
Exposure to Shear Stress
THP-1 monocytes were either infected with mock PBS (Mock) or C. pneumoniae EB (Infected) at MOI 2 for 36 h. 36 h post infection the cells were incubated on ice for 5 min, gently scraped, and separated from the supernatant by centrifugation. The cells were resuspended in fresh media containing 2 μg/mL gentamicin at a concentration of 6 × 106 cells/mL and incubated for 1 h for equilibration. 500 μL of this cell suspension was sheared for 1 h at either 0 (static) or 7.5 dyn/cm2 (shear), using a cone-and-plate viscometer (DVII + Pro, Brookfield Instruments, Middleboro, MA) with the sample cup maintained at 37 °C using a circulating water bath (TC-650, Brookfield Instruments). Gentamicin was added to prevent potential contamination during shear exposure. The static controls were maintained at conditions similar to shear treatment except for the rotation of the cone. Under these experimental conditions, the evaporation (<10%) and pH change (<0.3 units) were minimal and similar for both static and shear conditions. Immediately post shear, the cells were spun down by centrifugation (5 min, 160g), and the supernatant was collected. The supernatant was filter-sterilized (0.22 μm filter), supplemented with recommended 1× concentration of Halt protease inhibitor cocktail, and stored at −80°C for further use. The protease inhibitor was added to prevent the degradation of chemokines during storage. The cells collected post shear were tested for viability by trypan blue exclusion assay and further incubated to 72 h post infection to analyze infectivity as described above.
Endothelial Adhesion Under Flow
Primary human aortic endothelial cells (HAECs) extracted from the aorta of cadaver of individual with no known cardiovascular abnormalities was obtained, and used within five passages following manufacturer's protocol (Life Technologies, Grand Island, NY). The cells cultured to confluence in ibiTreat Vl0.1. slides (Ibidi GmbH, Munich, Germany) were treated for 4 h with supernatants from uninfected or infected monocytes obtained from experiments described above. The flow chamber was assembled on top of an inverted microscope attached to a time-lapse digital camera (FX-360, Leica Microsystems). A monocyte suspension consisting 106 cells/mL was then drawn through the perfusion chamber using a syringe pump (Harvard Apparatus, Holliston, MA, USA) at a constant flow rate corresponding to a wall shear stress of 1 dyn/cm2. After 1 min of perfusion, the adhesion of monocytes to HAECs was captured by bright-field microscopy at 20× magnification for 4 min in 5 different fields of view (0.1 × 0.1 mm2). The images were analyzed offline using Image J (NIH).
Chemotaxis Assays
200 μL of supernatant was collected after the exposure of THP-1 cells to shear stress and diluted to 600 μL with fresh media. The diluted supernatants were added to the lower well of the Boyden chamber (BD Biosciences, San Jose, CA). 100 μL of uninfected THP-1 cells at a density of 5 × 106 cells per mL was added to the top well containing a 5 μm pore-filter. The setup was incubated for 2 h at 37 °C, and number of cells transmigrated through the filter to the lower well were counted from 10 μL of cell suspension drawn from the lower well using an automated cell counter (Countess, Life Technologies).
Statistics
All the experiments were performed in triplicate and each experiment was repeated at least two times under independent conditions. The results are represented as mean ± SD from one representative experiment in the plots. Statistical differences between treatments were evaluated either using two-tailed Student's t test, or two-way ANOVA (GraphPad Prism, La Jolla, CA), and the results were considered significant if p < 0.05.
Results
C. pneumoniae Infection of Monocytes
C. pneumoniae is an obligate intracellular bacterium with tropism to various cell types including epithelial cells, monocytes or macrophages, endothelial cells, and smooth muscle cells.17 In this work, we used the well-established THP1 cell line as a model for primary human monocytes.38 C. pneumoniae infects THP1 monocytes and forms inclusions, which grow over a period of 72 h (Fig. 1). In general, we observed some variation in the number and size of chlamydial inclusions in the monocytes with some inclusions large enough to occupy half the cell volume by pushing the nucleus to one side, and with others that are much smaller represented as dots distributed through the cell (Fig. 1a). Such multiple inclusions are commonly observed in C. pneumoniae infection of various cell types.14 The infectivity at the end of 72 h was 80–90% as measured by counting the number of cells with inclusions, and also confirmed by flow cytometry (Fig. 1b). The infected cells attached to the surface, and the viability of these cells remained ∼90% through most of the infection cycle. We also observed that the infection is associated with substantial redistribution of actin cytoskeleton as the monocytes adhere to the tissue culture flask. The infected monocytes take on a ‘fried-egg’ appearance characteristic of macrophage-like phenotype, an observation that is consistent with previous reports.50
Figure 1.
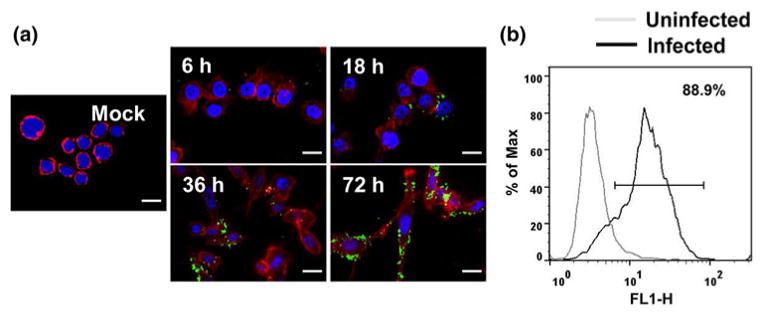
Chlamydia pneumoniae infection. (a) THP1 monocytes were infected with Chlamydia pneumoniae (TW183) at an MOI 2 and were incubated for 72 h. The adherent cells were fixed at different time points, and stained with anti-Chlamydia pneumoniae antibody, Alexa Fluor 660 phalloidin and DAPI. Chlamydial inclusions (green), THP1 actin (red) and nucleus (blue) are shown. The uninfected cells were cytospun and stained. Scale bar = 15 μM. (b) Flow cytometric analysis of monocyte infectivity.
Kinetics of Chemokine Secretion from Infected Monocytes
Next, we followed the release of chemokines macrophage inflammatory proteins (MIP-1α, MIP-1β), monocyte chemoattractant protein (MCP-1), interleukin (IL-8), RANTES, and Interferon-γ induced protein of 10 kDa (IP-10) over the life cycle of infection, i.e., 72 h. We observed that following an initial delay of 6 h, most of the chemokine levels increase, and after 18 h of infection, MIP-1α, MIP-1β, MCP-1, and IL-8 reach their peak levels whereas RANTES and IP-10 reach their peak levels by 36 h post infection (Fig. 2). All chemokines except MIP-1α remained at high levels till 72 h. In contrast, uninfected cells did not produce any detectable levels of chemokines.
Figure 2.

Kinetics of chemokine production by C. pneumoniae-infected cells. THP1 monocytes (1 × 106 cells/mL) were infected with mock (only media) or Chlamydia pneumoniae (TW183) at an MOI 2. The supernatants were collected at 2, 6, 18, 36, and 72 h and analyzed. The results are expressed as mean ± SD of one representative experiment performed in triplicate, and the experiments were performed twice.
Effect of Shear Stress on Chemokine Secretion
Having established that chlamydial infection triggers the release of chemokines, we sought to examine the effect of shear stress on this process since the infected monocytes experience shear stress while in circulation. To this end, after 36 h of infection, the supernatant was discarded, and adherent and non-adherent monocytes were resuspended in fresh media for 1 h. The cells were then subjected to a physiological shear stress of 7.5 dyn/cm2 for 1 h, and chemokine secretion was measured. We chose 36 h as representative time point since it is the mid-point in the infection cycle, and all chemokines are expressed at this stage. 7.5 dyn/cm2 was used as representative bulk shear stress experienced by cells in circulation since the shear stress varies between 1 and 5 dyn/cm2 in veins and 5–20 dyn/cm2 in arteries. Uninfected monocytes subjected to shear, and infected monocytes under static conditions were used as controls. We observed that shear stress does not alter the infectivity or viability of the cells as evidenced by intact inclusion and maintenance of cell membrane integrity (Fig. 3). There was a modest decrease in viability due to chlamydial infection compared to uninfected cells. The chemokine release profiles are shown in Fig. 4. We observe that the uninfected cells, under static or shear conditions, do not release any chemokines indicating that shear stress alone does not trigger chemokine release. We also observe that the infected cells under static conditions release substantial quantities of fresh chemokines within a short duration suggesting that the presence of chlamydial infection promotes sustained release of chemokines even after 36 h post-infection. As the infected cells were sheared, all the chemokines measured in this study, namely, IL-8, RANTES, MCP-1, MIP-1α, MIP-1β and IP-10, were upregulated 2–4 fold in 1 h. We observed that the actual levels of chemokines varied between experiments because of inherent variations in infectivity, the number of C. pneumoniae inclusions per infected cell, size of the inclusions, and hence the degree of inflammatory response.37 However, the upregulation due to shear stress compared to static levels was consistent irrespective of the absolute amounts of chemokines released following infection. This data suggests that shear stress enhances pro-inflammatory response only in the presence of infection.
Figure 3.
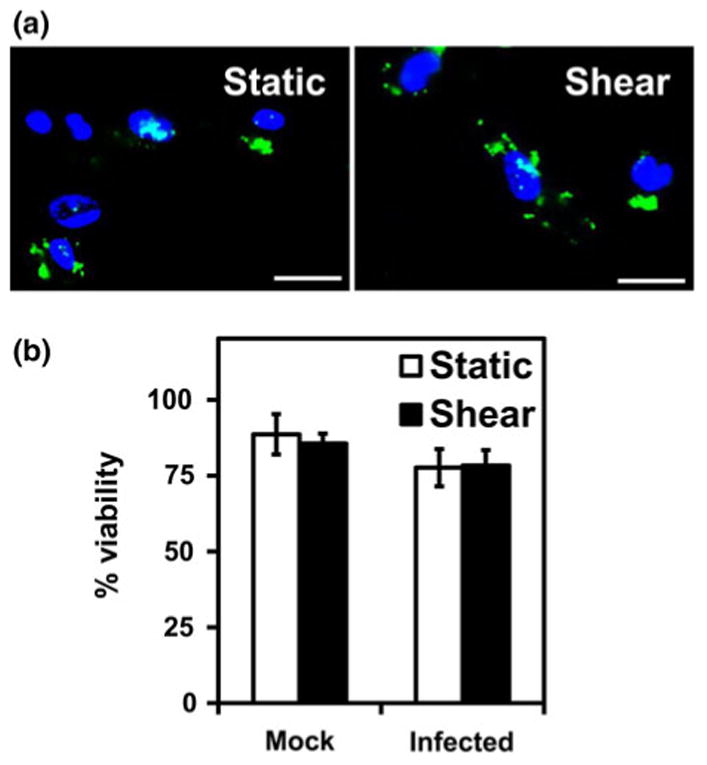
Effect of shear stress on infectivity and viability of monocytes. THP1 monocytes were infected with mock (only media) or Chlamydia pneumoniae at MOI 2 for 36 h. The cells were resuspended in fresh media, and sheared at 0 (static) or 7.5 dyn/cm2 (shear) for 1 h. (a) The cells were incubated for an additional 32 h to finish 72 h of infection cycle and then fixed and stained with anti-Chlamydia pneumoniae antibody and DAPI. Chlamydial inclusions (green) and THP1 nucleus (blue) are shown. Scale bar = 20 μM. (b) The viability was assessed by trypan blue exclusion. The results are mean ± SD of one representative experiment performed in triplicate, and the experiments were repeated three times.
Figure 4.

Effect of shear stress on chemokine secretion from infected monocytes. Uninfected or infected THP1 cells were incubated under static conditions or subjected to shear stress as described in Fig. 3. The supernatants were collected immediately, and analyzed for chemokines. The results are mean ± SD of one representative experiment performed in triplicate, and the experiments were repeated three times. The * and § represent statistically significant change (p<0.05, ANOVA) in chemokine production by due to infection only under static conditions, or due to shear stress only in infected cells compared to respective controls.
Effect of Chemokine Secretion on Monocyte Migration
Infected monocytes from circulation can migrate into the subendothelium, and the chemokines released due to infection can attract fresh monocytes from circulation that form foam cells and eventually result in atherosclerotic plaque.26 As a simplified model of this process, we used supernatants collected from infected monocytes and evaluated its effects on monocyte recruitment to endothelium and infiltration. We observed that supernatant from infected, but not uninfected monocytes, can activate endothelium to support monocyte adhesion (Fig. 5a), which can then migrate to the subendothelium. We studied the effect of chemokine release on monocyte migration by incubating fresh, uninfected THP-1 monocytes in the upper well of Boyden chamber, and supernatants collected from infected or uninfected monocyte under static or shear conditions to the lower well, and quantify the monocytes migrated to the lower chamber after 2 h of incubation. As shown in Fig. 5b, the supernatant from uninfected cells did not result in any chemotaxis, consistent with the absence of chemokines (Fig. 4). On the other hand, supernatant from infected monocytes under static conditions profoundly increases the number of monocytes migrated. This increase is further enhanced by nearly 3-fold in supernatants from infected monocytes exposed to shear stress. This increase correlates well with the 2–4 fold increase in the chemokine levels due to shear stress.
Figure 5.

(a) Effect chlamydial infection on monocyte recruitment to endothelium. 106 monocytes/mL was perfused over confluent HAECs activated with uninfected or infected monocyte supernatant at a wall shear stress of 1 dyn/cm2. After 1 min of perfusion, the interactions were captured by bright-field microscopy at 20× magnification for 4 min in 5 different fields of view (0.1 × 0.1 mm2). The results are expressed as mean ± SD of one representative experiment performed in triplicate, and the experiments were performed two times. The * represent statistically significant change (p<0.05, Student's t test) in monocyte adhesion due to infection compared to uninfected control. (b) Effect of infection and shear stress on monocyte chemotaxis. Supernatants from experiments described in Fig. 3 were diluted 3-fold, added to the lower portion of the Boyden chamber. Fresh, uninfected THP1 cells at a density of 5 × 106 cells/mL were added to the top portion of the Boyden chamber with 5 μm pore-filter. The setup was incubated for 2 h at 37 °C, and the number of cells transmigrated through the pore to the bottom of the well were counted. The results are expressed as mean ± SD of the number of transmigrated cells per μL of suspension in the lower well from one representative experiment performed in triplicate, and the experiments were performed three times. The * and § represent statistically significant change (p<0.05, ANOVA) in monocyte migration by due to infection only under static conditions, or due to shear stress only in infected cells compared to respective controls.
Discussion
Atherosclerosis is a multi-factorial chronic inflammatory disease. An emerging paradigm based on multiple epidemiological studies is that infectious agents including bacteria/viruses may contribute to the pathogenesis either via direct infection of vascular cells or indirect effects due to cytokines or acute phase proteins.22 In particular, C. pneumoniae gained attention after its isolation from atherosclerotic plaques,20,29 and since then numerous studies have confirmed that C. pneumoniae infection as a highly likely risk factor for atherosclerosis.22,42 However, the role of C. pneumoniae infection at various stages of atherosclerotic disease progression is not well-understood. We report here for the first time that C. pneumoniae infection and shear stress synergistically exacerbates atherosclerotic process by altering the inflammatory microenvironment.
C. pneumoniae forms multiple inclusions within monocytes and remained viable without initiating host cell lysis for at least up to 72 h. During this long infection cycle, C. pneumoniae induces the production of several pro-inflammatory cytokines including TNFα, and IL1β.23 It has also previously been shown that C. pneumoniae infection of monocytes results in the upregulation of chemokines MCP-1 and MIP1α,36 and their genes and the chemokine receptor CCR2.48 We show here that, in addition to these chemokines, others including IL-8, MIP1β, RANTES and IP-10 are secreted from infected monocytes over the long infection cycle. The infected monocytes experience shear stress due to blood flow as they travel through circulation from the lungs to the atherosclerotic foci. It is now well-established that shear stress alters cellular and molecular inflammatory responses in vascular cells.49 In this work, we have demonstrated that shear stress increases the release of several chemokines from C. pneumoniae-infected monocytes. Since chemokine upregulation was concomitant with shear exposure, it is likely that this shear-induced enhancement is mediated at the translational level or released from existing stores. Together with our previous study,16 this work underscores the importance of the interaction between biochemical and biomechanical stimuli in determining the inflammatory response.
The infected monocytes/macrophages from circulation transmigrate into the arterial wall and lodge in the subendothelium, where they continue to release cytokines and chemokines.26,43 The cytokines from infected monocytes can activate the endothelium to recruit leukocytes from the blood.16 The chemokine gradient triggers leukocyte migration into the plaque site, which is a critical step in inflammation. The increase in chemokine gradient due to infection and shear exposure correlates well with the increase in monocyte migration across the barrier. It has been shown that most of the chemokines analyzed in this work are present at higher levels in atherosclerotic lesions.6,39 High expression levels MCP-1 are found within atherosclerotic lesions,35 and knock down of MCP-112 has shown a significant reduction in the atherosclerotic plaque size due to decrease in leukocyte trafficking which forms a major part of the lesion. Higher serological level of MCP-1 is also correlated in patients at risk for coronary artery disease.32 RANTES, MIP-1P and MIP-1β are also found in the atherosclerotic lesions, and blocking RANTES inhibit lesion formation and leukocyte infiltration in the lesions.46,47 IP-10 levels are also high in the lesions and it participates in atherosclerosis by modulating the local balance of regulator and effector T-cell populations.24 IL-8 is produced by macrophages and is believed to be central in macrophage accumulation in advanced lesions.5 Further, these proteins are not only involved in inflammatory response during atherosclerosis but also in other important processes that contribute to disease progression including smooth muscle cell migration, MMP release, LDL uptake.2,18
In summary, our results show that C. pneumoniae infection triggers an inflammatory response in monocytes that is exacerbated by shear stress due to blood flow resulting in leukocyte migration to the plaque site, which is a principal component in atheroprogression. Our model provides avenues to explore the connection between infection, inflammation, and biomechanical forces relevant in vivo. Taken together, we show mechanical forces may play an important role in hastening disease progression by modulating vascular inflammatory response during systemic infections, and in a larger context, our results warrant the consideration of biomechanical factors as potent regulators of cellular and molecular responses in disease pathophysiology.
Acknowledgments
This study was supported by grants from the NIH HL112629. We would like to thank Aswathi Cheeniyil for technical assistance.
Footnotes
Shankar J. Evani and Shatha F. Dallo contributed equally to this work.
References
- 1.Anderson JL. Infection, antibiotics, and atherothrombosis—end of the road or new beginnings? N Engl J Med. 2005;352:1706–1709. doi: 10.1056/NEJMe058019. [DOI] [PubMed] [Google Scholar]
- 2.Baker CS, Gupta S. Chemokines: the link between inflammation, restenosis and atherosclerosis? Int J Cardiol. 2001;80:107–108. doi: 10.1016/s0167-5273(01)00484-3. [DOI] [PubMed] [Google Scholar]
- 3.Balin BJ, Little CS, Hammond CJ, Appelt DM, Whittum-Hudson JA, Gerard HC, Hudson AP. Chlamydophila pneumoniae and the etiology of late-onset Alzheimer's disease. J Alzheimer's Dis. 2008;13:371–380. doi: 10.3233/jad-2008-13403. [DOI] [PubMed] [Google Scholar]
- 4.Blessing E, Lin TM, Campbell LA, Rosenfeld ME, Lloyd D, Kuo CC. Chlamydia pneumoniae induces inflammatory changes in the heart and aorta of nomocholesterolemic C57BL/6J mice. Infect Immun. 2000;68:4765–4768. doi: 10.1128/iai.68.8.4765-4768.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boisvert WA, Rose DM, Johnson KA, Fuentes ME, Lira SA, Curtiss LK, Terkeltaub RA. Up-regulated expression of the CXCR2 ligand KC/GRO-alpha in atherosclerotic lesions plays a central role in macrophage accumulation and lesion progression. Am J Pathol. 2006;168:1385–1395. doi: 10.2353/ajpath.2006.040748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braunersreuther V, Mach F, Steffens S. The specific role of chemokines in atherosclerosis. Thromb Haemost. 2007;97:714–721. [PubMed] [Google Scholar]
- 7.Campbell LA, Kuo CC. Chlamydia pneumoniae—an infectious risk factor for atherosclerosis. Nat Rev Microbiol. 2004;2:23–32. doi: 10.1038/nrmicro796. [DOI] [PubMed] [Google Scholar]
- 8.Campbell LA, Kuo CC. Cultivation and laboratory maintenance of Chlamydia pneumoniae. Curr Protoc Microbiol. 2009;Chapter 11(Unit11):B1. doi: 10.1002/9780471729259.mc11b01s12. [DOI] [PubMed] [Google Scholar]
- 9.Cannon CP, Braunwald E, McCabe CH, Grayston JT, Muhlestein B, Giugliano RP, Cairns R, Skene AM. Pravastatin or Atorvastatin E, Infection Therapy-Thrombolysis in Myocardial Infarction I Antibiotic treatment of Chlamydia pneumoniae after acute coronary syndrome. N Engl J Med. 2005;352:1646–1654. doi: 10.1056/NEJMoa043528. [DOI] [PubMed] [Google Scholar]
- 10.Carter JD, Inman RD. Chlamydia-induced reactive arthritis: hidden in plain sight? Best Pract Res Clin Rheumatol. 2011;25:359–374. doi: 10.1016/j.berh.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Choi EY, Kim BK, Hong H, Kwon M, Song YG, Byun KH, Park HY, Whang KC, Kim HS. Upregulation of extracellular matrix metalloproteinase inducer (EMMPRIN) and gelatinases in human atherosclerosis infected with Chlamydia pneumoniae: the potential role of Chlamydia pneumoniae infection in the progression of atherosclerosis. Exp Mol Med. 2002;34:391–400. doi: 10.1038/emm.2002.56. [DOI] [PubMed] [Google Scholar]
- 12.Combadiere C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito G, Merval R, Proudfoot A, Tedgui A, Mallat Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C (lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 13.Davidson M, Kuo CC, Middaugh JP, Campbell LA, Wang SP, Newman WP, III, Finley JC, Grayston JT. Confirmed previous infection with Chlamydia pneumoniae (TWAR) and its presence in early coronary atherosclerosis. Circulation. 1998;98:628–633. doi: 10.1161/01.cir.98.7.628. [DOI] [PubMed] [Google Scholar]
- 14.Dumrese C, Maurus CF, Gygi D, Schneider MK, Walch M, Groscurth P, Ziegler U. Chlamydia pneumoniae induces aponecrosis in human aortic smooth muscle cells. BMC Microbiol. 2005;5:2. doi: 10.1186/1471-2180-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esposito G, Blasi F, Allegra L, Chiesa R, Melissano G, Cosentini R, Tarsia P, Dordoni L, Cantoni C, Arosio C, Fagetti L. Demonstration of viable Chlamydia pneumoniae in atherosclerotic plaques of carotid arteries by RT-PCR. Ann Vasc Surg. 1999;13:421–425. doi: 10.1007/s100169900277. [DOI] [PubMed] [Google Scholar]
- 16.Evani SJ, Murthy AK, Mareedu N, Montgomery RK, Arulanandam BP, Ramasubramanian AK. Hydrodynamic regulation of monocyte inflammatory response to an intracellular pathogen. PLoS One. 2011;6:e14492. doi: 10.1371/journal.pone.0014492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaydos CA, Summersgill JT, Sahney NN, Ramirez JA, Quinn TC. Replication of Chlamydia pneumoniae in vitro in human macrophages, endothelial cells, and aortic artery smooth muscle cells. Infect Immun. 1996;64:1614–1620. doi: 10.1128/iai.64.5.1614-1620.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerszten RE, Mach F, Sauty A, Rosenzweig A, Luster AD. Chemokines, leukocytes, and atherosclerosis. J Lab Clin Med. 2000;136:87–92. doi: 10.1067/mlc.2000.108154. [DOI] [PubMed] [Google Scholar]
- 19.Gieffers J, van Zandbergen G, Rupp J, Sayk F, Krüger S, Ehlers S, Solbach W, Maass M. Phagocytes transmit Chlamydia pneumoniae from the lungs to the vasculature. Eur Respir J. 2004;23:506–510. doi: 10.1183/09031936.04.00093304. [DOI] [PubMed] [Google Scholar]
- 20.Grayston JT, Kuo CC, Coulson AS, Campbell LA, Lawrence RD, Lee MJ, Strandness ED, Wang SP. Chlamydia pneumoniae (TWAR) in atherosclerosis of the carotid artery. Circulation. 1995;92:3397–3400. doi: 10.1161/01.cir.92.12.3397. [DOI] [PubMed] [Google Scholar]
- 21.Gupta S, Leatham EW, Carrington D, Mendall MA, Kaski JC, Camm AJ. Elevated Chlamydia pneumoniae antibodies, cardiovascular events, and azithromycin in male survivors of myocardial infarction. Circulation. 1997;96:404–407. doi: 10.1161/01.cir.96.2.404. [DOI] [PubMed] [Google Scholar]
- 22.Gurfinkel E, Lernoud V. The role of infection and immunity in atherosclerosis. Expert Rev Cardiovasc Ther. 2006;4:131–137. doi: 10.1586/14779072.4.1.131. [DOI] [PubMed] [Google Scholar]
- 23.Heinemann M, Susa M, Simnacher U, Marre R, Essig A. Growth of Chlamydia pneumoniae induces cytokine production and expression of CD14 in a human monocytic cell line. Infect Immun. 1996;64:4872–4875. doi: 10.1128/iai.64.11.4872-4875.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heller EA, Liu E, Tager AM, Yuan Q, Lin AY, Ahluwalia N, Jones K, Koehn SL, Lok VM, Aikawa E, Moore KJ, Luster AD, Gerszten RE. Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation. 2006;113:2301–2312. doi: 10.1161/CIRCULATIONAHA.105.605121. [DOI] [PubMed] [Google Scholar]
- 25.Hoymans V, Bosmans J, Ursi D, Dubois F, Van Marck E, Altwegg M, Ieven M, Vrints C. Systemic inflammation, Chlamydia pneumoniae DNA in circulating leukocytes and coronary atherosclerosis. Acta Cardiol. 2002;57:213–219. doi: 10.2143/AC.57.3.2005391. [DOI] [PubMed] [Google Scholar]
- 26.Kalayoglu MV, Perkins BN, Byrne GI. Chlamydia pneumoniae-infected monocytes exhibit increased adherence to human aortic endothelial cells. Microbes Infect. 2001;3:963–969. doi: 10.1016/s1286-4579(01)01458-7. [DOI] [PubMed] [Google Scholar]
- 27.Kasirer-Friede A, Ruggeri ZM, Shattil SJ. Role for ADAP in shear flow-induced platelet mechanotransduction. Blood. 2010;115:2274–2282. doi: 10.1182/blood-2009-08-238238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krull M, Maass M, Suttorp N, Rupp J. Chlamydophila pneumoniae Mechanisms of target cell infection and activation. Thromb Haemost. 2005;94:319–326. doi: 10.1160/TH05-04-0261. [DOI] [PubMed] [Google Scholar]
- 29.Kuo CC, Grayston JT, Campbell LA, Goo YA, Wissler RW, Benditt EP. Chlamydia pneumoniae (TWAR) in coronary arteries of young adults (15–34 years old) Proc Natl Acad Sci U S A. 1995;92:6911–6914. doi: 10.1073/pnas.92.15.6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacIntyre A, Abramov R, Hammond CJ, Hudson AP, Arking EJ, Little CS, Appelt DM, Balin BJ. Chlamydia pneumoniae infection provmotes transmigration of monocytes through human brain endothelial cells. J Neurosci Res. 2003;71:740–750. doi: 10.1002/jnr.10519. [DOI] [PubMed] [Google Scholar]
- 31.Mamata Y, Hakki A, Newton C, Burdash N, Klein TW, Friedman H. Differential effects of Chlamydia pneumoniae infection on cytokine levels in human T lymphocyte- and monocyte-derived cell cultures. Int J Med Microbiol. 2007;297:109–115. doi: 10.1016/j.ijmm.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 32.Martinovic I, Abegunewardene N, Seul M, Vosseler M, Horstick G, Buerke M, Darius H, Lindemann S. Elevated monocyte chemoattractant protein-1 serum levels in patients at risk for coronary artery disease. Circ J. 2005;69:1484–1489. doi: 10.1253/circj.69.1484. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell MJ, King MR. Shear-induced resistance to neutrophil activation via the formyl peptide receptor. Biophys J. 2012;102:1804–1814. doi: 10.1016/j.bpj.2012.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moazed TC, Kuo Cc, Grayston JT, Campbell LA. Evidence for systemic dissemination of Chlamydia pneumoniae via macrophages in the mouse. J Infect Dis. 1998;177:1322–1325. doi: 10.1086/515280. [DOI] [PubMed] [Google Scholar]
- 35.Nelken NA, Coughlin SR, Gordon D, Wilcox JN. Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest. 1991;88:1121–1127. doi: 10.1172/JCI115411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Netea MG, Selzman CH, Kullberg BJ, Galama JM, Weinberg A, Stalenhoef AF, Van der Meer JW, Dinarello CA. Acellular components of Chlamydia pneumoniae stimulate cytokine production in human blood mononuclear cells. Eur J Immunol. 2000;30:541–549. doi: 10.1002/1521-4141(200002)30:2<541::AID-IMMU541>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 37.Poikonen K, Lajunen T, Silvennoinen-Kassinen S, Paldanius M, Leinonen M, Saikku P. Susceptibility of human monocyte-macrophages to Chlamydia pneumoniae infection in vitro is highly variable and associated with levels of soluble CD14 and C pneumoniae IgA and human HSP-IgG antibodies in serum. Scand J Immunol. 2008;67:279–284. doi: 10.1111/j.1365-3083.2007.02061.x. [DOI] [PubMed] [Google Scholar]
- 38.Qin Z. The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis. 2012;221:2–11. doi: 10.1016/j.atherosclerosis.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 39.Reape TJ, Rayner K, Manning CD, Gee AN, Barnette MS, Burnand KG, Groot PH. Expression and cellular localization of the CC chemokines PARC and ELC in human atherosclerotic plaques. Am J Pathol. 1999;154:365–374. doi: 10.1016/S0002-9440(10)65283-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenfeld ME, Blessing E, Lin TM, Moazed TC, Campbell LA, Kuo CC. Chlamydia, inflammation, and atherogenesis. J Infect Dis. 2000;181:S492–S497. doi: 10.1086/315618. [DOI] [PubMed] [Google Scholar]
- 41.Rothstein NM, Quinn TC, Madico G, Gaydos CA, Lowenstein CJ. Effect of azithromycin on murine arteriosclerosis exacerbated by Chlamydia pneumoniae. J Infect Dis. 2001;183:232–238. doi: 10.1086/317941. [DOI] [PubMed] [Google Scholar]
- 42.Roulis E, Polkinghorne A, Timms P. Chlamydia pneumoniae: modern insights into an ancient pathogen. Trends Microbiol. 2013;21(3):120–128. doi: 10.1016/j.tim.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 43.Rupp J, Koch M, van Zandbergen G, Solbach W, Brandt E, Maass M. Transmission of Chlamydia pneumoniae infection from blood monocytes to vascular cells in a novel transendothelial migration model. FEMS Microbiol Lett. 2005;242:203–208. doi: 10.1016/j.femsle.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Schmid-Schonbein GW, Kistler EB, Hugli TE. Mechanisms for cell activation and its consequences for biorheology and microcirculation: multiorgan failure in shock. Biorheology. 2001;38:185–201. [PubMed] [Google Scholar]
- 45.Vasconcelos EM, Degasperi GR, de Oliveira HC, Vercesi AE, de Faria EC, Castilho LN. Reactive oxygen species generation in peripheral blood monocytes and oxidized LDL are increased in hyperlipidemic patients. Clin Biochem. 2009;42:1222–1227. doi: 10.1016/j.clinbiochem.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Veillard NR, Kwak B, Pelli G, Mulhaupt F, James RW, Proudfoot AE, Mach F. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res. 2004;94:253–261. doi: 10.1161/01.RES.0000109793.17591.4E. [DOI] [PubMed] [Google Scholar]
- 47.Veillard NR, Steffens S, Burger F, Pelli G, Mach F. Differential expression patterns of proinflammatory and antiinflammatory mediators during atherogenesis in mice. Arterioscler Thromb Vasc Biol. 2004;24:2339–2344. doi: 10.1161/01.ATV.0000146532.98235.e6. [DOI] [PubMed] [Google Scholar]
- 48.Virok D, Loboda A, Kari L, Nebozhyn M, Chang C, Nichols C, Endresz V, Gonczol E, Berencsi K, Showe MK, Showe LC. Infection of U937 monocytic cells with Chlamydia pneumoniae induces extensive changes in host cell gene expression. J Infect Dis. 2003;188:1310–1321. doi: 10.1086/379047. [DOI] [PubMed] [Google Scholar]
- 49.Weinbaum S, Duan Y, Thi M, You L. An integrative review of mechanotransduction in endothelial, epithelial (renal) and dendritic cells (osteocytes) Cell Mol Bioengineering. 2011;4:510–537. doi: 10.1007/s12195-011-0179-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamaguchi H, Haranaga S, Widen R, Friedman H, Yamamoto Y. Chlamydia pneumoniae infection induces differentiation of monocytes into macrophages. Infect Immun. 2002;70:2392–2398. doi: 10.1128/IAI.70.5.2392-2398.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zaragoza C, Marquez S, Saura M. Endothelial mechanosensors of shear stress as regulators of atherogenesis. Curr Opin Lipidol. 2012;23:446–452. doi: 10.1097/MOL.0b013e328357e837. [DOI] [PubMed] [Google Scholar]