

Abstract
Biological nitrogen fixation is an essential function of acid mine drainage (AMD) microbial communities. However, most acidophiles in AMD environments are uncultured microorganisms and little is known about the diversity of nitrogen-fixing genes and structure of nif gene cluster in AMD microbial communities. In this study, we used metagenomic sequencing to isolate nif genes in the AMD microbial community from Dexing Copper Mine, China. Meanwhile, a metagenome microarray containing 7,776 large-insertion fosmids was constructed to screen novel nif gene clusters. Metagenomic analyses revealed that 742 sequences were identified as nif genes including structural subunit genes nifH, nifD, nifK and various additional genes. The AMD community is massively dominated by the genus Acidithiobacillus. However, the phylogenetic diversity of nitrogen-fixing microorganisms is much higher than previously thought in the AMD community. Furthermore, a 32.5-kb genomic sequence harboring nif, fix and associated genes was screened by metagenome microarray. Comparative genome analysis indicated that most nif genes in this cluster are most similar to those of Herbaspirillum seropedicae, but the organization of the nif gene cluster had significant differences from H. seropedicae. Sequence analysis and reverse transcription PCR also suggested that distinct transcription units of nif genes exist in this gene cluster. nifQ gene falls into the same transcription unit with fixABCX genes, which have not been reported in other diazotrophs before. All of these results indicated that more novel diazotrophs survive in the AMD community.
Introduction
Biological nitrogen fixation occurs in more than 100 genera distributed among several of the major phylogenetic divisions of Bacteria and Archaea [1]. Bacterial nif genes are known to encode the components of the nitrogenase enzyme complex. The structural subunit of dinitrogenase reductase and the 2 subunits of dinitrogenase are encoded by the nifH, nifD, and nifK genes, respectively. In many diazotrophs like Azotobacter vinelandii [2], Herbaspirillum seropedicae [3], Pseudomonas stutzeri [4], and Bradyrhizobium japonicum [5], these proteins have similar sequences and common structures and functions. Furthermore, genetic and biochemical analyses revealed that many additional nif genes, including nifE, nifN, nifX, nifQ, nif W, nifV, nifA, nifB, nifZ, and nifS, play roles in the regulation of nif genes and maturation processes of inactive products, such as electron transport and FeMo-cofactor biosynthesis and assembly [6], [7]. However, the linkage and arrangement of nif gene cluster vary considerably in many diazotrophs. The apparent conservation of gene arrangement suggests that they serve some important function, perhaps in regulation of nitrogen fixation[8]. In addition, the fixABCX genes first identified in Rhizobium meliloti [9] and subsequently in other diazotrophs were reported to encode a membrane complex participating in electron transfer to nitrogenase [10].
Acid mine drainage (AMD) is the outflow of acidic water from metal or coal mines, which causes worldwide environmental problems. Despite the extreme acidity, heat, and high concentrations of toxic metals, a wide variety of microorganisms populate AMD environments. These organisms can sustain by the oxidation of sulfide minerals, CO2, O2, and N2 derived from air, and phosphate liberated by water-rock interaction [11]. Since the input of externally-derived fixed nitrogen is negligible, biological nitrogen fixation is an important function of AMD microbial communities. However, a few microorganisms in AMD environments are represented by isolates that have been cultivated and described [12] and only several species in three genera (Acidithiobacillus, Leptospirillum and Methylacidiphilum) have been proved to have nitrogen-fixing ability until now [13], [14]. Little is known about the diversity and structure of nitrogen-fixing genes of AMD microbial community.
Metagenomic sequencing and metagenomic libraries recently provide powerful tools to isolate novel genes or gene clusters from unexploited gene pools in uncultured microorganisms [15]. In this study, we found evidences of novel nif genes present in acid mine drainage from Dexing Copper Mine, China, using metagenomic sequencing. We also present the phylogenetic analysis of nif genes in AMD community. To understand the organization of nif gene clusters of uncultured microorganisms in AMD community, a metagenome microarray was constructed to screen nif-containing fosmids. A comparative analysis of the nitrogen-fixing gene cluster in different species revealed the presence of novel nitrogen-fixing gene cluster in AMD community. These results confirmed that there are more novel diazotrophs surviving in AMD community.
Materials and Methods
Sample permits
The studied locations are in a state owned by Jiangxi Copper Corporation. All necessary permits were obtained for the described field studies from Jiangxi Copper Corporation. Furthermore, our study did not harm the environment and did not involve endangered or protected species.
Site description and sample collection
The microbial community growing on the surface of effusion pool beside the mill tailings at Dexing Copper Mine, China, was sampled in August 2008. The mill tailings containing low grade chalcopyrite and pyrites is the largest tailing heap in China. A total of 50 L of the original water sample was obtained at 0 to 10 cm below the water surface. The original water sample was filtered through 0.22 µm pore size filter (Millipore) immediately, and the filters were stored at −80°C until DNA extraction.
Bacterial strains and plasmids
EPI300-T1R (Epicentre, Madison, WI) was used as the host strain for fosmid library cloning. Escherichia coli BL21 (Tiangen) was used as the host strain for nif gene cloning and expression. The plasmids pCC2FOS (Epicentre, Madison, WI) and pET-28a (+) (Novagen) were used as the cloning vector and expression vector, respectively.
DNA extraction, metagenomic sequencing and metagenomic library construction
Total DNA was extracted using the liquid nitrogen grinding method [16] and finally suspended in MilliQ water. The DNA sample was further determined in 1% (w/v) agarose gels, and NanoDrop measurements gave a concentration of 350 ng/µl with A260/A280 of 1.90. A total of 5 µg of total DNA was pyrosequenced using Roche 454 GS FLX system (Majorbio, China). Since the length of generated reads were long enough to annotate (90% reads >400 base pairs), assembly of the raw sequences was not performed. The unassembled metagenomic dataset was subjected to further analysis. At the same time, a metagenomic library was constructed using total DNA and CopyControl™ Fosmid Library Production Kit (Epicentre, Madison, WI) according to the manufacture's protocol. The collection of the metagenomic library contained total of 7,776 large-insert fosmid clones.
NifK gene amplification
NifK sequences were PCR amplified with self-designed universal nifK primers sxnif_K1 (5′-CCTGGATGACCGAAGACGC-3′) and sxnif_K2 (5′-GGTGCCGCCTTCATACAT-3′). Amplification was performed in 50 µl reaction mixtures containing 1 µl of DNA extracts, 1 µl each of 10 µM forward and reverse primers, 25 µl of universal Taq PCR Master Mix(Tiangen Biotech, China),22 µl of deionized water. The PCR conditions for amplification were as follows: 94°C for 4 min, then 32 cycles of 94°C for 30 s, 51°C for 30 s, and 72°C for 45 s, followed by a final extension at 72°C for 10 min. PCR products of nifK were visualized on 2% agarose gels in TAE buffer and purified directly with the QIAquick PCR purification kit (Qiagen, Germany).
Metagenome microarray construction, hybridization, sequencing and assembly
To screen nif-containing fosmids, metagenome microarray was constructed using metagenomic library mentioned above. Each clone was incubated in a shaking incubator at 37°C and 170 r.p.m in the presence of chloramphenicol and an inducer (Epicentre). After overnight incubations, cells were harvested and the fosmid DNA was extracted using QIAprep spin miniprep kit (Qiagen, Germany) according to the manufacturer's protocol. The fosmid DNAs were stored in final concentration of 40 ng·µl−1. 10 µl fosmid DNA of each clone was transferred to a 384-well microplate, and diluted 1∶1 (V/V) in 40% dimethyl sulfoxide (Sigma, USA). Then, the fosmid DNA of each clone was arrayed on microarray with two replicates using Genemachines OmniGrid Accent microarrayer (Genomic Solutions, USA). In addition, the following controls were spotted to check by hybridization, printing, and data analysis: (i) environmental DNA as positive controls, (ii) negative controls with E. coli genomic DNA, and (iii) blanks. The microarrays were post-treated as described previously[17], and stored in room temperature.
Here, we chose PCR products of nifK genes as probe for microarray hybridization, since NifK is indispensable in all diazotrophs. They were labeled with the Bioprime DNA Labeling kit (Invitrogen, Carlsbad, CA) according to the manufacture's protocol. Then, the labeled DNA was purified using a QIAquick PCR purification kit (Qiagen, Germany), concentrated into crystallization in a SpeedVac, and finally resuspended in 20 µl MilliQ water. And labeled DNA was mixed with hybridization solution. The hybridization solution contained 20 µl of labeled DNA, 65 µl of formamide (50%,v/v), 19.5 µl of 20×SSC (1×SSC containing 150 mM NaCl and 15 mM trisodium citrate), 9.1 µl of Herring Sperm DNA(10 mg/mL)(Promega, Madison, WI), 3.9 µl of 10% sodium dodecyl sulfate (SDS), 1.1 µl of DTT(0.1 M) in a total volume of 130 µl. Finally, the mixed solution was incubated at 98°C for 3 min, and then kept at 65°C. Microarray hybridization was performed at 54°C using the HS 4800Pro Hybridization station (TECAN, Switzerland). After hybridization, the microarray was visualized using a GenePix 4100A Microarray Scanner (Axon, USA). The normalized intensity of each spot was calculated as described previously[18], and positive clones in metagenomic library can be obtained based on the positive spots in metagenome microarray. Each positive clone was tested using PCR amplification with universal nifK primers mentioned above.
The verified positive fosmids were individually isolated using QIAprep spin miniprep kit (Qiagen), and pyrosequenced using Roche 454 GS FLX system (Majorbio, China). A total of 5 µg of fosmid DNA from each clone was tagged individually using a multiplex identifier adaptor containing a unique 10 base pair sequence that is recognized by the sequencing analysis software. Finally, each fosmid had ∼1.2 Mb of sequencing data for assembly, equivalent to the >30X clone coverage. And the average length of reads was 410 bp. Each of the fosmids was assembled into one single contig with the program Newbler [19].
Annotation and analysis of genome fragments
Metagenomic sequencing yielded a total of 640,892 reads and 300 Mb of raw sequence. The individual metagenomic sequences >400 bp (approximately 90% sequences) were annotated using the non-redundant (NR) database, KEGG database, COG database. The sequences annotated as nif genes were selected to further analyze (Table.1). Protein-coding genes of fosmids were predicted using GLIMMER [20] and the RAST server [21], and further curated (Table.2). Identified ORFs were compared to known proteins in the non-redundant (NR) database, KEGG database and COG database using BLASTX [22], and all hits with e-value >1e-5 were considered nonsignificant. Other unassigned ORFs were annotated using the hmmpfam program of the HMMER package [23]. The hidden Markov models for the protein domains were obtained from the Pfam database 26.0 (http://pfam.sanger.ac.uk/). For comparative analysis, BLASTN and BLASTX searches among fosmids and different bacterial genomes were performed, leading to the identification of regions of similarity. To allow for the interactive visualization of genomic fragment comparisons, we used Artemis Comparison Tool [24]. Phylogenetic trees were constructed by the use of Molecular Evolutionary Genetics Analysis 4.0 (MEGA 4.0) software[25].
Table 1. detection of nif genes in metagenomic sequencing of acid mine drainage.
| Gene | Function | sequence number |
| nifH | nitrogenase reductase | 82 |
| nifD | nitrogenase molybdenum-iron protein subunit alpha | 127 |
| nifK | nitrogenase molybdenum-iron protein subunit beta | 93 |
| nifE | nitrogenase molybdenum-cofactor biosynthesis protein | 85 |
| nifN | nitrogenase molybdenum-cofactor biosynthesis protein | 75 |
| nifX | iron-molybdenum cofactor processing protein | 26 |
| nifA | Nif-specific regulatory protein | 89 |
| nifB | FeMo cofactor biosynthesis protein | 78 |
| nifQ | molybdenum ion binding protein | 1 |
| nifS | cysteine desulfurase | 11 |
| nifT | nitrogen fixation protein | 0 |
| nifU | Fe-S cluster assembly protein | 2 |
| nifV | homocitrate synthase | 62 |
| nifW | nitrogenase stabilizing/protective protein | 2 |
| nifZ | iron-sulfur cofactor synthesis protein | 1 |
| nifJ | pyruvate-flavodoxin oxidoreductase | 6 |
| nifL | nitrogen fixation negative regulator | 1 |
| nifP | serine acetyltransferase | 1 |
| total | 742 |
Table 2. List of ORFs from fosmid DX-1A-14, gene length, and similar genes in GenBank.
| ORF | Gene length (bp) | Close relative (protein, [organism], identitya) |
| 1 | 594 | Nitrogenase MoFe protein [Herbaspirillum seropedicae SmR1] 80% |
| 2 | 1515 | MoFe cofactor biosynthesis protein NifE [Burkholderia vietnamiensis G4] 79% |
| 3 | 1362 | MoFe cofactor biosynthesis protein NifN [Herbaspirillum seropedicae SmR1] 68% |
| 4 | 405 | MoFe cofactor biosynthesis protein NifX [Herbaspirillum seropedicae SmR1] 74% |
| 5 | 462 | Hypothetical protein [Herbaspirillum seropedicae SmR1] 71% |
| 6 | 204 | Hypothetical protein [Beijerinckia indica subsp. indica ATCC 9039] 58% |
| 7 | 315 | 4Fe-4S ferredoxin [Rubrivivax benzoatilyticus JA2] 70% |
| 8 | 576 | NifQ family protein [Candidatus Accumulibacter phosphatis clade IIA] 52% |
| 9 | 294 | Ferredoxin protein [Herbaspirillum seropedicae SmR1] 74% |
| 10 | 1299 | Ferredoxin protein [Herbaspirillum seropedicae SmR1] 79% |
| 11 | 1089 | Ferredoxin protein [Herbaspirillum seropedicae SmR1] 80% |
| 12 | 849 | Ferredoxin protein [Herbaspirillum seropedicae SmR1] 80% |
| 13 | 363 | Nitrogenase stabilizing protein [Herbaspirillum seropedicae SmR1] 50% |
| 14 | 1131 | Homocitrate synthase [Herbaspirillum seropedicae SmR1] 63% |
| 15 | 1587 | Transmembrane protein [Ralstonia eutropha H16] 59% |
| 16 | 240 | Conserved hypothetical protein [Methylococcus capsulatus str. Bath] 55% |
| 17 | 1659 | Nif-specific regulatory protein [Herbaspirillum seropedicae SmR1] 61% |
| 18 | 1872 | Fe-S protein assembly chaperone HscA [Methylovorus sp. SIP3-4] 69% |
| 19 | 357 | Iron-sulfur cluster insertion protein ErpA [Nitrosomonas europaea] 69% |
| 20 | 1563 | FeMo cofactor biosynthesis protein [Herbaspirillum seropedicae SmR1] 83% |
| 21 | 753 | Conserved hypothetical protein [Herbaspirillum seropedicae SmR1] 60% |
| 22 | 240 | Putative NifZ protein [Methylococcus capsulatus str. Bath] 60% |
| 23 | 1161 | Aminotransferase class V [Beijerinckia indica subsp. indica ATCC 9039] 58% |
| 24 | 267 | Conserved hypothetical protein [Rhodopseudomonas palustris BisB18] 43% |
| 25 | 1629 | Rhodanese domain protein [Beijerinckia indica subsp. indica ATCC 9039] 55% |
| 26 | 414 | Oxygen-binding (globin) protein [Herbaspirillum seropedicae SmR1] 68% |
| 27 | 426 | Two component regulator protein [Herbaspirillum seropedicae SmR1] 60% |
| 28 | 4557 | Methyl-accepting chemotaxis transducer [Acidovorax delafieldii 2AN] 51% |
| 29 | 990 | RuBisCO operon transcriptional regulator [Beggiatoa sp. PS] 54% |
| 30 | 1422 | RubisCO formI large subunit [Acidithiomicrobium sp.] 91% |
| 31 | 333 | RubsiCO small subunit [Acidithiobacillus ferrivorans SS3] 70% |
| 32 | 2382 | Carboxysome shell polypeptide [Halothiobacillus neapolitanus c2] 46% |
| 33 | 1497 | Carboxysome shell carbonic anhydrase [Halothiobacillus neapolitanus c2] 58% |
Nucleotide identity of fosmid DX-1A-14 gene to the gene of the organism to which it is most related.
Transcriptional unit prediction, cloning, expression and reverse transcription PCR
The program FindTerm (http://linux1.softberry.com/berry.phtml) was used to analyze potential transcription terminators. Locations of significant σN, NifA recognition sites in the upstream of genes and of potential transcriptional terminators are presented in Table.3. Reverse transcription PCR was performed to confirm the co-transcription of nifQ and fixX. The contiguous fixX and nifQ gene sequence was amplified from fosmid DX-1A-14 by polymerase chain reaction (PCR), using the oligonucleotides Fix_nif_F 5′-CGCGCTAGCATGAGCGGACTGCGTGTCGAAG-3′ (NheI site is underlined) and Fix_nif_R 5′-CAGGAATTCTCAGGCCGCCCGCAGGGCTTG-3′ (EcoRI site is underlined). The amplified DNA fragment was cleaved with restriction endonuclease NheI and EcoRI, and ligated in the NheI/EcoRI-digested plasmid pET-28a (Novagen, Nadison, WI). Then, the resulting plasmid pET-28NIF containing fixX and nifQ was transformed into E. coli strain BL21. The E. coli strain BL21 with plasmid pET-28NIF was grown in Terrific Broth. The growth was achieved aerobically at 37°C in 20 ml of media in a 50-mililiter flask, and cells were induced with 0.1 mM isopropyl β-D-thiogalactoside at A600 ∼0.6. After one hour incubation, cells were harvested by centrifugation at 4°C at 10,000 rpm for 2 min in a 5415R centrifuge (Eppendorf AG, Hamburg, Germany). Total RNA was extracted using Total RNA Isolation Kit (Omega Bio-Tek, Doraville, USA) and then treated with RNase-free DNase I (QIAGEN, Carlsbad, CA) to digest residual chromosomal DNA. Total RNA was quantified at A260 and A280 with NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, USA). Reverse transcription PCR was performed using ImProm-II™ Reverse Transcription System, using the above mentioned primers. PCR products were visualized on 2% agarose gels in TAE buffer, purified directly with the QIAquick PCR purification kit (Qiagen, Germany) and sequenced bidirectionally.
Table 3. Gene products in the nif-fix cluster of fosmid DX-1A-14.
| Gene | Product size (kDa) | Function | Regulatory feature | Organisms with closest matcha | % Identityb |
| nifK | 22.5 | Nitrogenase structure; Fe-Mo protein beta | H. seropedicae SmR1 | 80 | |
| nifE | 55.3 | Fe-Mo cofactor synthesis | σN-binding sites | B. vietnamiensis G4 | 79 |
| nifN | 49.3 | Fe-Mo cofactor synthesis | H. seropedicae SmR1 | 68 | |
| nifX | 14.9 | Fe-Mo cofactor synthesis | H. seropedicae SmR1 | 74 | |
| nrf1 | 17.3 | NifX-associated protein | H. seropedicae SmR1 | 71 | |
| orf1 | 7.5 | Unknown | B. indica ATCC 9039 | 58 | |
| fdxB | 11.5 | Ferredoxin | R.benzoatilyticus JA2 | 63 | |
| nifQ | 21.4 | Fe-Mo cofactor synthesis | ρ-independent terminator | C.AccumulibacterUW-1 | 52 |
| fixX | 10.6 | Electron transfer; ferredoxin-like protein | H. seropedicae SmR1 | 74 | |
| fixC | 47.8 | Electron transfer; electron transfer flavoprotein quinone oxidoreductase | H. seropedicae SmR1 | 79 | |
| fixB | 39.3 | Electron transfer; electron transfer flavoprotein alpha subunit | H. seropedicae SmR1 | 80 | |
| fixA | 30.4 | Electron transfer; electron transfer flavoprotein beta subunit | H. seropedicae SmR1 | 80 | |
| nifW | 13.5 | Protection o f the Fe-Mo protein; Maturation and activation | H. seropedicae SmR1 | 50 | |
| nifV | 40.9 | Fe-Mo cofactor synthesis | NifA-,σN-binding sites | H. seropedicae SmR1 | 63 |
| Aer | 55.9 | Aerotaxis sensor receptor | σN-binding sites; ρ-independent terminator | R. eutropha H16 | 59 |
| orf2 | 8.6 | Unknown | M.capsulatus str. Bath | 54 | |
| nifA | 61.2 | Transcriptional activator | NifA-,σN-binding sites | H. seropedicae SmR1 | 61 |
| hscA | 66.4 | Fe-S protein assembly; molecular chaperone | Methylovorus sp. SIP3 | 69 | |
| erpA | 12.6 | Iron-sulfur cluster insertion | σN-binding sites | N. europaea | 69 |
| nifB | 56.9 | Fe-Mo cofactor synthesis | NifA-,σN-binding sites | H. seropedicae SmR1 | 82 |
| orf3 | 28.3 | Unknown | H. seropedicae SmR1 | 59 | |
| nifZ | 8.4 | Activation and Maturation | M.capsulatus str. Bath | 60 | |
| nifS | 40.2 | Cysteine desulfurase; Maturation and activation | B. indica ATCC 9039 | 57 | |
| orf4 | 9.6 | Unknown | R. palustris BisB18 | 43 | |
| sseA | 59.2 | Rhodanese; activation of apoferredoxins | B. indica ATCC 9039 | 54 | |
| nrf2 | 15.6 | Hemoglobin; oxygen-binding protein | H. seropedicae SmR1 | 68 | |
| nrf3 | 15.0 | Two component response regulator | σN-binding sites | H. seropedicae SmR1 | 59 |
| mcpA | 54.4 | Chemotaxis | A. delafieldii 2AN | 48 |
Organism in which the gene products most similar to that of the nif-fix cluster was found. Organism: Herbaspirillum seropedicae, Burkholderia vietnamiensis, Beijerinckia indica, Rubrivivax benzoatilyticus, Candidatus Accumulibacter phosphatis clade IIA, Ralstonia eutropha, Methylococcus capsulatus, Methylovorus sp., Nitrosomonas europaea, Rhodopseudomonas palustris, Acidovorax delafieldii.
Identity of the deduced anomic acid sequence of gene product to the gene product of the organism to which it is most related.
Nucleotide sequence accession numbers
The bioproject of metagenomic sequencing has been registered in NCBI database, and assigned accession number PRJNA202393. And the genomic DNA sequences of fosmid DX-1A-14 and DX-4H-17 containing nif gene cluster were submitted to GenBank and have been compiled under accession number JX308284 and JQ815896.
Results and Discussion
Detection of nif genes in acid mine drainage
To obtain as many nif genes as possible in AMD environment, we performed metagenomic sequencing of the AMD sample from Dexing Copper Mine in China. Metagenomic sequencing yielded a total of 640,892 reads and 300 Mb of raw sequence with the average length of 471 bp. One of the important objectives in this metagenomic study is to determine the microbial community structure of nitrogen-fixing microorganisms in the AMD sample. Since nif genes are rather conservative and indispensable in nitrogen-fixing microorganisms, they are often used to get reliable phylogenetic affiliation [1]. In our case, 742 sequences were identified as nif genes including nifH, nifD, nifK genes and various additional genes from the metagenomic sequencing data (Table. 1). And they were classified into different phyla based on the similarities to the known sequences. The result showed that the community is massively dominated by γ-proteobacteria (90%), followed in smaller amounts by α-, β-proteobacteria and Verrucomicrobia (Fig.1). Furthermore, almost all of nif genes classified in γ-proteobacteria have their best hits (>90% amino acid identity) with Acidithiobacillus ferrooxidans or Acidithiobacillus ferrivorans. Thus, it is reasonable that the genus Acidithiobacillus greatly contribute to nitrogen fixation in the AMD community. Except the nif genes classified in γ-proteobacteria, the other nif gene sequences are most similar to known microorganisms, ranging from 30% to 85% identity. The discovery of these novel nif genes suggested that more diazotrophs survive in the AMD community.
Figure 1. Classification of total nif genes obtained from metagenomic sequencing reads.

742 nif sequences were classified into different phyla based on the similarities to the known sequences. The community is massively dominated by γ-proteobacteria, followed in smaller amounts by α-, β-proteobacteria and Verrucomicrobia.
Here, we also used the phylogenetic tree of nifK gene to analyze the taxonomic distribution of nitrogen-fixing microorganisms. A total of 93 nifK sequences were aligned and fell into five distinct groups, a to e (Fig. 2). The amino acid sequence heterogeneity within each group was moderate, while the differences of amino acid sequences among groups were significant. Group a sequences had best similarities within Acidithiobacillus genera, and 92% of nifK sequences were classified into this group. This result fitted quite well with the analysis of community structure using nif genes. Group b, sharing 68% amino acid similarity with group a, was assigned in Gammaproteobacteria group in the nifK phylogenetic tree. These nifK genes in group b have their best similarities with Methylococcus capsulatus. Only one sequence fell into group c, but its taxonomic status can not be determined. And group d and e were assigned into Betaproteobacteria group in the nifK phylogenetic tree. They have best hits within Herbaspirillum and Burkholderia genera, separately. These results suggested that the phylogenetic diversity of nitrogen-fixing microorganisms is much higher than previously thought in the AMD community.
Figure 2. Phylogenetic tree of nifK genes.

The phylogenetic tree was constructed by the neighbor-joining method using MEGA, version 4.0 with 1000 bootstrap repetitions. The sequences obtained from metagenomic sequencing of acid mine drainage are designated DX_SY_, followed by their number in library. These sequences are shown in bold. Only some representatives of 93 nifK sequences are shown here. The scale represents the number of amino acid substitutions per site.
Gene annotation and sequence analysis
After microarray hybridization and sequencing, we found that most of nifK genes in positive clones had best hits with Acidithiobacillus ferrooxidans, which is consistent with the result of metagenomic sequencing. However, the nifK gene of fosmid DX-1A-14 showed novel sequence characteristics. The full-length fosmid sequencing confirmed that the fosmid harbored a novel nif gene cluster. Meanwhile, fosmid DX-4H-17 which harbored a large overlap sequence containing no nifK gene (approximately 25 kbp) with fosmid DX-1A-14 was chosen by nonspecific binding of nifK probe and fully-sequenced. Thus, a 32.5-kb nucleotide sequence with 33 open reading frames (ORFs) was obtained based on the assembly of fosmid DX-1A-14 and DX-4H-17. Gene annotation indicated that most ORFs showed sequence homologies with nif, fix and associated genes, and 5 ORFs in the end of sequence were identified as RubisCO associated genes. Gene length, Gene annotation, closest strain hits and percent similarity in fosmid DX-1A-14 are summarized in Table.2.
The nifH, nifD, and nifK genes are required for the functional nitrogenase in almost all diazotrophs. Unfortunately, only a truncated nifK gene was located in the end of the nucleotide sequence without nifH and nifD genes. It is highly possible that nifH, nifD and part of nifK sequence were truncated when fosmid library was constructed. Furthermore, iron-sulfur (FeS) cluster is an important component for nitrogenase and both NifS and NifU are required for the formation of iron-sulfur cluster in nitrogenase in Azotobacter vinelandii [26]. However, we could identify a nifS gene but nifU in the nucleotide sequence. It is reported that NifU might act as a scaffold for the assembly of the Fe-S cluster required for the maturation of the nitrogenase complex [27]. Two ORFs encoding Fe-S cluster assembly accessory proteins HscA and ErpA were located between nifA and nifB (Fig. 3). HscA is reported to be a molecular chaperone of Fe-S cluster assembly in the ISC (iron-sulfur cluster) system [28], and ErpA is a Fe-S cluster insertion protein, which is required for the delivery of Fe-S clusters. Besides, a gene encoding rhodanese was located in the genomic region neighboring the nifS gene. There is evidence to suggest that rhodanese can mobilize sulfur from thiosulfate for in vitro formation of Fe-S clusters [29]. Besides, these genes are always present in nif gene clusters in other diazotrophs [13], [14]. It is possible that these proteins mentioned above were involved in the assembly Fe-S cluster for nitrogenase.
Figure 3. Comparison of the physical organization of nif, fix, and associated genes from fosmid DX-1A-14 with those from three closest organisms.
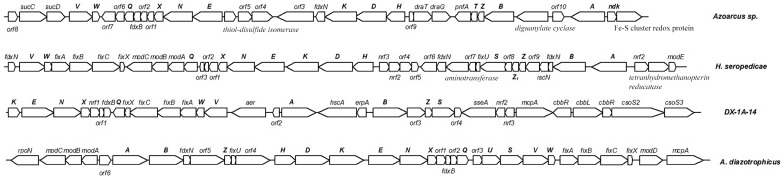
Organisms: Azoarcus sp., Herbaspirillum seropedicae, Acetobacter diazotrophicus. The nif genes are highlighted in bold. nrf, nif associated genes; orf, hypothetical protein. The structure of nif gene cluster differs greatly from those of unknown microorganisms. The nif gene cluster does not contain modABC genes occurring in H. seropedicae, and RubisCO gene cluster can not be identified in the nif gene cluster region of H. seropedicae. nifQ gene is clustered together with nifENX and fdxB genes in A. diazotrophicus, while nifV, nifW, fixABCX, and nifQ genes constitute a single operon in fosmid DX-1A-14.
Sequence comparison with known nif gene clusters in diazotrophs revealed that many genes in fosmid DX-1A-14 were generally most similar to those found in β-proteobacteria, with 15 genes products being most like those of Herbaspirillum seropedicae, ranging from 50% (for nifW) to 82% (for nifB) identity (Table. 2). NifZ was most similar to the gene product of Methylococcus species, member of γ-proteobacteria (60% identity). And NifE was most similar to gene products of Burkholderia vietnamiensis (79% identity). NifS and NifQ had low levels of sequence identity of 57% with Beijerinckia indica subsp. indica ATCC 9039 and 52% with Candidatus Accumulibacter phosphatis clade IIA str. UW-1, separately. And all fix gene products were most similar to those of H. seropedicae. It is worth pointing out that all of these species mentioned above were not reported to survive in AMD environment and that gene identities are rather low. This may result from the insufficient knowledge of nitrogen-fixing genes in AMD environment. Besides, the phylogeny of RubisCO gene cluster in fosmid DX-1A-14 is significantly inconsistent with nif gene cluster. Most gene products of RubisCO gene cluster were generally most similar to those found in γ-proteobacteria (Table.2). Thus, taxonomic status of the nucleotide sequence cannot be assign into known microorganisms.
Organization of nitrogen-fixing genes
In many genomes of nitrogen-fixing bacteria, nif genes are always organized into several clusters. In Klebsiella pneumoniae, a total of 20 nif-specific genes are organized in a single cluster spanning approximately 24 kbp genomic region [30]. In Rhizobium meliloti and Azotobacter vinelandii, the nif genes are also highly clustered, forming several small nif gene clusters in the chromosome [2], [31]. Here, comparative genome analysis with the nif gene clusters of closest diazotrophs was performed (Fig.3).
One portion of the nif-fix cluster of this nucleotide sequence (nifK to nifW) is most like that of H. seropedicae. In almost all diazotrophs, the structural genes nifHDK for the nitrogenase complex were distinctly clustered. Transcriptional analysis determined that the whole nifHDKENXorf1orf2orf3 operon is transcribed from a single promoter located upstream of the nifH gene in H. seropedicae [32]. In fosmid DX-1A-14, a high GC sequence was present, which exhibited strong homology to the nif promoter consensus sequence (GG-N10-GC) [33], in intergenic spacing between nifK and nifE gene, suggesting that the clustered genes starting from nifE may form a new operon. Another primary divergence in gene organization of this nucleotide sequence and H. seropedicae is that the nifQ and modABC genes formed a cluster in the downstream from nifHDKENXorf 1 orf 2 orf 3 operon and constitute a single operon in H. seropedicae [34], but modABC genes were not found in this nucleotide sequence (Fig. 3). The modABC genes encode a high-affinity molybdate transport system in H. seropedicae [35]. Another organization of nifQ gene that is different from that of H. seropedicae is found in A. diazotrophicus (Fig.3). nifQ gene was clustered together with nifENX and fdxB genes in the nif-fix cluster of A. diazotrophicus [7]. Unexpectedly, the nifQ gene in this nucleotide sequence had a divergent orientation with nifENX and fdxB genes, although nifQ is also located in the immediate downstream of fdxB gene (Table.2). Further sequence analysis suggested that nifV, nifW, fixABCX, and nifQ genes that had continuous arrangement in this nucleotide sequence were organized in a single gene cluster without intergenic spacing (Fig.3).
On the other hand, many additional nif genes including nifA, nifB, nifS, and nifZ interspersed in the upstream of nifHDK in A. diazotrophicus and H. seropedicae, however, more additional nif genes were located in the downstream of nifHDK in fosmid DX-1A-14 (Fig.3). And nifA gene in this nucleotide sequence is not linked to the other nif genes like that in A. diazotrophicus. The majority of the nif gene promoters is of σN-dependent type and activated by NifA protein [36]. Thus, nifA gene is always considered to be the most important regulatory gene in nitrogen fixation process. In many nitrogen-fixing bacteria, nifB forms a nif gene cluster with other genes involved in Fe-Mo cofactor synthesis such as nifQ, nifW, and fdxB genes [37], but these genes are not adjacent to the nifB gene in fosmid DX-1A-14. In fact, the nifB, orf3, nifZ, nifS, and orf 4 genes was organized in a single gene cluster without intergenic spacing in fosmid DX-1A-14. However, nifS genes were always clustered with nifU, nifV, nifW in most nitrogen-fixing bacteria [26].
Except these nif-fix genes, another important feature of the nif-fix cluster was that some other genes interspersed in the fragment region of fosmid DX-1A-14. For example, two chemotaxis genes, Aer and mcpA, were found in fosmid DX-1A-14. These chemotaxis genes may be responsible for chemotactic responses to oxygen levels or nutrition [38]. What's interesting is that a truncated RubisCO gene cluster including cbbR, cbbL, cbbR, csoS2, and csoS3 genes is located in the immediate downstream of nif-fix cluster (Table. 2). The analysis of fosmid DX-4H-17 revealed that a complete RubisCO gene cluster occurs in the organism to which the fosmid DX-1A-14 sequence belongs.
Transcriptional organizations of the nif-fix cluster
The transcriptional organizations were predicted by sequence analysis, which revealed NifA-, σN-binding sites upstream of operons required, respectively, for nif gene transcriptional activation and for nif promoter recognition in all proteobacterial diazotrophs [35]. The transcriptional organizations of the nif-fix gene cluster in fosmid DX-1A-14 showed several interesting features (Table.3). The cotranscription of nifENX nrf1 orf1 fdxB was identified by a typical σN-binding site (GG-N10-CG) in the upstream of nifE. The continuous arrangement and suitable intergenic spacing from nifHDK genes also suggested the six genes possibly compose an operon. However, there was no consensus sequences (TGT-N10-ACA) [33] for the typical NifA binding sites in the promoter region. But in the upstream of the nifV gene, two typical NifA binding site consensus sequences were detected from position -216 to -200: TGTATCAAACCATACA and -126 to -110: TTTACGAAGGAAAACA. This finding suggested that there could be an independent transcriptional unit starting from the nifV gene in fosmid DX-1A-14. The continuous arrangement and unusual overlaps among the 3′ and 5′ ends of adjacent genes indicated the cotranscription of nifV to nifQ. Reverse transcription PCR also confirmed that the nifQ gene fell into the same transcription unit with fixX gene (Fig.4). This has not been reported in other diazotrophs before. Locations of possibly significant NifA, σN recognition sites upstream of genes and potential transcription terminators downstream of genes were also presented in Table.3.
Figure 4. Reverse transcription PCR of fixX and nifQ gene fragment.
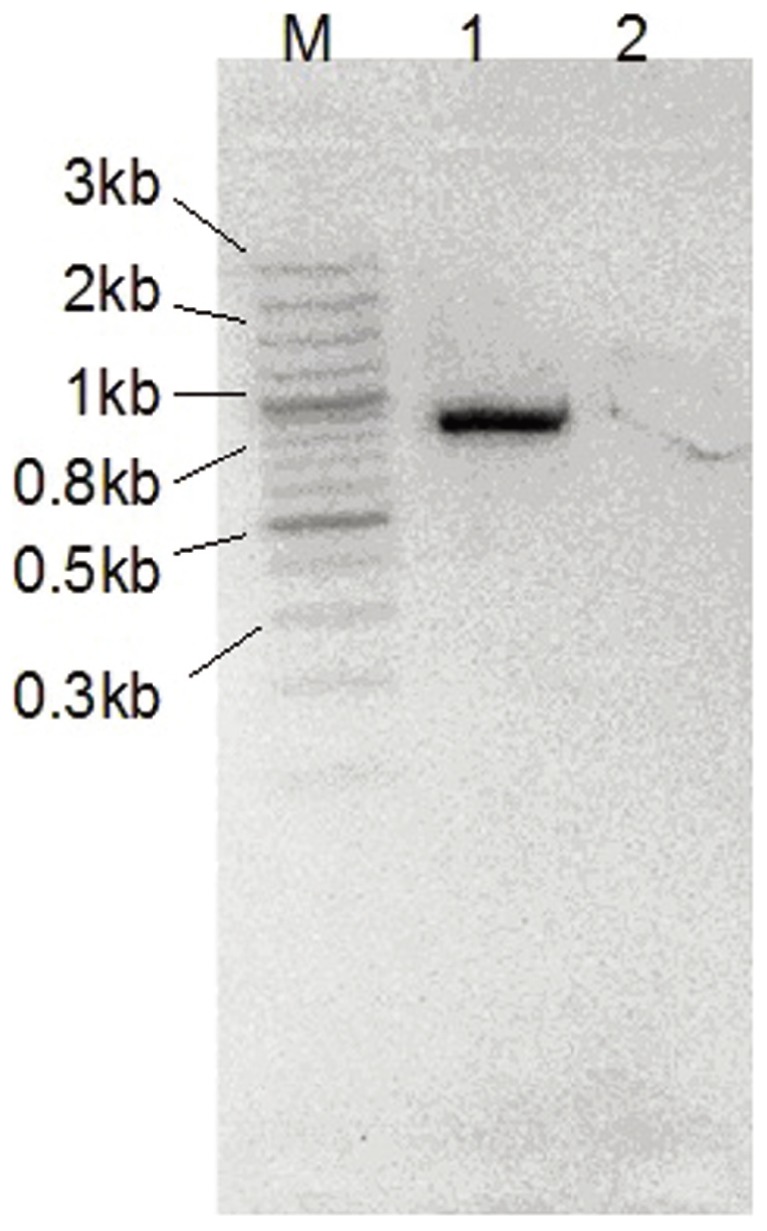
Lane M, 100; lane 1, PCR products with the template of total RNA extraction; lane 2, PCR products with the template of total RNA extracting digested by RNase.
In conclusion, we isolated 742 nif genes using metagenomic sequencing from AMD community. Metagenomic analysis suggested that the AMD community is massively dominated by the genus Acidithiobacillus, but the phylogenetic diversity of nitrogen-fixing microorganisms is much higher than previously thought in the AMD community. To understand the structure of nif gene clusters of uncultured microorganisms in AMD community, a metagenome microarray was constructed to screen novel nif gene clusters. A 32.5-kb genomic sequence harboring nif-fix gene cluster was isolated from the metagenome of AMD community. Most of nif, fix genes were individually similar to those of H. seropedicae, but the organization of the nif-fix cluster of this nucleotide sequence showed some distinct features. The NifA-, σN-binding sites in the promoter region indicated that distinct transcription units of nif genes exist in the gene cluster. These results provided a sketch about the structure of nitrogen-fixing gene cluster in the uncultured microorganisms of AMD community.
Acknowledgments
This work would like to thank Dr. Zhili He in University of Oklahoma for helpful discussion and advices.
Funding Statement
This work was supported by the National Basic Research Program (No. 2010CB630901) and the National Natural Science Foundation of China (No. 30770051). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Young J (1992) Phylogenetic classification of nitrogen-fixing organisms. Biological nitrogen fixation 23: 43–86. [Google Scholar]
- 2. Jacobson M, Brigle K, Bennett L, Setterquist R, Wilson M, et al. (1989) Physical and genetic map of the major nif gene cluster from Azotobacter vinelandii . Journal of Bacteriology 171: 1017–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pedrosa F, Teixeira KRS, Machado I, Steffens M, Klassen G, et al. (1997) Structural organization and regulation of the nif genes of Herbaspirillum seropedicae . Soil Biology and Biochemistry 29: 843–846. [Google Scholar]
- 4. Yan Y, Yang J, Dou Y, Chen M, Ping S, et al. (2008) Nitrogen fixation island and rhizosphere competence traits in the genome of root-associated Pseudomonas stutzeri A1501. Proceedings of the National Academy of Sciences 105: 7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Adams TH, McClung CR, Chelm BK (1984) Physical organization of the Bradyrhizobium japonicum nitrogenase gene region. Journal of Bacteriology 159: 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Masepohl B, Drepper T, Paschen A, Gross S, Pawlowski A, et al. (2002) Regulation of nitrogen fixation in the phototrophic purple bacterium Rhodobacter capsulatus . Journal of molecular microbiology and biotechnology 4: 243–248. [PubMed] [Google Scholar]
- 7. Lee S, Reth A, Meletzus D, Sevilla M, Kennedy C (2000) Characterization of a major cluster of nif, fix, and associated genes in a sugarcane endophyte, Acetobacter diazotrophicus . Journal of Bacteriology 182: 7088–7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kallas T, Coursin T, Rippka R (1985) Different organization of nif genes in nonheterocystous and heterocystous cyanobacteria . Plant molecular biology 5: 321–329. [DOI] [PubMed] [Google Scholar]
- 9. Earl C, Ronson C, Ausubel F (1987) Genetic and structural analysis of the Rhizobium meliloti fixA, fixB, fixC, and fixX genes. Journal of Bacteriology 169: 1127–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Edgren T, Nordlund S (2004) The fixABCX genes in Rhodospirillum rubrum encode a putative membrane complex participating in electron transfer to nitrogenase. Journal of Bacteriology 186: 2052–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baker BJ, Banfield JF (2003) Microbial communities in acid mine drainage. FEMS Microbiology Ecology 44: 139–152. [DOI] [PubMed] [Google Scholar]
- 12. Bond PL, Smriga SP, Banfield JF (2000) Phylogeny of microorganisms populating a thick, subaerial, predominantly lithotrophic biofilm at an extreme acid mine drainage site. Applied and Environmental Microbiology 66: 3842–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Levicán G, Ugalde JA, Ehrenfeld N, Maass A, Parada P (2008) Comparative genomic analysis of carbon and nitrogen assimilation mechanisms in three indigenous bioleaching bacteria: predictions and validations. BMC genomics 9: 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cárdenas JP, Valdés J, Quatrini R, Duarte F, Holmes DS (2010) Lessons from the genomes of extremely acidophilic bacteria and archaea with special emphasis on bioleaching microorganisms. Applied microbiology and biotechnology 88: 605–620. [DOI] [PubMed] [Google Scholar]
- 15. Handelsman J (2004) Metagenomics: application of genomics to uncultured microorganisms. Microbiology and Molecular Biology Reviews 68: 669–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou J, Bruns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Applied and Environmental Microbiology 62: 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo X, Yin H, Cong J, Dai Z, Liang Y, et al. (2013) RubisCO Gene Clusters Found in a Metagenome Microarray from Acid Mine Drainage. Applied and Environmental Microbiology 79: 2019–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rhee S-K, Liu X, Wu L, Chong SC, Wan X, et al. (2004) Detection of genes involved in biodegradation and biotransformation in microbial communities by using 50-mer oligonucleotide microarrays. Applied and Environmental Microbiology 70: 4303–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chaisson MJ, Pevzner PA (2008) Short read fragment assembly of bacterial genomes. Genome research 18: 324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Delcher AL, Harmon D, Kasif S, White O, Salzberg SL (1999) Improved microbial gene identification with GLIMMER. Nucleic acids research 27: 4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aziz R, Bartels D, Best A, DeJongh M, Disz T, et al. (2008) The RAST Server: rapid annotations using subsystems technology. BMC genomics 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eddy SR (2008) A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS computational biology 4: e1000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, et al. (2005) ACT: the Artemis comparison tool. Bioinformatics 21: 3422–3423. [DOI] [PubMed] [Google Scholar]
- 25. Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Molecular biology and evolution 24: 1596–1599. [DOI] [PubMed] [Google Scholar]
- 26. Jacobson MR, Cash VL, Weiss MC, Laird NF, Newton WE, et al. (1989) Biochemical and genetic analysis of the nifUSVWZM cluster from Azotobacter vinelandii . Molecular and General Genetics MGG 219: 49–57. [DOI] [PubMed] [Google Scholar]
- 27. Agar J, Yuvaniyama P, Jack R, Cash V, Smith A, et al. (2000) Modular organization and identification of a mononuclear iron-binding site within the NifU protein. Journal of Biological Inorganic Chemistry 5: 167–177. [DOI] [PubMed] [Google Scholar]
- 28. Johnson DC, Dean DR, Smith AD, Johnson MK (2005) Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem 74: 247–281. [DOI] [PubMed] [Google Scholar]
- 29.Jutabha P (2001) Biochemical and genetic characterization of mercaptopyruvate sulfurtransferase and paralogous putative sulfurtransferases of Escherichia coli: Virginia Polytechnic Institute and State University.
- 30. Arnold W, Rump A, Klipp W, Priefer UB, Pühler A (1988) Nucleotide sequence of a 24,206-base-pair DNA fragment carrying the entire nitrogen fixation gene cluster of Klebsiella pneumoniae . Journal of molecular biology 203: 715–738. [DOI] [PubMed] [Google Scholar]
- 31. Corbin D, Ditta G, Helinski D (1982) Clustering of nitrogen fixation (nif) genes in Rhizobium meliloti . Journal of Bacteriology 149: 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Machado I, Yates M, Machado H, Souza E, Pedrosa F (1996) Cloning and sequencing of the nitrogenase structural genes nifHDK of Herbaspirillum seropedicae . Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas/Sociedade Brasileira de Biofisica[et al] 29: 1599. [PubMed] [Google Scholar]
- 33. Pawlowski K, Klosse U, De Bruijn F (1991) Characterization of a novel Azorhizobium caulinodans ORS571 two-component regulatory system, NtrY/NtrX, involved in nitrogen fixation and metabolism. Molecular and General Genetics MGG 231: 124–138. [DOI] [PubMed] [Google Scholar]
- 34. Pedrosa FO, Monteiro RA, Wassem R, Cruz LM, Ayub RA, et al. (2011) Genome of Herbaspirillum seropedicae strain SmR1, a specialized diazotrophic endophyte of tropical grasses. PLoS genetics 7: e1002064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pedrosa F, Benelli E, Yates M, Wassem R, Monteiro R, et al. (2001) Recent developments in the structural organization and regulation of nitrogen fixation genes in Herbaspirillum seropedicae . Journal of biotechnology 91: 189–195. [DOI] [PubMed] [Google Scholar]
- 36. Fischer HM (1994) Genetic regulation of nitrogen fixation in rhizobia . Microbiological reviews 58: 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liang Y, Kaminski P, Elmerich C (1991) Identification of a nifA-like regulatory gene of Azospirillum brasilense Sp7 expressed under conditions of nitrogen fixation and in the presence of air and ammonia. Molecular Microbiology 5: 2735–2744. [DOI] [PubMed] [Google Scholar]
- 38. Ferrández A, Hawkins AC, Summerfield DT, Harwood CS (2002) Cluster II che genes from Pseudomonas aeruginosa are required for an optimal chemotactic response. Journal of Bacteriology 184: 4374–4383. [DOI] [PMC free article] [PubMed] [Google Scholar]