

Abstract
Streptococcus agalactiae, long recognized as a mammalian pathogen, is an emerging concern with regard to fish. In this study, we used a mouse model and in vitro cell infection to evaluate the pathogenetic characteristics of S. agalactiae GD201008-001, isolated from tilapia in China. This bacterium was found to be highly virulent and capable of inducing brain damage by migrating into the brain by crossing the blood–brain barrier (BBB). The phagocytosis assays indicated that this bacterium could be internalized by murine macrophages and survive intracellularly for more than 24 h, inducing injury to macrophages. Further, selective capture of transcribed sequences (SCOTS) was used to investigate microbial gene expression associated with intracellular survival. This positive cDNA selection technique identified 60 distinct genes that could be characterized into 6 functional categories. More than 50% of the differentially expressed genes were involved in metabolic adaptation. Some genes have previously been described as associated with virulence in other bacteria, and four showed no significant similarities to any other previously described genes. This study constitutes the first step in further gene expression analyses that will lead to a better understanding of the molecular mechanisms used by S. agalactiae to survive in macrophages and to cross the BBB.
Introduction
Streptococcus agalactiae, commonly known as group B streptococcus (GBS), has a broad host range and is pathogenic to mammals, reptiles, amphibians, and fish [1]. It has been reported to cause neonatal pneumonia and meningitis in humans, mastitis in cows and meningoencephalitis in fish [2], [3], [4]. Recently, numerous outbreaks of S. agalactiae infections have been described in multiple fish farms, especially tilapia farms [5], [6], [7]. Since 2009, an outbreak of severe infectious GBS disease has occurred in tilapia culture farms in the south of China, causing large economic losses due to high mortality in the infected fish [8].
The pathogenesis of S. agalactiae infection in tilapias is not yet fully described or understood. It is known that once GBS injures or penetrates cellular barriers to reach the bloodstream or deeper tissues, an immunologic response is triggered to clear the organism. Central to this response are host phagocytic cells, including neutrophils and macrophages [9]. The ability of GBS to remain in the host as a commensal organism and to establish infection in susceptible individuals suggests that the organism may be able to subvert the host immune system [10]. It has been reported that when GBS is engulfed by professional phagocytic cells, such as macrophages and neutrophils, the organism can remain viable for a long period of time [11], [12], [13], although the mechanism of survival is unknown. The intracellular localization of GBS in macrophages may protect the organism from more active antimicrobial molecules in the blood and thus may be important in establishing bacteremia and subsequent meningitis. In line with this, in the closely related organism and extracellular pathogen Streptococcus pyogenes, it has been suggested that an important mechanism to establish infection is the survival of the organism in phagocytic cells [14]. Macrophages have been suggested to serve as vectors in the blood-borne spread of Listeria monocytogenes to cerebral endothelial cells, followed by further spread into the brain parenchyma [15], [16], [17], [18]. An early “Trojan Horse” theory suggested that Streptococcus suis is taken up by macrophages, allowing the bacteria to survive and travel intracellularly in the circulation [19].
One way to understand host-pathogen interactions is to dissect their molecular mechanisms by studying gene expression [20]. Selective capture of transcribed sequences (SCOTS) is a PCR-based RNA analytical method that has been used with success in many bacteria [20], [21], [22], [23]. In this study, we investigated the virulence of S. agalactiae isolated from fish using animal and cell culture models and used the SCOTS approach to identify genes preferentially expressed by S. agalactiae upon interactions with murine macrophages RAW264.7, a process that might be essential for the establishment of infection by this pathogen. To our best knowledge, this is the first report to broadly define transcripts expressed by S. agalactiae in macrophages.
Results
Experimental infection of mice
To determine the virulence of S. agalactiae GD201008-001, we performed the bacterial infection in BALB/c mice. Surprisingly, all mice injected intraperitoneally with 101–104 CFU in 100 µl of PBS died within 36 h post infection, while no mice died in the PBS control group (Table 1). It suggests that the bacterial strain GD201008-001 is highly virulent to BALB/c mice by intraperitoneal administration, with LD50 values of less than 10 CFU.
Table 1. Determination of LD50 in BALB/c mice challenged with S. agalactiae GD201008-001.
| Dose of challenge CFU/(0.1 ml) | Number of death/total | ||
| 12 h.p.i. | 24 h.p.i. | 36 h.p.i. | |
| 1×104 | 0/10 | 10/10 | 10/10 |
| 1×103 | 0/10 | 8/10 | 10/10 |
| 1×102 | 0/10 | 6/10 | 10/10 |
| 1×101 | 0/10 | 5/10 | 10/10 |
| PBS | 0/10 | 0/10 | 0/10 |
| LD50 | <1×101CFU | ||
Twenty-five mice were inoculated intraperitoneally with 2×102 CFU of the bacterial strain in 100 µl of PBS. Bacterial isolation was carried out to determine bacterial loads in the main tissues from infected mice (Fig. 1).
Figure 1. Recovery of bacteria from liver, spleen, brain and blood of mice infected with GD201008-001.
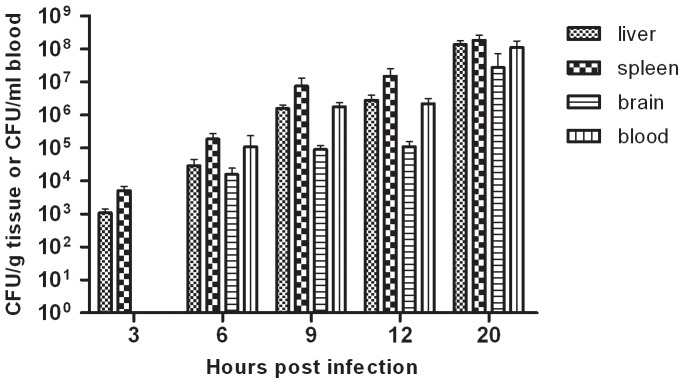
Recovered bacteria are expressed as CFU per gram of liver, spleen or brain and CFU per milliliter of blood.
At 3 hours post-infection (h.p.i.), bacterial loads exceeded 103 CFU/g in liver and spleen whilst this bacterium could not be isolated from brains or blood at this time. Three hours after, at 6 h.p.i, bacterial loads exceeded 104 CFU/ml in blood and 104 CFU/g in liver, spleen and brain. Bacterial number continued to increase with time over the course of the infection. At 20 h.p.i., bacterial loads were more than 108 CFU/ml in blood, 108 CFU/g in liver and spleen, and more than 107 CFU/g in the brain.
Histopathology and immunohistochemistry for brain tissues
To determine the pathological damage to infected mouse brains, we chose brains from infected mice at 20 h.p.i. to perform histopathological and immunohistochemical analyses.
Histopathologically, all the brains from infected mice showed significant lesions. Meningeal hemorrhage and erythrocyte aggregation were prominent (Fig. 2A). Microglial cell numbers increased and cell volumes enlarged significantly. Also, glial nodules were found (Fig. 2B). The hippocampus was lytic and necrotic (Fig. 2C). And signs of damage were apparent in the nerve fibers (Fig. 2D). No alterations were found in the brains from the control group (Fig. 2E and Fig. 2F).
Figure 2. Histopathological changes shown by H&E staining in collected brains from mice infected with GD201008-001 at 20 h.p.i.

me = meninges;mi = microglia;gn = glial nodules;hc = Hippocampus;nf = nerve fibers. (A) Prominent meningeal hemorrhage and erythrocyte aggregation in the meninges (400×). (B) Microglial cells showing an increase in the number and volume. Glial nodules were distributed (400×). (C) Lytic and necrotic hippocampus (400×). (D) Nerve fibers showing severe damage (400×). (E) and (F) No histopathological changes in sham infection control (injected with PBS) (400×). Scale bar = 20 µm.
Using immunohistochemistry, bacterial antigens were demonstrated in the brains of all infected mice. Antigen could be detected in the microglia. The closer to the blood vessel the microglias were, the more antigens were found inside the cells (Fig. 3A). Antigen staining was also found in the glial nodules (Fig. 3B). A large amount of bacterial antigens were detected in the hippocampus (Fig. 3C) and nerve fibers (Fig. 3D). No evidences of positive staining were found in the negative control group (injected with GBS, stained with unimmunized rabbit serum) (Fig. 3E) and sham infection control group (injected with PBS, stained with immunized rabbit serum) (Fig. 3F).
Figure 3. Immunohistochemical detection of bacterial antigen in collected brains from mice infected with GD201008-001at 20 h.p.i..

bv = blood vessel;mi = microglia;gn = glial nodules;hc = Hippocampus;nf = nerve fibers (A) Positive stain in the microglia (400×). (B) Bacterial antigens were detected in the glial nodules (400×). (C) Intense antigen staining in the hippocampus. (D) A large amount of bacterial antigens were detected in the nerve fibers (400×). (E) Negative control (injected with GBS, stained with unimmunized rabbit serum) showed clean background (400×). (F) Sham mice (injected with PBS, stained with immunized rabbit serum) showed no antigen staining (400×). Scale bar = 20 µm.
S. agalactiae interactions with murine macrophages
Temporal observation of the behavior of S. agalactiae GD201008-001 after phagocytosis by RAW264.7 was made using GFP as an intrinsic label to track the bacterial cells following ingestion by macrophages. These observations revealed that the GFP-expressing S. agalactiae could be visualized within the macrophages (Fig. 4).
Figure 4. Fluorescence microscopy-based phagocytosis of GFP- expressing S. agalactiae.
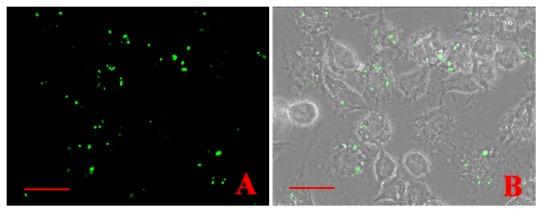
GFP expressing bacteria were internalized by RAW264.7(1000×). Scale bar = 20 µm.
To determine the ability of macrophages to internalize S. agalactiae, RAW264.7 macrophages were infected at MOI = 1, 10 or 100 with S. agalactiae for 1 h. After antibiotic treatment to kill the extracellular bacteria, intracellular bacteria were plated onto Todd-Hewitt agar to be cultured overnight. As shown in Fig. 5, we were able to show that entry of S. agalactiae into RAW264.7 macrophages occurs in a dose-dependent manner. The MOI = 10 and MOI = 100 groups were significantly (p<0.05) more internalized by macrophages than bacteria at MOI = 1.
Figure 5. Phagocytosis of GD201008-001 by RAW264.7 macrophages.
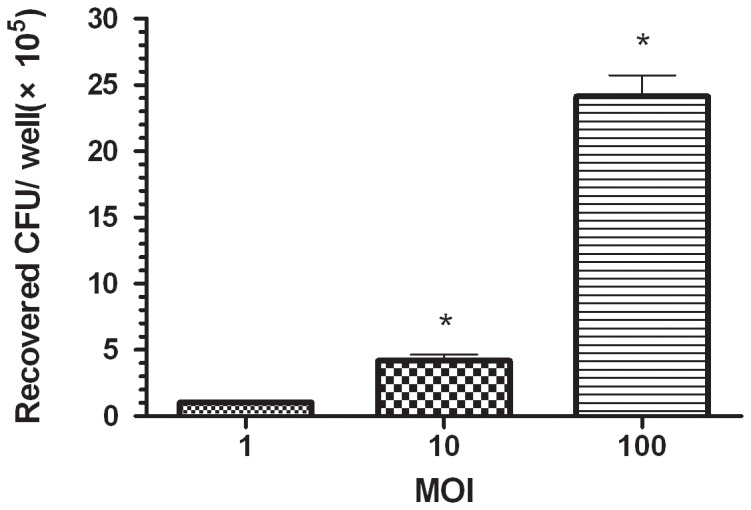
At 1(means ± S.D. obtained from three independent experiments, n = 4). An asterisk indicates a significant difference versus the phagocytosis values from the group treated at an MOI of 1 (P<0.05).
To analyze the intracellular fate of the bacteria once internalized, we modified the phagocytosis assay in order to quantify intracellular bacterial survival over time. After exactly 60 min of incubation of S. agalactiae with macrophages to allow internalization, antibiotics were added and the treatment was lengthened for different times up to 24 h. As shown in Fig. 6, after 60 min of phagocytosis, the numbers of viable intracellular bacteria showed an obvious decrease within phagocytic cells over the first 3 h.p.i., then increased slightly up to 7 h.p.i, but then descended again. Finally the numbers of viable intracellular bacteria rate decreased to about 34% of their maximum at 24 h.p.i.
Figure 6. Intracellular survival of S. agalactiae within RAW264.7 macrophages.
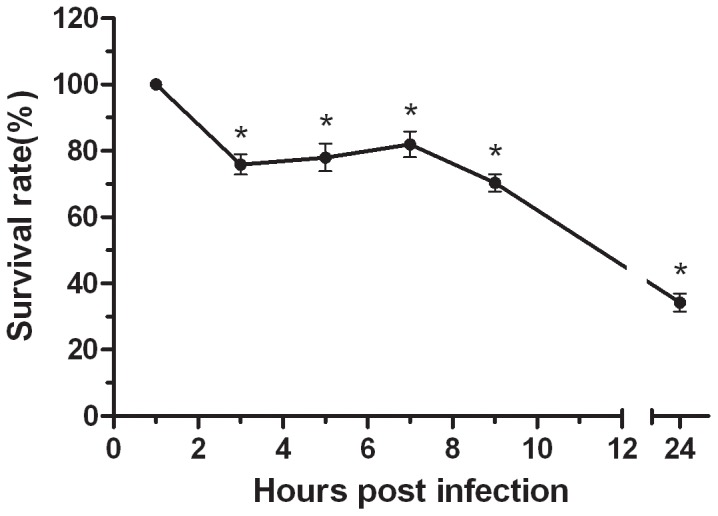
RAW264.7 cells were infected with S. agalactiae (MOI = 1) and phagocytosis was left to proceed for 1 h. Antibiotics were then added for a period of 1 h. This initial antibiotic-treatment was extended for different times up to 24 h and cells were lysed to quantify the intracellular bacteria by viable plate counting. The results are expressed as the intracellular survival rates of the bacteria (means± SD obtained from three independent experiments, n = 4). An asterisk indicates a significant difference versus the intracellular bacteria survival rate of the 1 h.p.i group (P<0.05).
In order to determine if S. agalactiae could be cytotoxic to RAW264.7, lactose dehydrogenase (LDH) release was measured. As shown in Fig. 7, GD201008-001 was found to be cytotoxic to macrophages in a concentration-dependent manner. At an MOI of 1, the cells showed almost no toxicity, even at 4 h.p.i. However, in contrast to the MOI = 1 group, the cytotoxicity levels at MOIs of 10 and 100 were significantly higher (P<0.05). In particular, the level of cytotoxicity was up to 82% at an MOI of 100.
Figure 7. Cytotoxic effect of S. agalactiae on RAW264.7 macrophages.
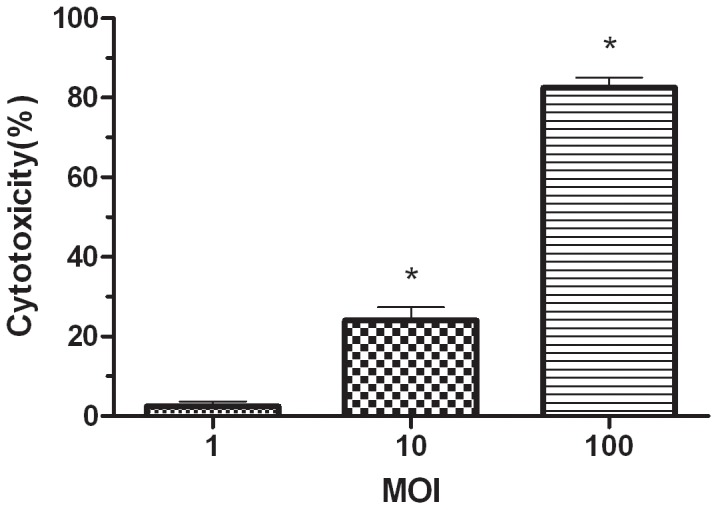
The percentage of macrophage cytotoxicity was determined by measuring LDH release in the presence of different concentrations (MOI of 1, 10 or 100) of S. agalactiae GD201008-001 after a 4 h incubation at 37°C. An asterisk indicates that the bacterial cytotoxicity is significantly different (p<0.05) from the cytotoxicity value at an MOI of 1. (n = 4).
Selective capture of S. agalactiae transcripts from infected macrophages
To identify S. agalactiae genes preferentially expressed under interaction with macrophages using the SCOTS approach, a total of sixty clones that hybridized to a cDNA probe from S. agalactiae surviving in macrophages but did not hybridize to a cDNA probe prepared from bacteria grown in culture medium, were chosen for further characterization analysis (Fig. 8). Among the 60 clones, four showed no homology to genes with known function. The remaining 56 genes could be characterized into 6 functional categories (Table 2): (1) Nine genes were involved in cell envelope biogenesis/outer membrane synthesis, responsible for the construction of peptidoglycans, cell surface proteins, or components of bacterial secretory systems. Among these, dltA and clone 20 (D-alanyl-D-alanine carboxypeptidase) were associated with cell wall biogenesis and four genes belonged to cell membrane proteins; (2) Thirty-two genes were involved in metabolism/stress response, including enzymes involved in the biosynthesis and metabolism of sugar, fatty acids, proteins, and nucleic acids. The up-regulated genes were an adaptation to oxidative stress inside the macrophages and an environment lacking nutritional resources; (3) Four transporters genes were identified, including the components of ABC-type transport systems and chloride channels; (4) Five genes were involved in cell division/replication, including genes associated with the replication and genetic regulation of the bacteria. DivIB and rpoC were involved in cell replication. UvrA was involved in cell damage repair; (5) Three genes were involved in protein sorting, including molecular chaperones and signal peptidase; (6) Four genes possessed regulatory functions, including transcriptional regulation. Examples included hrcA, encoding a heat-inducible transcription repressor, and covR, encoding the response regulator of the two-component CovR/S regulatory system.
Figure 8. Southern dot blot analysis of selected SCOTS clones.

Southern dot blot showing (I) SCOTS clones (each dot corresponds to one clone in Table 2) and (II) 0.1 µg (top) or 1 µg (bottom) of genomic S. agalactiae DNA as positive controls, hybridized to a digoxigenin (DIG)-labeled probe generated from RAW264.7-derived (a) or culture-derived cDNA (b) after three rounds of normalization.
Table 2. Genes identified by SCOTS that are differentially expressed by S. agalactiae upon interaction with RAW264.7.
| Function and clone | Gene and/or possible function | GenBank Identification | Reference(s) |
| Cell envelope biogenesis, outer membrane | |||
| Clone A1 | cfa, | YP_330368.1 | |
| cyclopropane-fatty-acyl-phospholipid synthase (S.agalactiae A909) | |||
| Clone A2 | dltA, | ZP_08649940.1 | [29], [30], [31], [32], [33], [34] |
| D-alanine-D-alanyl carrier protein ligase subunit 1 (S.agalactiae ATCC 13813) | |||
| Clone A3 | D-alanyl-D-alanine carboxypeptidase (S. agalactiae 2603V/R) | NP_687183.1 | |
| Clone A4 | cell wall surface anchor family protein (S.agalactiae A909) | YP_329518.1 | |
| Clone A5 | secA, | ZP_08650039.1 | [35], [36] |
| preprotein translocase subunit SecA (S.agalactiae ATCC 13813) | |||
| Clone A6 | polysaccharide biosynthesis protein (S.agalactiae A909) | YP_330055.1 | |
| Clone A7 | cstA, | NP_735499.1 | |
| hypothetical protein gbs1050 (S. agalactiae NEM316) | |||
| Clone A8 | hypothetical protein FSLSAGS3026_12085 (S. agalactiae FSL S3-026) | EGS27102.1 | |
| Clone A9 | Can B domain-containing protein (S.agalactiae FSL S3-026) | EGS27960.1 | |
| Metabolism/stress response | |||
| Clone A10 | hypothetical protein SAG0894 (S. agalactiae 2603V/R) | NP_687908.1 | |
| Clone B1 | hylB, | YP_329897.1 | [9] |
| hyaluronate lyase (S. agalactiae A909) | |||
| Clone B2 | clpE, | NP_734999.1 | [40], [41] |
| hypothetical protein gbs0535 (S. agalactiae NEM316) | |||
| Clone B3 | peptidase, M20/M25/M40 family (S. agalactiae H36B) | ZP_00784166.1 | |
| Clone B4 | metallopeptidase, zinc binding (S. agalactiae COH1) | ZP_00784711.1 | |
| Clone B5 | endopeptidase O, putative (S. agalactiae CJB111) | ZP_00788097.1 | |
| Clone B6 | hypothetical protein SAG0872 (S. agalactiae 2603V/R) | NP_687886.1 | |
| Clone B7 | pulA, | ZP_00786133.1 | [46], [47], [48], [49], [50] |
| pullulanase, putative (S. agalactiae COH1) | |||
| Clone B8 | mannosyl-glycoprotein endo-beta-N-acetylglucosamidase family protein (S. agalactiae FSL S3-026) | EGS28355.1 | |
| Clone B9 | ciaH, | NP_735468.1 | [37], [38], [39] |
| sensor histidine kinase CiaH (S. agalactiae NEM316) | |||
| Clone B10 | serB, | NP_687649.1 | |
| phosphoserine phosphatase SerB (S. agalactiae 2603V/R) | |||
| Clone C1 | acetyltransferase (S. agalactiae 2603V/R) | NP_688337.1 | |
| Clone C2 | pyridine nucleotide-disulfide oxidoreductase family protein (S. agalactiae 2603V/R) | NP_688351.1 | |
| Clone C3 | htpX, | NP_688618.1 | |
| heat shock protein HtpX (S. agalactiae 2603V/R) | |||
| Clone C4 | CsbD family protein (S. agalactiae A909) | YP_329332.1 | |
| Clone C5 | fhs, | YP_329765.1 | |
| formate—tetrahydrofolate ligase (S. agalactiae A909) | |||
| Clone C6 | folP, | YP_329821.1 | |
| dihydropteroate synthase (S. agalactiae A909) | |||
| Clone C7 | aroA, | ZP_08650982.1 | |
| 3-phosphoshikimate-1-carboxyvinyltransferase (S. agalactiae ATCC 13813) | |||
| Clone C8 | nox, | ZP_00783526.1 | [42] |
| NADH oxidase (S. agalactiae H36B) | |||
| Clone C9 | morphine 6-dehydrogenase (S. agalactiae ATCC 13813) | ZP_08650234.1 | |
| Clone C10 | carB, | ZP_08650601.1 | [43] |
| carbamoyl-phosphate synthase, large subunit (S. agalactiae ATCC 13813) | |||
| Clone D1 | msrB, | ZP_00785672.1 | [44], [45] |
| methionine sulfoxide reductases B (S. agalactiae COH1) | |||
| Clone D2 | nrdI, | YP_330497.1 | |
| flavoprotein NrdI (S. agalactiae A909) | |||
| Clone D3 | pyrH, | NP_688507.1 | |
| uridylate kinase (S. agalactiae 2603V/R) | |||
| Clone D4 | sdhA, | YP_330694.1 | |
| L-serine dehydratase, iron-sulfur-dependent subunit alpha (S. agalactiae A909) | |||
| Clone D5 | aroD, 3-dehydroquinate dehydratase (S. agalactiae 2603V/R) | NP_688377.1 | |
| Clone D6 | hypothetical protein SAK_0322 (S. agalactiae A909) | YP_328974.1 | |
| Clone D7 | fabK, | NP_687380.1 | |
| enoyl-ACP reductase (S. agalactiae 2603V/R) | |||
| Clone D8 | cylI, | EGS28236.1 | |
| cylI protein (S. agalactiae FSL S3-026) | |||
| Clone D9 | gap, | NP_688758.1 | [51], [52] |
| glyceraldehyde-3-phosphate dehydrogenase (S. agalactiae 2603V/R) | |||
| Clone D10 | glycosyl hydrolase (S. agalactiae A909) | YP_329453.1 | |
| Clone E1 | ccpA, | YP_329455.1 | [53] |
| catabolite control protein A (S. agalactiae A909) | |||
| Transport | |||
| Clone E2 | voltage-gated chloride channel family protein (S. agalactiae 2603V/R) | NP_687570.1 | |
| Clone E3 | ABC transporter ATP-binding protein (S. agalactiae 2603V/R) | NP_689021.1 | |
| Clone E4 | oligopeptide ABC transporter oligopeptide-binding protein (S. agalactiae 2603V/R) | NP_687222.1 | |
| Clone E5 | thiW protein (S. agalactiae ATCC 13813) | ZP_08650771.1 | |
| Cell division/replication | |||
| Clone E6 | divIB, | NP_687507.1 | |
| cell division protein DivIB (S. agalactiae 2603V/R) | |||
| Clone E7 | uvrA, | ZP_00783998.1 | |
| excinuclease ABC, A subunit (S. agalactiae H36B) | |||
| Clone E8 | MutS2 family protein (S. agalactiae FSL S3-026) | EGS26693.1 | |
| Clone E9 | IS861, transposase OrfB (S. agalactiae 2603V/R) | NP_688077.1 | |
| Clone E10 | rpoC, | EGS28475.1 | |
| DNA-directed RNA polymerase subunit beta (S. agalactiae FSL S3-026) | |||
| Protein sorting | |||
| Clone F1 | dnaJ, | ZP_00783391.1 | |
| dnaJ protein (S. agalactiae H36B) | |||
| Clone F2 | lepB, | ZP_00788470.1 | |
| Signal peptidase I (S. agalactiae CJB111) | |||
| Regulatory | |||
| Clone F3 | hrcA, | YP_328819.1 | |
| heat-inducible transcription repressor (S. agalactiae A909) | |||
| Clone F4 | MerR family transcriptional regulator (S. agalactiae A909) | YP_330526.1 | [54] |
| Clone F5 | LysR family transcriptional regulator (S. agalactiae NEM316) | NP_735874.1 | [55] |
| Clone F6 | covR, | YP_004478477.1 | [10] |
| response regulator protein (S. parauberis KCTC 11537) | |||
| Unknown function | |||
| Clone F7 | unknown (S. agalactiae H36B) | ZP_00783887.1 | |
| Clone F8 | hypothetical protein SAG1491 (S. agalactiae 2603V/R) | NP_688485.1 | |
| Clone F9 | yobC, | AAL96904.1 | |
| hypothetical protein spyM18_0084 (S.pyogenes MGAS8232) | |||
| Clone F10 | hypothetical protein FSLSAGS3026_02113 (S.agalactiae FSL S3-026) | EGS28676.1 |
Validation of SCOTS results by q-PCR
The SCOTS approach, as used in this study, should result in the identification of genes that are upregulated by S. agalactiae upon interaction with macrophages [24]. Therefore, to validate our SCOTS results, we used quantitative PCR (q-PCR) to measure the expression levels of the genes from Table 2. In order to ensure that one gene from each functional category (the genes of unknown functions were not included) was selected for Real-time PCR analysis, we randomly selected one from the functional category (≤5 genes), three (>5 and ≤10 genes), and five (>10 genes). Therefore, a total of 12 genes were tested. The q-PCR analysis for these selected genes showed that they were indeed upregulated by S. agalactiae upon interaction with RAW264.7 (Fig. 9), with changes ranging from 1.7 to 11.69 fold.
Figure 9. qRT-PCR determination of the genes up-regulated during intracellular survival of S. agalactiae.
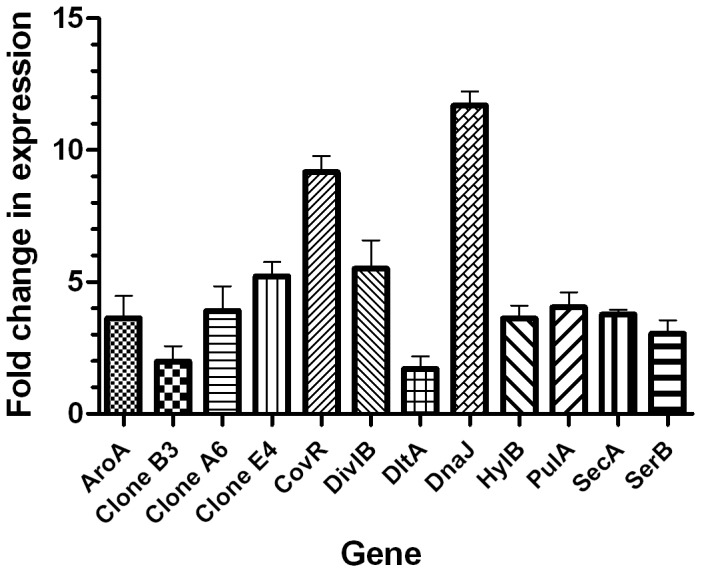
Real-time RT-PCR analysis of differentially expressed genes of S. agalactiae vis a vis survival in RAW264.7 and in THB cultures. Twelve genes, aroA, clone B3 (peptidase), clone A6 (polysaccharide biosynthesis protein), clone E4 (oligopeptide ABC transporter oligopeptide -binding protein), covR, divIB, dltA, dnaJ, hylB, pulA, secA and serB were selected and amplified using real-time RT-PCR. Real time RT-PCR data for each gene is relative to that obtained for the 16S rRNA control. Data points represent the means ± SD of three independent experiments.
Discussion
S. agalactiae is a recognized pathogen in global aquaculture and disease outbreaks have resulted in significant economic losses in fish. In the present study, an experimental infection of S. agalactiae GD201008-001, a strain isolated from moribund cultural tilapia in China, was performed in mice. We found that this bacterial strain was highly virulent (LD50 <101 CFU). The infected mice developed clinical signs of central nervous system (CNS) infection characteristic of brain damage. And the pathological findings in the brains correlated with the clinical behavioural abnormalities, such as lethargy and loss of orientation. These findings supported that S. agalactiae was neurotropic as described earlier by Eldar et al. [7] and Abuseliana et al. [25].
Our understanding of the pathogenesis of GBS infection remains limited, particularly concerning the mechanisms by which this bacterial species travels and disseminates in the bloodstream to reach the central nervous system. Efforts should be made to characterize the underlying pathogenic mechanisms that are essential for crossing the BBB. Histological investigations in the giant Queensland grouper, Epinephelus lanceolatus (Bloch), and other wild fish have shown that macrophages may act as a vehicle for S. agalactiae, allowing the bacterium to cross the BBB and enter the CNS and thereby be more easily disseminated to other organs and tissues, manifesting as a bacterial septicemia [26]. For this reason, we used RAW264.7 murine macrophages as a model to study the interactions between S. agalactiae and macrophages. Our study showed that S. agalactiae could be phagocytosed in large numbers by murine macrophages in the absence of complement and antibodies in a dose dependent manner. GBS not only entered macrophages very efficiently but also survived intracellularly for more than 24 h, a period that in vivo would be more than sufficient to maintain a bacteremia required for developing meningitis [13]. Our results indicated that the numbers of viable intracellular GBS in RAW264.7 decreased at first, then increased slightly up to 7 h.p.i., and then descended again afterwards. These dynamic changes probably reveal the process of interaction between GBS and macrophages. However, in general, the survival of the bacteria showed a downward trend. Mancuso et al. [27] showed that encapsulated GBS survived longer in mouse bone marrow-derived dendritic cells compared to mouse macrophages. The present study also showed that macrophages were injured by S. agalactiae. It is possible S. agalactiae may induce apoptosis or necrosis in macrophages, enabling the bacterium to overcome and then inhibit the host's immune defense system. Studies on murine macrophages have shown that human S. agalactiae induces macrophage apoptosis, affording a novel way to overcome the host immune system [28]. Liu et al. [12] have reported that β-hemolysin/cytolysin of GBS could produce direct cytolytic injury to macrophages and induce macrophage apoptosis over a longer interval. This led to us to speculate that hemolysin may play an important role in the mechanism of killing phagocytes in S. agalactiae GD201008-001. Nevertheless, this hypothesis remains to be defined in future studies.
Using the SCOTS differential cDNA cloning approach, we identified 60 differentially expressed S. agalactiae transcripts and q-PCR analysis showed that the genes were indeed upregulated upon interaction with RAW264.7 murine macrophages. Some of the genes identified by SCOTS might be considered, on the basis of their functions in other organisms, potential candidates as S. agalactiae virulence factors. For instance, the dltA gene (dltA belongs to an operon comprising four genes, dltA, dltB, dltC, and dltD), which is present in all genomes of low G+C bacteria determined so far [29]. In all species where this operon has been studied, all four of the genes are required to catalyze the incorporation of D-alanine residues into lipoteichoic acids (LTAs). D-alanylation of LTAs allows gram-positive bacteria to modulate their surface charge, to regulate ligand binding, and to control the electromechanical properties of the cell wall [29]. In addition, formation of D-alanyl-LTAs is required to resist the action of antimicrobial peptides in L. monocytogenes [30], S. pneumoniae [31], Staphylococcus aureus [32] and S. pyogenes (group A Streptococcus) [33]. The S. agalactiae D-alanyl-LTAs has been shown to have a role in resistance to defensins and phagocytic cells [34]. Therefore, S. agalactiae might modulate the degree of D-alanylation of its LTAs by upregulating dltA upon interaction with murine macrophages.
Limia A et al. [35] have shown that survival within phagocytic cells is associated with significant abnormalities in the maturation of phagosomes and trafficking in the host cell. Their data suggest the intracellular life of M. avium results in translocation of proteins by the Sec pathway [35]. Studies by Owens et al. [36] have shown that secA is critical to the protein export process in other bacteria such as M. tuberculosis. In this regard, it may be of interest to further evaluate the hypothesis that secA plays an important role in survival in macrophages by S. agalactiae by interfering with phagosome maturation.
It is known that all pathogens engulfed by macrophages suffer a barrage of bactericidal attacks from exposure to reactive oxygen and nitrogen species, antimicrobial peptides, proteases, peptidoglycan-degrading enzymes, and metal deprivation. In this study, ciaH, encoding the histidine kinase involved in many stress responses, was upregulated. Gene expression in prokaryotes is controlled by two-component systems that comprise a sensor histidine kinase and a cognate DNA binding response regulator. Pathogenic bacteria utilize two-component systems to regulate expression of their virulence factors and for adaptive responses to the external environment [37]. Qi et al. [38] reported that inactivation of ciaH in S. mutans abolished mutacin production, diminished competence development, altered sucrose-dependent biofilm formation, and significantly reduced acid tolerance. CiaH and ciaR are in the same genomic organization as the ciaRH operon in S. pneumoniae [39]. It has been reported that the two-component regulatory system CiaRH of S. pneumoniae affects β-lactam susceptibility, autolysis, bacteriocin production, competence development, host colonization and virulence [39]. It might be interesting to evaluate whether this sensor contributes to the adaption to the external environment.
Additionally, in this study we identified several upregulated genes of enzymes involved in the stress response, such as clpE, nox, carB and msrB. ATP-dependent caseinolytic proteases (Clp) play a fundamental role in stress tolerance and virulence in many pathogenic bacteria. The results of Zhang et al. [40] indicated that Clp ATPase (clpE) affected pneumococcal pathogenesis by modulating the expression of some important virulence determinants and metabolism-related factors in S. pneumoniae. Nair et al. [41] indicated that clpE is a novel member of the HSP100 family, involved in cell division and virulence in L. monocytogenes. The nox gene, encoding NADH oxidase, was previously reported to play an important role in the oxidative and acid stress responses of S. mutans [42]. CarB encodes carbamoyl-phosphate synthase. Park et al. [43] used a hypersensitive Escherichia coli genetic system to identify genes involved in resistance to nitrosative stress imposed by reactive nitrogen intermediates; one of the ten candidate genes was a carbamoyl-phosphate synthase. Methionine sulfoxide reductases A and B (msrA and msrB) are antioxidant repair enzymes that reduce S- and R-diastereomers of methionine sulfoxides back to methionine, respectively. Zhao et al. [44] reported that Msr repair enzymes are important for the oxidative stress response, macrophage survival, and persistent infection with Enterococcus faecalis. Ehrt et al. [45] suggested that msrA and msrB contribute to the resistance of M. tuberculosis against reactive nitrogen intermediates (RNI) and reactive oxygen intermediates (ROI). Msr can repair oxidative damage by reducing methionine sulfoxide (MetSO) back to methionine (Met) using electrons derived from thioredoxin, thioredoxin reductase, and NADPH [44]. We also found there is a thioredoxin reductase region in the clone C2 protein (pyridine nucleotide-disulfide oxidoreductase family protein) by using the BLAST search from NCBI (data not shown). Increased clpE, nox, CarB and msrB mRNA levels suggest that the encoded enzymes may be involved in the maintenance of function in stress response to adverse environmental conditions.
Interestingly, one of the genes identified by SCOTS (clone B1), hylB, encodes hyaluronate (HA) lyase. The degradation of polymeric HA and other glycosaminoglycans by HA lyases plays an essential role in many biological processes. For example, HA lyases can help S. agalactiae spread through host tissues [9]. It is thus tempting to speculate that upregulating the expression of the HA lyases identified in this work might, in vivo, be useful to increase the permeability of the BBB and thereby contribute to the migration of S. agalactiae to the CNS. Another gene, pulA, encodes pullulanase. It has previously been reported that pulA is likely to be involved in bacterial adhesion to host cells and suggested that in vivo immunization with this antigen could prevent streptococcal colonization [46], [47], [48], [49]. Gourlay et al. [50] also found that both recombinant forms of pullulanase from GBS appear to bind to ME180 cervical epithelial cells. Similarly, the gap gene encodes glyceraldehyde-3-phosphate dehydrogenase (GAPDH), possibly implicated in the adhesion of the bacteria. Brassard et al. [51] found the GAPDH protein of S. suis seemed to be involved in the first steps of bacterial adhesion to host cells. Also, Oliveira et al. provided evidence for a novel function of secreted GAPDH as an inducer of apoptosis in murine macrophages [52]. It might therefore be plausible that after GBS enters the CNS via a “Trojan horse” or “modified” Trojan horse mechanism, this bacterium may escape from the “Trojan horse” with the help of GAPDH and go on to promote the development of meningitis.
CcpA encodes catabolite control protein A. Willenborg et al. [53] demonstrated that ccpA has a significant effect on capsule synthesis and the virulence properties of S. suis. Deletion of ccpA altered expression of surface-associated virulence factors and revealed a markedly reduced thickness of the capsule. This phenotype correlated with enhanced binding to porcine plasma proteins and a reduced resistance to killing by porcine neutrophils. It is well accepted that the capsule of GBS enables avoidance of immunologic clearance by host phagocytic cells, including neutrophils and macrophages [9]. The role of ccpA during macrophage infection in GBS is unclear; however, considering that ccpA is involved in the formation of the capsule, we hypothesized that upregulation of ccpA expression may contribute to the resistance of S. agalactiae to phagocytosis and survival in RAW264.7 cells. Further studies are required to evaluate this hypothesis.
Regulatory systems play important roles in the adaptive responses of many bacteria to environmental cues such as cell contact and entry. MerR-type and lysR-type transcriptional regulators (LTTRs) regulate a diverse set of genes, including those involved in virulence, metabolism, quorum sensing and motility [54], [55]. CovR is a response regulator protein. Recently, CovR/S two-component global regulatory system has received considerable attention. Sendi et al. [56] have shown the critical involvement of CovR/S in virulence of S. agalactiae. Mutations in the cov gene lead to an increased β-hemolytic activity with lower capsule expression. And the phenotype showed low intracellular survival in human neutrophils in contrast to the low hemolytic variant with high capsule expression. The findings of Cumley et al. [10] showed that CovS/R is required for survival of S. agalactiae inside the phagosome. A recent study by Sagar et al. [57] demonstrated that the absence of β-hemolysin may enable S. agalactiae to survive in higher numbers inside professional phagocytes, and highlighted the importance of CovR/S regulator due to its suppression effect on β-hemolysin expression. In addition, a recent publication reported that the expression of the cov regulator was increased in S. pyogenes recovered from the intracellular environment of macrophages [58]. MerR, lysR and covR were also found in our SCOTS result. Therefore, it is hypothesized that transcriptional regulators might play an important role in survival within phagosomes and hence the virulence of GBS.
Globally, our SCOTS results indicate that internalization of GBS by macrophages causes major changes in the expression of genes associated with signal transduction mechanisms and transcription, showing that GBS senses the phagosomal environment and activates mechanisms necessary for survival in a new niche. Metabolic adaptation accounts for more than 50% (32/60) of differentially regulated genes in intracellular bacteria. This agrees with the observation for other pathogens [59], [60] that some of the most highly expressed genes in intracellular bacteria are those involved in metabolism. The changes may reflect the ability of S. agalactiae to utilize resources present in macrophages.
In conclusion, we investigated the virulence of S. agalactiae isolated from fish in mouse and macrophage models. For the first time, we identified genes that were up-regulated during the interaction between GBS and macrophages. Characterizing these genes and their products and deciphering their roles in intracellular growth will enable us to gain a better understanding of S. agalactiae pathogenesis. Allele inactivation studies addressing the roles of several of these genes are now under way.
Materials and Methods
Ethics statement
Animal experiments were carried out according to animal welfare standards and approved by the Ethical Committee for Animal Experiments of Nanjing Agricultural University, China. All animal experiments complied with the guidelines of the Animal Welfare Council of China.
Cell lines, bacterial strains, plasmids and culture conditions
The cell lines, bacterial strains and plasmids used in this study are listed in Table 3. A green fluorescent protein (GFP) expression plasmid, pSL5.28 [61], was provided by the Vaccine and Infectious Disease Organization, University of Saskatchewan. RAW264.7 cells (ATCC) were maintained in Dulbecco's modified Eagle medium (DMEM) with high glucose (Gibco, Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS). Cells were used from passages 5 to 20.
Table 3. Characteristics of cell lines, bacterial strains, plasmids and primers used in this study.
| Cell line, strain, plasmid or primer primer | Characteristics and/or sequences | Source/reference |
| Cell line | ||
| RAW264.7 | murine (BALB/c) macrophage cell line | ATCC |
| Strains | ||
| GD20101008-001 | S.agalactiae, serotype Ia, isolated from cultured tilapia in China. | Our lab |
| DH5α | Escherichia coli | TaKaRa |
| Plasmids | ||
| pSL5.28 | green fluorescent protein (GFP) expression | [61] |
| pMD18-T | Vector for cloning Taq polymerase-amplified PCR products | TaKaRa |
| pMD18-T16S | pMD18-T containing the 16S rRNA sequence (1507bp) of S.agalactiae | This work |
| pMD18-T23S1 | pMD18-T containing the 5′end 1500 bp fragment of 23S rRNA of S.agalactiae | This work |
| pMD18-T23S2 | pMD18-T containing the 3′end 1403 bp fragment of 23S rRNA of S.agalactiae | This work |
| Primers | ||
| SCOTS-N6-01 | 5′ GACACTCTCGAGACATCACCGGTACCNNNNNN 3′ | This work |
| SCOTS-N6-02 | 5′ TGCTCTAGACGTCCTGATGGTTCNNNNNN 3′ | This work |
| SCOTS01 | 5′ GACACTCTCGAGACATCACCGGTACC 3′ | This work |
| SCOTS02 | 5′ TGCTCTAGACGTCCTGATGGTTC 3′ | This work |
| 16S01 | 5′ AGAGTTTGATCCTGGCTCAGG 3′ | |
| 16S02 | 5′ TACCTTGTTACGACTTCACC 3′; amplifies the 16S rRNA sequence(1507 bp) | This work |
| 23SN01 | 5′ GGTTAAGTTAATAAGGGCGCAC 3′ | |
| 23SN02 | 5′ TTGACCAGACACTTCCAATCGT 3′; amplifies the 5′ end of 23S rRNA sequence(1500 bp) | This work |
| 23SC01 | 5′ ACAGTGAGGTGTGATATGAGTC 3′ | |
| 23SC02 | 5′ TTGGATAAGTCCTCGAGCTATT 3′; amplifies the 3′ end of 23S rRNA sequence(1403 bp) | This work |
| 16S-L | 5′CGACGATACATAGCCGACCT3′ | |
| 16S-R | 5′CCGTCACTTGGTAGATTTTCC3′; amplifies S.agalactiae 16S rRNA sequence (210 bp) | This work |
| AroA-L | 5′GATCGGATTCAGGTCGTTGT3′ | |
| AroA-R | 5′CTTTGACCAAGAGAGCAGCA3′; amplifies the sequence of aroA (172 bp) | This work |
| Clone B3-L | 5′CAATCAAGCTCTCAGCCAAA3′ | |
| Clone B3-R | 5′GTGGTATGAATGGCGTCGTA3′; amplifies the sequence of clone B3 (210 bp) | This work |
| Clone A6-L | 5′ATTGCGGGAGTGGTAGGAGTT3′ | |
| Clone A6-R | 5′TGTACGTCTTGAAGCCAGTAG3′; amplifies the sequence of clone A6 (162 bp) | This work |
| Clone E4-L | 5′CGTCAGCAGTAACAGGCTCA3′ | |
| Clone E4-R | 5′CGATCCAAAGGACCGTTATG3′; amplifies the sequence of clone E4 (194 bp) | This work |
| CvoR-L | 5′TCCAATAAATGTTCGCGTGTC3′ | |
| CvoR-R | 5′CAGCTGTAGCAGAAGAAAGTG3′; amplifies the sequence of clone covR (189 bp) | This work |
| DivIB-L | 5′GACTATTTTCCGCAACCTGAC3′ | |
| DivIB-R | 5′AGAAAGACTTCCTTTTTACAA3′; amplifies the sequence of clone divIB (185 bp) | This work |
| DltA-L | 5′GTTCCTAACATTTCATCAGTA3′ | |
| DltA-R | 5′GCTACTATCTAACGATTTCAA3′; amplifies the sequence of clone dltA(198 bp) | This work |
| DnaJ-L | 5′TCAATGGTGGGCTTTATGGT3′ | |
| DnaJ-R | 5′TGACGTTTCTACCGCTCCAT3′; amplifies the sequence of clone dnaJ(169 bp) | This work |
| HylB-L | 5′CGCTACTTATCGTCGTTTGGA3′ | |
| HylB-R | 5′ATTGAGCGAGGGACACCGATT3′; amplifies the sequence of clone hylB (195 bp) | This work |
| PulA-L | 5′CGAGAAGCAACATATTTACCG3′ | |
| PulA-R | 5′TAACTTGGTCTGGGGCATTGT3′; amplifies the sequence of clone pulA (188 bp) | This work |
| SecA-L | 5′AGATAACTCCCAGTGCTAAAC3′ | |
| SecA-R | 5′AACTGAAGATACTTATCGCCC3′; amplifies the sequence of clone secA(206 bp) | This work |
| SerB-L | 5′TCCTACCCCAGCGGATTTGAT3′ | |
| SerB-R | 5′AAAGCGAATCGTCCTGGTGTC3′; amplifies the sequence of clone serB(189 bp) | This work |
S. agalactiae strain GD201008-001, β-hemolysin/cytolysin positive, which belongs to serotype Ia, MLST type ST-7, was isolated from farmed tilapia with meningoencephalitis in the Guangdong province of China in 2010 [62]. Its genome sequence has been deposited in the GenBank database under accession number CP003810. The bacterial strain was grown using either Todd-Hewitt broth (THB) or agar (THA) (Becton Dickinson, MD, USA) or sheep blood agar plates at 37°C.
Determination of virulence of GD201008-001
BALB/c mice (22–24 g, 50 days old) were purchased from the Experimental Animal Center, Yangzhou University. The mice were divided into 5 groups with 10 mice in each group. Mid-exponential phase bacteria were washed twice in PBS. Bacteria were then resuspended in PBS to 1×105 CFU/ml and a 10× dilution serial with the lowest concentration being 1×102 CFU/ml was prepared. Four groups of mice were injected intraperitoneally (i.p) with 100 µl of the bacterial suspension of different dilutions respectively. The 5th group was injected with 100 µl PBS and served as control. Death of mice was recorded.
Experimental infection of mice
BALB/c mice (22–24 g, 50 days old) were divided into 6 groups with 5 mice in each group. Mid-exponential phase bacteria were washed twice in PBS. Bacteria were then resuspended in PBS to 2×103 CFU/ml. Five groups of mice were injected i.p with 100 µl of the bacterial suspension. The 6th group was injected with 100 µl PBS and served as control. Samples were taken at 3 h, 6 h, 12 h and 20 h post infection. At each designated time, one mouse from each infection group and one from the control group were anesthetized with CO2. Blood was collected and the brains, livers, and spleens were obtained aseptically. In order to avoid possible surface contamination, livers, and spleens were sterilized with 75% alcohol,and then washed with PBS. The organs (0.05 g/organ) were trimmed, placed in 500 µl of PBS, and homogenized in a high-speed homogenizer. Then, 200 µl of 102, 103, 104 or 105 dilutions of the homogenate in PBS were plated onto blood agar plates. Blood samples (200 µl) were also plated. The blood agar plates were incubated overnight at 37°C. Colonies were counted and expressed as CFU/g for organ samples or CFU/ml for blood samples.
Histopathology and immunohistochemistry
At 20 h.p.i., the infected mice were euthanized and the brains was collected and placed into 10% neutral buffered formalin. After fixation the organs were embedded in paraffin, 4 mm sections were taken, and these were stained with hematoxylin and eosin for histological evaluation. For immunohistochemistry, sequential slides were stained using an immunoperoxidase method as follows. First, sections were blocked with 3% H2O2 for 30 min, and then non-specific background staining was blocked by incubating the sections for 30 min with normal rabbit serum. The sections were then incubated for 1 h with rabbit anti-GBS polyclonal serum (1∶1000, prepared in our laboratory using the GBS strain GD201008-001 from this study) or unimmunized rabbit serum (1∶1000) as a control, followed by biotinylated goat anti-rabbit immunoglobulin (Ding-Guo, China) diluted 1/100 at 37°C for 30 min and subsequent incubation with HRP conjugated streptavidin (DingGuo, China) at 37°C for 30 min. Finally, the sections were developed with the HRP-DAB chromogenic substrate kit (Tiangen, China) for 10 min, and then the slides were counterstained with hematoxylin.
Phagocytosis and intracellular survival assays
Green fluorescent protein (GFP)-expressing S. agalactiae GD201008-001 was prepared as previously described [61]. The phagocytosis assay was performed as described above, with some modifications. RAW264.7 cells were grown on 24-well tissue culture plates. After an initial 1 h exposure to the bacteria (at an MOI of 1, 10 or 100), gentamicin and penicillin were added. After antibiotic treatment, the cell monolayers were washed three times and the medium was replaced with 1 ml of sterile distilled water to lyse the macrophages. After vigorous pipetting to ensure complete cell lysis, viable intracellular streptococci were determined by quantitative plating of serial dilutions of the lysates on THB agar. After overnight incubation at 37°C, the number of bacteria was calculated.
For intracellular survival studies, an internalization assay was performed as described above, except that after a 1 h initial exposure to the bacteria, gentamicin and penicillin were added and the treatment was continued for different times up to 24 h. At different time points, cells were processed as described above and bacteria counted. The results come from at least three independent experiments.
Cellular injury assays
A lactate dehydrogenase (LDH) release measurement assay was used to measure cytotoxicity levels (Promega CytoTox96, Promega Corporation, Madison, WI, USA) as previously described [63] with some modifications. Briefly, bacteria were grown and diluted as described above, and cells grown in 96-well plates were infected with 100 µL aliquots of a bacterial suspension at different concentrations (MOIs of 1, 10 or 100). The plates were centrifuged at 800×g for 10 min to bring the bacteria to the surface of each monolayer and were then incubated for 4 h at 37°C with 5% CO2 (see Results). Non-infected cells and bacteria alone were used as negative controls, whereas cells lysed with the lysis solution from the Promega CytoTox96 kit (Promega Corporation, Madison, WI) were used as positive controls (100% toxicity). At the end of the incubation time, the plates were centrifuged at 300 g for 20 min to pellet the bacteria and the LDH concentration was measured as previously described [64].
Experimental infection, RNA isolation, cDNA synthesis and amplification
For RNA isolation, macrophage infection was performed as described above with some modifications. Cells were seeded at 2×106 cells per well in 6-well tissue culture dishes. Bacteria were grown in THB to log-phase, washed in PBS and then added to the cell monolayer at an MOI of 100∶1 before centrifugation for 10 min at 800×g to synchronize phagocytosis. After incubation and antibiotic treatment, total RNA was isolated from the infected cells or from broth-grown bacteria using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. RNA samples were treated with RNase-free DNase I (Fermentas) and RNA concentrations and integrity were evaluated by A260/A280 spectrophotometer readings and agarose gel electrophoresis, respectively. Total RNA isolated from infected macrophages or from broth-grown bacteria was converted to first strand cDNA by random priming with Superscript II reverse transcriptase (Invitrogen). Random priming was performed as described [66] using oligonucleotides containing the SCOTS-N6-01 and SCOTS-N6-02 terminal sequences at the 5′ end for cDNAs from the infected macrophages and from broth-grown bacteria, respectively, and a random hexamer at the 3′ end. The cDNA was then converted to double-stranded form with Klenow fragment (Fermentas) as described previously [65]. Then, individual cDNAs were amplified by PCR with 30 cycles of amplification (95°C for 30 s, 66°C for 60 s and 72°C for 60 s).
Selective capture of transcribed sequences (SCOTS)
Genomic DNA from GD20101008-001 was biotinylated as described previously [22]. The biotinylated S. agalactiae genomic DNA (12 µg) was mixed with 100 µg of pMD18-T16S, pMD18-T23S1 and pMD18-T23S2 plasmid DNA (S. agalactiae rrnA DNA cloned into pMD18-T). The mixture was sonicated using a microprobe for 5 s at an output power of 25 W (Fisher Scientific 100). After sonication, the mixture was precipitated with 1/10 volume of 3 M sodium acetate and an equal volume of isopropanol, and re-suspended in 160 µl of 10 mM N-(2-hydroxyethyl) piperazine-N-(3- propanesulfonic acid) (EPPS)-1 mM EDTA. The mixture was then divided into 20 samples of 8 µl each. For each round of SCOTS, a sample of the mixture (containing 0.6 µg of S. agalactiae genomic DNA and 5 µg of rrnA DNA) was denatured by incubation at 98°C for 3 min. Two microlitres of 1 M NaCl were added to the mixture and it was incubated at 67°C for 30 min. This step allowed the plasmid rrnA DNA to hybridize to the rrnA sites on the S. agalactiae genomic DNA, thereby rendering these sites unavailable for hybridization with ribosomal DNA present in the cDNA mixtures. At the same time, total amplified cDNA, either from infected cells or from broth grown bacteria in 8 µl of 10 mM EPPS-1 mM EDTA was also denatured at 98°C for 3 min followed by the addition of 2 µl of 1 M NaCl. The denatured cDNA mixture was added to the biotinylated chromosomal DNA–rrnA pre-hybridized mixture, and hybridization was continued at 67°C for 24 h. cDNA was captured by streptavidin-coated magnetic beads (Dynal M280) according to the manufacturer's instructions. The process of elution was performed as described previously [22]. For each growth condition, in the first round of SCOTS, 10 separate samples of the cDNA mixtures were captured by hybridization to rDNA-blocked genomic DNA in parallel reactions.
Enrichment for cDNA molecules in macrophages
To identify cDNA molecules representing transcripts from genes whose expression is either specific to or upregulated during the interaction between S. agalactiae and macrophages, an additional step was included in the experiments. Preparations of cDNA mixtures from S. agalactiae that had interacted with macrophages were obtained by three rounds of SCOTS and added to biotinylated genomic DNA that had been pre-hybridized with both rDNA and cDNA preparations from broth-grown S. agalactiae (also obtained following three rounds of SCOTS). Hybridization proceeded for 24 h at 67°C, and the hybridized molecules were recovered by binding to streptavidin-coated beads as described above.
Dot blot hybridization and analysis of individual cDNA clones
Macrophage-specific or upregulated cDNAs were ligated into the cloning vector pMD18-T (TaKaRa). For verification, cloned inserts were amplified by PCR. PCR amplicons of positive SCOTS clones were transferred to a positively charged membrane (Roche). Samples of S. agalactiae genomic DNA and cDNA mixtures generated from infected macrophages and bacteria cultured under normal growth conditions were labeled with DIG-dUTP (Roche) and used as probes. Dot blot hybridization using DIG Easy Hyb (Roche) was used in accordance with the manufacturer's instructions. S. agalactiae genomic DNA (1 or 0.1 µg) spotted on nylon membranes was used as a positive control exposed alongside the corresponding SCOTS clone dot blots. Hybridization signals stronger than 1 µg of genomic DNA or weaker than 0.1 µg of genomic DNA were deemed strong or weak signals, respectively. The positive sequences of these clones satisfied the requirement that their signals be weaker or absent when hybridized to normalized culture-specific cDNA, and strong on the blot hybridizing to normalized macrophage-specific cDNA, indicating that these sequences are either absent or present in much less abundance in cDNA prepared from S. agalactiae grown under standard culture conditions. The inserts of positive cDNA clones from the library were sequenced using the standard Sanger method. Database searches as well as DNA and protein similarity comparisons were carried out using the BLAST algorithm from the National Center for Biotechnology Information at the National Library of Medicine. http://www.ncbi.nlm.nih.gov/BLAST/Blast.cgi).
Real-time qRT-PCR
To validate the SCOTS results, we used quantitative PCR (q-PCR) to measure the level of expression of randomly selected genes on a new series of infected replicates. Infection of RAW264.7, growth of S. agalactiae in broth, and RNA extraction from both samples were performed as described above. cDNAs were synthesized in triplicate using Super-Script II with random hexamers (Roche, Laval, Quebec,Canada). q-PCR was performed using the QuantiTect SybrGreen PCR kit (QIAGEN, Mississauga, Ontario, Canada) according to the manufacturer's instructions. For each sample, a no-reverse transcription reaction was run as a control. The primers used are described in Table 1. For each q-PCR run, the calculated threshold cycle (CT) was normalized to the CT of the 16S rRNA internal control amplified from the corresponding sample, and the n-fold change was calculated by the 2−ΔΔCT method as previously described [66].
Statistical analysis
Each cell internalization or injury experiment was repeated in at least three independent test series with a minimum of four parallels each. All statistics were performed using an unpaired two-tailed t-test with a 95% confidence interval. A P-value <0.05 was considered to indicate a significant difference. In all experiments, error bars were denoted using the standard deviation (±SD) of independent experiments.
Funding Statement
The study was funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Elliott JA, Facklam RR, Richter CB (1990) Whole-cell protein-patterns of nonhemolytic group-B, type Ib, streptococci isolated from humans, mice, cattle, frogs, and fish. J Clin Microbiol 28: 628–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jones N, Bohnsack JF, Takahashi S, Oliver KA, Chan MS, et al. (2003) Multilocus sequence typing system for group B streptococcus. J Clin Microbiol 41: 2530–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eldar A, Bejerano Y, Livoff A, Horovitcz A, Bercovier H (1995) Experimental streptococcal meningoencephalitis in cultured fish. Vet Microbiol 43: 33–40. [DOI] [PubMed] [Google Scholar]
- 4. Evans JJ, Klesius PH, Glibert PM, Shoemaker CA, Al Sarawi MA, et al. (2002) Characterization of beta-haemolytic Group B Streptococcus agalactiae in cultured seabream, Sparus auratus L., and wild mullet, Liza klunzingeri (Day), in Kuwait. J Fish Dis 25: 505–513. [Google Scholar]
- 5. Evans JJ, Klesius PH, Pasnik DJ, Bohnsack JF (2009) Human Streptococcus agalactiae isolate in Nile Tilapia (Oreochromis niloticus). Emerg Infect Dis 15: 774–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suanyuk N, Kong FR, Ko D, Gilbert GL, Supamattaya K (2008) Occurrence of rare genotypes of Streptococcus agalactiae in cultured red tilapia Oreochromis sp. and Nile tilapia O. niloticus in Thailand-relationship to human isolates? Aquaculture 284: 35–40. [Google Scholar]
- 7. Eldar A, Bejerano Y, Livoff A, Horovitcz A, Bercovier H (1995) Experimental streptococcal meningo-encephalitis in cultured fish. Vet Microbiol 43: 33–40. [DOI] [PubMed] [Google Scholar]
- 8. Chen M, Li LP, Wang R, Liang WW, Huang Y, et al. (2012) PCR detection and PFGE genotype analyses of streptococcal clinical isolates from tilapia in China. Vet Microbiol 159: 526–530. [DOI] [PubMed] [Google Scholar]
- 9. Doran KS, Nizet V (2004) Molecular pathogenesis of neonatal group B streptococcal infection: no longer in its infancy. Mol Microbiol 54: 23–31. [DOI] [PubMed] [Google Scholar]
- 10. Cumley NJ, Smith LM, Anthony M, May RC (2012) The CovS/CovR acid response regulator is required for intracellular survival of group B streptococcus in macrophages. Infect Immun 80: 1650–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cornacchione P, Scaringi L, Fettucciari K, Rosati E, Sabatini R, et al. (1998) Group B streptococci persist inside macrophages. Immunology 93: 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu GY, Doran KS, Lawrence T, Turkson N, Puliti M, et al. (2004) Sword and shield: Linked group B streptococcal beta-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc Natl Acad Sci U S A 101: 14491–14496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. ValentinWeigand P, Benkel P, Rohde M, Chhatwal GS (1996) Entry and intracellular survival of group B streptococci in J774 macrophages. Infect Immun 64: 2467–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaplan EL, Chhatwal GS, Rohde M (2006) Reduced ability of penicillin to eradicate ingested group A streptococci from epithelial cells: Clinical and pathogenetic implications. Clin Infect Dis 43: 1398–1406. [DOI] [PubMed] [Google Scholar]
- 15. Drevets DA (1999) Dissemination of Listeria monocytogenes by infected phagocytes. Infect Immun 67: 3512–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Drevets DA, Jelinek TA, Freitag NE (2001) Listeria monocytogenes-infected phagocytes can initiate central nervous system infection in mice. Infect Immun 69: 1344–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Drevets DA, Sawyer RT, Potter TA, Campbell PA (1995) Listeria monocytogenes infects human endothelial cells by two distinct mechanisms. Infect Immun 63: 4268–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greiffenberg L, Goebel W, Kim KS, Weiglein I, Bubert A, et al. (1998) Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: InlB-Dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect Immun 66: 5260–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gottschalk M, Segura M (2000) The pathogenesis of the meningitis caused by Streptococcus suis: the unresolved questions. Vet Microbiol 76: 259–272. [DOI] [PubMed] [Google Scholar]
- 20. Daigle F, Graham JE, Curtiss R (2001) Identification of Salmonella typhi genes expressed within macrophages by selective capture of transcribed sequences (SCOTS). Mol Microbiol 41: 1211–1222. [DOI] [PubMed] [Google Scholar]
- 21. Graham JE, Clark-Curtiss JE (1999) Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc Natl Acad Sci U S A 96: 11554–11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hou JY, Graham JE, Clark-Curtiss JE (2002) Mycobacterium avium genes expressed during growth in human macrophages detected by selective capture of transcribed sequences (SCOTS). Infect Immun 70: 3714–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fittipaldi N, Gottschalk M, Vanier G, Daigle F, Harel J (2007) Use of selective capture of transcribed sequences to identify genes preferentially expressed by Sreptococcus suis upon interaction with porcine brain microvascular endothelial cells. Appl Environ Microbiol 73: 7802–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daigle F, Hou JY, Clark-Curtiss JE (2002) Microbial gene expression elucidated by selective capture of transcribed sequences (SCOTS). Methods Enzymol 358: 108–122. [DOI] [PubMed] [Google Scholar]
- 25. Abuseliana AF, Mohd Daud HH, Aziz SA, Bejo SK, Alsaid M (2011) Pathogenicity of Streptococcus agalactiae isolated from a fish farm in Selangor to juvenile red tilapia (Oreochromis sp.). J Anim Vet Adv 10: 914–919. [Google Scholar]
- 26. Bowater RO, Forbes-Faulkner J, Anderson IG, Condon K, Robinson B, et al. (2012) Natural outbreak of Streptococcus agalactiae (GBS) infection in wild giant Queensland grouper, Epinephelus lanceolatus (Bloch), and other wild fish in northern Queensland, Australia. J Fish Dis 35: 173–186. [DOI] [PubMed] [Google Scholar]
- 27. Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, et al. (2009) Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol 10: 587–594. [DOI] [PubMed] [Google Scholar]
- 28. Fettucciari K, Rosati E, Scaringi L, Cornacchione P, Migliorati G, et al. (2000) group B streptococcus induces apoptosis in macrophages. J Immunol 165: 3923–3933. [DOI] [PubMed] [Google Scholar]
- 29. Neuhaus FC, Baddiley J (2003) A continuum of anionic charge: Structures and functions of D-Alanyl-Teichoic acids in gram-positive bacteria. Microbiol Mol Biol Rev 67: 686–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abachin E, Poyart C, Pellegrini E, Milohanic E, Fiedler F, et al. (2002) Formation of D-alanyl-lipoteichoic acid is required for adhesion and virulence of Listeria monocytogenes. . Mol Microbiol 43: 1–14. [DOI] [PubMed] [Google Scholar]
- 31. Kovacs M, Halfmann A, Fedtke I, Heintz M, Peschel A, et al. (2006) A functional dlt operon, encoding proteins required for incorporation of D-alanine in teichoic acids in gram-positive bacteria, confers resistance to cationic antimicrobial peptides in Streptococcus pneumoniae . J Bacteriol 188: 5797–5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, et al. (2005) D-alanylation of teichoic acids promotes group A streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J Bacteriol 187: 6719–6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Poyart C, Pellegrini E, Marceau M, Baptista M, Jaubert F, et al. (2003) Attenuated virulence of Streptococcus agalactiae deficient in D-alanyl-lipoteichoic acid is due to an increased susceptibility to defensins and phagocytic cells. Mol Microbiol 49: 1615–1625. [DOI] [PubMed] [Google Scholar]
- 34. Weidenmaier C, Peschel A, Kempf VAJ, Lucindo N, Yeaman MR, et al. (2005) DltABCD- and MprF-mediated cell envelope modifications of Staphylococcus aureus confer resistance to platelet microbicidal proteins and contribute to virulence in a rabbit endocarditis model. Infect Immun 73: 8033–8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Limia A, Sangari FJ, Wagner D, Bermudez LE (2001) Characterization and expression of secA in Mycobacterium avium. . FEMS Microbiol Lett 197: 151–157. [DOI] [PubMed] [Google Scholar]
- 36. Owens MU, Schmidt MG, King CH, Quinn FD (1996) Identification of export proteins from Mycobacterium tuberculosis that interact with SecA. Ann N Y Acad Sci 797: 240–241. [DOI] [PubMed] [Google Scholar]
- 37. Rajagopal L, Vo A, Silvestroni A, Rubens CE (2006) Regulation of cytotoxin expression by converging eukaryotic-type and two-component signalling mechanisms in Streptococcus agalactiae. Mol Microbiol 62: 941–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qi FX, Merritt J, Lux R, Shi WY (2004) Inactivation of the ciaH gene in Streptococcus mutans diminishes mutacin production and competence development, alters sucrose-dependent biofilm formation, and reduces stress tolerance. Infect Immun 72: 4895–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Muller M, Marx P, Hakenbeck R, Bruckner R (2011) Effect of new alleles of the histidine kinase gene ciaH on the activity of the response regulator CiaR in Streptococcus pneumoniae R6. Microbiology 157: 3104–3112. [DOI] [PubMed] [Google Scholar]
- 40. Zhang Q, Xu SX, Wang H, Xu WC, Zhang XM, et al. (2009) Contribution of ClpE to virulence of Streptococcus pneumoniae. . Can J Microbiol 55: 1187–1194. [DOI] [PubMed] [Google Scholar]
- 41. Nair S, Frehel C, Nguyen L, Escuyer V, Berche P (1999) ClpE, a novel member of the HSP100 family, is involved in cell division and virulence of Listeria monocytogenes . Mol Microbiol 31: 185–196. [DOI] [PubMed] [Google Scholar]
- 42. Derr AM, Faustoferri RC, Betzenhauser MJ, Gonzalez K, Marquis RE, et al. (2012) Mutation of the NADH oxidase gene (nox) reveals an overlap of the oxygen- and acid-mediated stress responses in Streptococcus mutans . Appl Environ Microbiol 78: 1215–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Park SH, Lee HW, Cao WG (2010) Screening of nitrosative stress resistance genes in Coxiella burnetii: Involvement of nucleotide excision repair. Microb Pathog 49: 323–329. [DOI] [PubMed] [Google Scholar]
- 44. Zhao C, Hartke A, La Sorda M, Posteraro B, Laplace JM, et al. (2010) Role of methionine sulfoxide reductases A and B of Enterococcus faecalis in oxidative stress and virulence. Infect Immun 78: 3889–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ehrt S, Schnappinger D (2009) Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol 11: 1170–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hytonen J, Haataja S, Finne J (2003) Streptococcus pyogenes glycoprotein-binding strepadhesin activity is mediated by a surface-associated carbohydrate-degrading enzyme, pullulanase. Infect Immun 71: 784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hytonen J, Haataja S, Finne J (2006) Use of flow cytometry for the adhesion analysis of Streptococcus pyogenes mutant strains to epithelial cells: investigation of the possible role of surface pullulanase and cysteine protease, and the transcriptional regulator Rgg. BMC Microbiol 6: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Bueren AL, Higgins M, Wang D, Burke RD, Boraston AB (2007) Identification and structural basis of binding to host lung glycogen by streptococcal virulence factors. Nat Struct Mol Biol 14: 76–84. [DOI] [PubMed] [Google Scholar]
- 49. Ferrando ML, Fuentes S, de Greeff A, Smith H, Wells JM (2010) ApuA, a multifunctional alpha-glucan-degrading enzyme of Streptococcus suis, mediates adhesion to porcine epithelium and mucus. Microbiology 156: 2818–2828. [DOI] [PubMed] [Google Scholar]
- 50. LJ, Santi I, Pezzicoli A, Grandi G, Soriani M, et al. (2009) Group B streptococcus pullulanase crystal structures in the context of a novel strategy for vaccine development. J Bacteriol 191: 3544–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brassard J, Gottschalk M, Quessy S (2004) Cloning and purification of the Streptococcus suis serotype 2 glyceraldehyde-3-phosphate dehydrogenase and its involvement as an adhesin. Vet Microbiol 102: 87–94. [DOI] [PubMed] [Google Scholar]
- 52. Oliveira L, Madureira P, Andrade EB, Bouaboud A, Morello E, et al. (2012) Group B streptococcus GAPDH is released upon cell lysis, associates with bacterial surface, and induces apoptosis in murine macrophages. PLoS ONE 7: e29963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Willenborg J, Fulde M, de Greeff A, Rohde M, Smith HE, et al. (2011) Role of glucose and CcpA in capsule expression and virulence of Streptococcus suis. . Microbiology 157: 1823–1833. [DOI] [PubMed] [Google Scholar]
- 54. Julian DJ, Kershaw CJ, Brown NL, Hobman JL (2009) Transcriptional activation of MerR family promoters in Cupriavidus metallidurans CH34. Antonie Van Leeuwenhoek 96: 149–159. [DOI] [PubMed] [Google Scholar]
- 55. Maddocks SE, Oyston PCF (2008) Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154: 3609–3623. [DOI] [PubMed] [Google Scholar]
- 56. Sendi P, Johansson L, Dahesh S, Van-Sorge NM, Darenberg J, et al. (2009) Bacterial phenotype variants in group B streptococcal toxic shock syndrome. Emerg Infect Dis 15: 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sagar A, Klemm C, Hartjes L, Mauerer S, van Zandbergen G, et al. (2013) The beta-hemolysin and intracellular survival of Streptococcus agalactiae in human macrophages. PLoS ONE 8: e60160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hertzén E, Johansson L, Kansal R, Hecht A, Dahesh S, et al. (2012) Intracellular Streptococcus pyogenes in human macrophages display an altered gene expression profile. PLoS ONE 7: e35218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jin H, Wan Y, Zhou R, Li LJ, Luo R, et al. (2008) Identification of genes transcribed by Haemophilus parasuis in necrotic porcine lung through the selective capture of transcribed sequences (SCOTS). Environ Microbiol 10: 3326–3336. [DOI] [PubMed] [Google Scholar]
- 60. Guo DC, Lu Y, Zhang AQ, Liu JS, Yuan DW, et al. (2012) Identification of genes transcribed by Pasteurella multocida in rabbit livers through the selective capture of transcribed sequences. FEMS Microbiol Lett 331: 105–112. [DOI] [PubMed] [Google Scholar]
- 61. Lun SC, Willson PJ (2004) Expression of green fluorescent protein and its application in pathogenesis studies of serotype 2 Streptococcus suis . J Microbiol Methods 56: 401–412. [DOI] [PubMed] [Google Scholar]
- 62. Liu GJ, Zhang W, Lu CP (2012) Complete genome sequence of Streptococcus agalactiae GD201008-001, isolated in China from Tilapia with meningoencephalitis. J Bacteriol 194: 6653–6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chabot-Roy G, Willson P, Segura M, Lacouture S, Gottschalk M (2006) Phagocytosis and killing of Streptococcus suis by porcine neutrophils. Microb Pathog 41: 21–32. [DOI] [PubMed] [Google Scholar]
- 64. Vanier G, Segura M, Friedl P, Lacouture S, Gottschalk M (2004) Invasion of porcine brain microvascular endothelial cells by Streptococcus suis serotype 2. Infect Immun 72: 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Froussard P (1992) A random-PCR method (rPCR) to construct whole cDNA library from low amounts of RNA. Nucleic Acids Res 20: 2900–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]