

Abstract
The liver is the first line of defense from environmental stressors in that hepatocytes respond to and metabolize them. Hence, hepatocytes can be damaged by stressors. Protection against hepatic cell damage and cell death is important for liver function and homeostasis. TAK1 (MAP3K7) is an intermediate of stressors such as bacterial moieties–induced signal transduction pathways in several cell types. Tak1 deficiency has been reported to induce spontaneous hepatocellular carcinoma. However, the regulatory mechanism of TAK1 activity in liver stress response has not yet been defined. Here we report that activation of TAK1 through TAK1 binding protein 2 (TAB2) is required for liver protection from stressors. We found that a bacterial moiety, lipopolysaccharides (LPS), activated TAK1 in primary hepatocytes, which was diminished by deletion of TAB2. Mice having hepatocyte-specific deletion of the Tab2 gene exhibited only late-onset moderate liver lesions but were hypersensitive to LPS-induced liver injury. Furthermore, we show that a chemical stressor induced greatly exaggerated liver injury in hepatocyte-specific Tab2-deficient mice. These results demonstrate that TAB2 is a sensor of stress conditions in the liver and functions to protect the liver by activating the TAK1 pathway.
Introduction
The liver is responsible for the first pass metabolism of absorbed exogenous chemicals and the major organ of clearance of toxicants and pathogens, and liver cells are constantly exposed to those foreign substances including bacterial moieties and food- and clinical drug-derived chemicals. These stressors directly impact on liver parenchymal cells, and indirectly through cytokines from activated circulating and residential immune cells such as Kupffer cells [1], [2]. Although the stressors and stressor-induced cytokines could activate cell death signaling in hepatocytes, they are normally resistant to those basal levels of stressors and functional under the homeostatic conditions. Excessive stressors derived from alcohol consumption and pathogens lead to liver injury. Furthermore, dysregulation of the protecting responses is likely to be associated with liver tumors in which cell death causes compensatory cell proliferation [3]. However, the mechanism by which hepatocytes respond to stressors and protect against cell death is still largely elusive.
Transcription factor NF-κB and mitogen-activated protein kinase, JNK, have been implicated in the regulatory mechanisms of hepatocyte death [4], [5]. NF-κB protects hepatocytes from concanavalin A-induced cell death, and ablation of NF-κB induces spontaneous hepatocyte death and compensatory proliferation [4], [6]. Activation of JNK is also closely associated with liver injury [5], [7], [8]. NF-κB transcriptionally activates several cell survival genes such as caspase inhibitors, cellular FLICE-like inhibitory protein (cFLIPL) and cellular inhibitor of apoptosis proteins (cIAPs), and anti-oxidant genes such as glutathione peroxidase and superoxide dismutase, which block proinflammatory cytokine TNF-induced cell death and oxidative damage [4], [9]-[11].
TAK1 (MAP3K7) is a member of the mitogen-activated protein kinase kinase kinase (MAP3K) family and an intermediate of stress-induced intracellular signaling pathways [12]–[14]. TAK1 activating stressors include proinflammatory cytokines TNF and IL-1 and Toll-like receptor ligands as well as chemical and physical stress, such as osmotic stress. TAK1 is the major upstream signaling mediator leading to activation of NF-κB and upregulation of antioxidant enzymes, thereby participating in cell survival in several tissues in vivo [15], [16]. Ablation of TAK1 causes cell death associated with oxidative damage and tissue injury in the epidermis, the intestinal epithelium, and blood vessels [16]–[18]. The major cause of cell death in Tak1-deficient tissues in vivo is TNF-induced cell death, and deletion of TNF receptor 1 largely rescues cell death and tissue injury in these Tak1-deficient tissues [16], [18]. Similar to these tissues, hepatocyte-specific deletion of the Tak1 gene causes cell death and liver injury, which further causes compensatory hepatocyte proliferation resulting in development of hepatocellular carcinoma [19], [20]. Thus, TAK1-mediated cell signaling presumably of NF-κB and antioxidant gene regulation is indispensable for hepatocyte protection from stressor-induced cell death. However, the pathway through which TAK1 is activated in hepatocytes in vivo has not yet been determined.
TAK1 binding partner proteins, TAK1 binding proteins 1, 2 and 3 (TAB1, TAB2 and TAB3), mediate activation of TAK1 through two different mechanisms. TAB1 mediates TAK1 oligomerization to induce autophosphorylation of TAK1 within the activation loop in the protein kinase domain, which catalytically activates TAK1 [13], [21], [22]. TAB2 and TAB3 are related proteins, both of which bind to polyubiquitin chains and recruit TAK1 to the polyubiquitin chain protein complexes [23]–[27]. Polyubiquitin-mediated oligomerization of TAK1 proteins facilitates TAK1 protein kinase activation. The TAB1-dependent mechanism is required for osmotic stress-induced TAK1 activation [13] and the basal TAK1 activity in the epidermis in vivo [28]. The polyubiquitin-mediated mechanism activates TAK1 in response to TNF, IL-1 and TLR ligands. Although both TAB2 and TAB3 can mediate the polyubiquitin mechanism, TAB2 plays a predominant role in TAK1 activation in several cell types including endothelial and immune cells [17], [23]. Double deficiency of Tab1 and Tab2 abolished TAK1 activity in the epidermis in vivo, which caused TNF-induced cell death and tissue damage resembling the phenotypes of TAK1 deficiency [28]. However, single deletion of either Tab1 or Tab2 does not cause any abnormalities in the epidermis, demonstrating that TAB1 and TAB2 are functionally redundant in the epidermis. Overall, TAB1 and TAB2 are major activators of TAK1 in several cell types, and contributions of TAB1- and TAB2-dependent mechanisms to TAK1 activity vary depending on cell types. In the current study, we investigated the role of TAB2 in hepatocytes and determined that TAB2 activates TAK1 in response to a TLR ligand lipopolysaccharide (LPS), which is essential for hepatocyte survival.
Materials and Methods
Mice
Tak1-floxed (Tak1flox/flox), Tab2-floxed (Tab2flox/flox), Tab1-floxed (Tab1flox/flox), mice were previously described [29]–[31]. Rosa26-CreERT [32] and Alb-Cre [33] transgenic mice were obtained from the Jackson Laboratories. All strains used were on a C57BL/6 background. Induction of Tab2 deletion in Rosa-CreER Tab2flox/flox was achieved by intraperitoneal (ip) injection of tamoxifen (50 mg/kg mouse weight) for 3 consecutive days. The tamoxifen injected mice were used for the experiments 3–6 weeks post injection to avoid potential acute toxicity derived from the transient Cre expression as described previously [34]. All animal experiments were conducted with the approval of the North Carolina State University Institutional Animal Care and Use Committee (IACUC protocol # 11-138B). All efforts were made to minimize animal suffering.
Reagents
Reagents used were tamoxifen (MP Biomedicals) and 4-hydroxytamoxifen (Sigma) LPS (From Salmonella minnesota, phenol extraction, Sigma) and diethylnitrosamine (DEN) (Sigma). Polyclonal antibodies used were TAK1, TAB1, and TAB2 described previously [14], [35], phosphorylated TAK1, cleaved caspase 3, phosphorylated JNK, JNK (Cell signaling), β-actin (Sigma-Aldrich), p65 and α-tubulin (Santa Cruz). An ALT assay kit (Biovision, Mountain View, CA) was used.
Primary Hepatocyte Culture
Primary hepatocytes were isolated with a standard collagenase procedure according to the instructions of Amaxa Biosystems (Amaxa, Gaithersburg, MD). Hepatocyte viability was assessed by the trypan blue dye assay, and only hepatocytes with 90% and above viability were used. Hepatocytes were plated on collagen-coated 60-mm dishes for protein isolation or 12-well plates for caspase 3 assay. Since Tak1 deficiency severely damages the liver even within one month of mouse age, we utilized an inducible deletion system to prepare Tak1+/+, Tak1 Δ/Δ, Tab1Tab2+/+ and Tab1Tab2DKO hepatocytes. Rosa-CreER Tak1flox/flox, Rosa-CreER Tab1flox/floxTab2flox/flox hepatocytes were immediately treated with 0.1 µM 4-hydroxytamoxifen or vehicle (ethanol), and the medium containing 4-hydroxytamoxifen or vehicle was replaced every 24 h for 72 h and subsequently stimulated with 1 µg/ml LPS. To prepare Tab2-deficient hepatocytes, we used hepatocyte-specific Tab2-deficient and control littermate mice. Alb-Cre Tab2flox/flox (Tab2-/-) and Tab2flox/flox (Tab2+/+) hepatocytes were cultured overnight and simulated with 1 µg/ml LPS.
Quantitative Real Time PCR Analysis
Total RNA was isolated from the liver using an RNeasy kit (Qiagen) and transcribed into cDNA using SuperScript VILO cDNA Synthesis Kit (Life Technologies). Expression levels of Tak1 and Tab2 were determined by quantitative real time PCR (qPCR) and normalized to the level of Gapdh. The following primers were used: Tab2-forward, 5′-GGATAGAATAAGCGAAGCCCGGAA-3′; Tab2-reverse, 5′-CTCTTTGAAGCCGTTCCATCCT-3′; Gapdh-forward, 5′-GAAGGTCGCTGTGAACGGA-3′; and Gapdh-forward, 5′-GTTAGTGGGGTCTCGCTCCT-3′. Primers for the Collagen type1 α1 (Col1a1), tissue inhibitor of metalloproteinase 1 (Timp1) and H19 genes were generated according to a previous report [19]. Cxcl2 and Ccl2 mRNAs were detected by using the TaqMan® system (Life Technologies)
Immunoblotting
Cell extracts were prepared using a lysis buffer (20 mM HEPES (pH 7.4), 150 mM NaCl, 12.5 mM β-glycerophosphate, 100 nM calyculin A, 1.5 mM MgCl2, 2 mM EGTA, 10 mM NaF, 2 mM DTT, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 20 µM aprotinin, and 0.5% Triton X-100). Liver extracts were prepared in the abovementioned lysis buffer containing a protease inhibitor cocktail (G-Biosciences, St Louis, MO). Proteins were resolved on SDS-PAGE and transferred to Hybond-ECL or Hybond-P membranes (GE Healthcare). The membranes were immunoblotted with various antibodies, and the bound antibodies were visualized with horseradish peroxidase-conjugated antibodies against rabbit or mouse IgG using the ECL or ECL advance Western blotting detection kit (GE Healthcare).
Cell Death Assays
Terminal dUTP nick-end labeling (TUNEL) assay was performed on formalin-fixed paraffin sections using an apoptotic cell death detection kit (Promega) according to the manufacturer's instructions. Seven to ten immunofluorescent images per mouse were randomly photographed, and at least 2000 DAPI-stained cells per mouse were counted. Quantitative results were generated from the counted numbers in 3-4 mice from independent experiments. 50 µg proteins from liver or hepatocyte extracts were used for Caspase 3 assay (Promega).
Electrophoresis Mobility Shift Assay (EMSA)
The binding mixture contained radiolabeled 32P-NF-κB oligonucleotide probe (Promega), 10 µg of cell extracts, 4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, 10 mM Tris-HCl (pH 7.5), 500 ng of poly (dI-dC) (GE Healthcare), and 10 µg of bovine serum albumin to a final volume of 15 µl. The reaction mixture was incubated at 25°C for 30 min, separated by 5% (w/v) polyacrylamide gel, and visualized by autoradiography.
Statistical Analysis
Results are expressed as either mean ± standard error of the mean (SEM) or mean ± standard deviation (SD). Differences between two groups were determined by two-tailed unpaired Student t test. Probability values of P are shown in figures. P≤0.05 was considered statistically significant.
Results
LPS Activates TAK1 through TAB2 in Primary Hepatocytes
Ablation of TAK1 in hepatocytes is known to spontaneously induce apoptosis in mouse liver (19,20). We hypothesized that TAK1 is activated through TAB1- and/or TAB2-dependent mechanisms by stressors and prevents stressor-induced apoptosis in the liver. To begin to test this hypothesis, we investigated whether stressors can activate TAK1 in hepatocytes. We found that LPS activates TAK1 in primary hepatocytes. TAK1 was activated and the activity peaked at 20–60 min following LPS challenge in primary hepatocytes (Fig. 1A). To determine whether TAB1 and/or TAB2 participate in LPS-induced TAK1 activation, we utilized primary hepatocytes having double deficiency of Tab1 and Tab2, and examined LPS-induced TAK1 activation. Tab1 and Tab2 double-deficient primary hepatocytes failed to activate TAK1 following LPS stimulation (Fig. 1B), demonstrating that TAB1- and/or TAB2-dependent mechanisms participate in the LPS signaling pathway. Furthermore, we found that Tab2 single deficient primary hepatocytes failed to activate TAK1 following LPS stimulation (Fig. 1C). The time course and the level of TAK1 activation upon LPS challenge were slightly varied on each primary preparation, but activation of TAK1 was always detected in wild type hepatocytes. In contrast, Tak1-, Tab1/Tab2 double- or Tab2 single-deficient hepatocytes did not exhibit any increase of TAK1 activity in all preparations. These results suggest that a bacterial moiety LPS activates TAK1 through the TAB2-dependent mechanism in hepatocytes. We also examined whether NF-κB activation, which is a well-known downstream target of TAK1, is altered by Tak1 or Tab2 single deletion in hepatocytes. We found that Tak1 deletion moderately reduced LPS-induced NF-κB activation in hepatocytes, suggesting that LPS activates NF-κB through multiple pathways including TAK1 pathway in hepatocytes (Fig. S1A). Similarly, Tab2 deletion partially reduced LPS-induced NF-κB activation (Fig. S1B). These results demonstrate that TAB2 is likely to function as an activator of TAK1 in response to LPS in hepatocytes, which may be important for prevention of liver injury. The LPS-TLR4 pathway has been reported to engage apoptosis and necrosis [36]–[38]. To begin to determine whether TAB2 is important for liver integrity, we asked whether deletion of Tab2 enhances LPS-induced cell death signaling. We examined caspase activity in LPS-treated Tab2-deficient primary hepatocytes. Tab2-deficient hepatocytes possessed slightly higher activity of caspase 3 compared to littermate control hepatocytes under unchallenged conditions, and caspase 3 activity was moderately but significantly upregulated by LPS treatment in Tab2-deficient hepatocytes (Fig. 1D). However, we did not observe any detectable increase in cell death in both wild type and Tab2-deficient hepatocytes following LPS stimulation. The weak activation of caspase 3 may be insufficient to induce cell death in Tab2-deficient hepatocytes. Thus, TAB2-dependent TAK1 activation is at least preventive to LPS-induced caspase 3 activation, and LPS is not a strong inducer of cell death in cultured hepatocytes. However, LPS is a strong inducer of liver injury in vivo, which is mediated not only through its direct effect on hepatocytes but through indirect stress through inflammatory responses. We speculate that that TAB2-dependent activation of TAK1 may be important for prevention of LPS-induced liver injury in vivo.
Figure 1. LPS activates TAK1 through TAB2 in primary hepatocytes.

Tak1- (A), Tab1 and Tab2 double- (B) or Tab2- (C and D) deficient and control primary hepatocytes were prepared as described in Material and Methods, and stimulated with 1 µg/ml LPS. Cell lysates were analyzed by immunoblotting. In these Tak1-floxed mice, Cre-mediated recombination resulted in deletion of 38 amino acids of TAK1, and the truncated TAK1 (TAK1Δ) was expressed at a low level presumably due to reduced protein stability as indicated in A. Asterisks indicate non-specific bands. (D) Tab2-deficient and control primary hepatocytes were stimulated with 1 µg/ml LPS for 24 h. Cell lysates were subjected to caspase 3 assay. Data are shown as means ± SD, n = 3.
TAB2 Knockout Mice Are Hypersensitive to LPS-Induced Liver Injury
To examine whether TAB2 plays a protective role in the liver in vivo, we generated TAB2-/- adult mice by using a ubiquitously expressing inducible Cre deleter strain, Rosa26.CreERT [32]. Tab2 deletion was induced by intraperitoneal injection of the CreERT activator, tamoxifen, and Tab2 mRNA and protein were greatly diminished at 3 weeks after tamoxifen injection (Fig. 2A). Although germline deletion of Tab2 is known to cause embryonic lethality around embryonic days 13-14 due to liver degeneration [31], Tab2 deletion in adult mice did not cause overt liver abnormalities. This suggests that TAB2 plays an indispensable role during embryogenesis which is not required for adult liver homeostasis at least under unchallenged conditions. We asked whether Tab2 deletion sensitizes the liver to LPS-induced injury in adult mice. Tab2 deletion alone did not induce cell death in the liver, but LPS challenge greatly upregulated caspase activity in Tab2-deficient liver but not in the control within 6 h (Figs. 2B and 2C). TUNEL-positive cells with LPS challenge were highly increased by Tab2-deficiency (Figs. 2D and 2E). JNK activity was upregulated in Tab2-deficient liver (Fig. 2F), which is known to be associated with liver damage [8] and has been also observed in Tak1-deficient liver in earlier studies [19], [20]. We note that, because Tab2-deficient liver was greatly damaged within 6 h post-LPS injection, we were not able to analyze the consequences beyond 6 h. These results demonstrate that TAB2 is indispensable in the protection of the liver from LPS-induced cell death.
Figure 2. LPS induces liver damage in Tab2-deficient mice.

(A–C) Adult Tab2+/+ and Tab2-/- mice were generated from Tab2flox/flox and Rosa-CreER Tab2flox/flox mice by ip injection of tamoxifen as described in Experimental Procedures. (A) Tab2 expression in the liver was determined by real-time PCR and immunoblotting at more than 3 weeks after tamoxifen injection. Tab2+/+ mice n = 5; Tab2-/- mice n = 6. Means ± SEM and P values are shown. β-actin is shown as a loading control. Arrow indicates the position of TAB2. (B, C) Tab2-/ - and control littermate or age matched mice were treated by ip injection of 8 mg/kg mouse weight LPS or vehicle (PBS) for 6 h, and the liver was isolated and protein extracts were subjected to caspase 3 assay (B) and analyzed by immunoblotting with anti-cleaved caspase 3 (C). Number of mice; untreated mice n = 3; LPS-treated Tab2+/+ mice n = 4, Tab2-/- mice n = 4. Means ± SEM and P values are shown (B). (D, E) Liver sections of samples described in B and C were analyzed by TUNEL staining (D). Scale bars, 20 µm. TUNEL-positive cells per more than 2000 total liver cells per mouse were counted (E). Number of mice; untreated mice n = 2; LPS-treated Tab2+/+ mice n = 3, Tab2-/- mice n = 3. Means ± SEM and P values are shown. (F) Liver proteins described in B and C were analyzed by immunoblotting with anti-phospho-JNK and anti-JNK. Each lane in C and F represents proteins from one animal.
Hepatocyte-Specific Deletion of Tab2 Induces Late Onset Liver Damage and Sensitizes Mice to LPS-Induced Liver Injury
To examine whether hepatocyte-derived TAB2 is responsible for the protection against stress-induced liver injury, we generated hepatocyte-specific Tab2-deficient (Tab2HepKO) mice by using hepatocyte-specific deleter, alb.Cre, transgenic mice [33]. The Tab2 expression in the liver but not lung, brain or heart extract was greatly diminished in the Tab2HepKO mice (Fig. 3A). We also generated Tak1HepKO mice, which are known to develop hepatocellular carcinoma [19], [20]. Tak1HepKO mice exhibited signs of highly increased liver damage including increased levels of serum aminotransferase (ALT), expression of chemokine (C-X-C motif) ligand 2 (Cxcl2), chemokine (C-C motif) ligand 2 (Ccl2), fibrosis markers, collagen, type I, α1 (Col1a1) and tissue inhibitor of metalloproteinase 1 (Timp1) as well as hepatocellular carcinoma marker, H19 gene within one month even in the absence of any exogenous stressors (Fig. S2), consistent with earlier studies. In contrast, Tab2HepKO mice developed normally and did not exhibit increased ALT and only marginal or moderate increases in inflammatory chemokine expression in the absence of exogenous stressors as shown in later in Fig. 4. Tab2HepKO liver was histologically indistinguishable from control littermate liver at least at several months of age (Fig. 3B). The lobular architecture was clear in both control and Tab2HepKO liver, although small foci of immune cell infiltration were occasionally observed in all genotypes (Fig. 3B). Aged Tab2HepKO liver exhibited moderately disorganized lobular architectures with relatively large infiltration foci as well as cytoplasmic eosinophilic materials (dashed line) around 10–12 months old of age, while Tak1HepKO showed severe structural damage including unclear lobular architecture in addition to immune cell infiltrations and profound increase of eosinophilic materials (Fig. 3C). Aged Tab2HepKO liver exhibited signs of fibrosis including increased Sirius Red staining, although it is much milder than that in Tak1HepKO (Fig. 3D). Aged Tab2HepKO liver expressed increased fibrosis marker genes, Col1a1 and Timp1 (Fig. 3E). These suggest that TAK1 basal activity but not TAB2-dependent TAK1 activation is required for liver integrity at least for several months, and that TAB2 may be important for long-term liver integrity by preventing accumulation of stress-induced liver lesions. To test acute stress-induced liver injury, Tab2HepKO mice were treated with LPS for 24 h. Caspase activity was upregulated in Tab2HepKO liver compared to control mice at 24 h post LPS injection (Fig. 3F); however, the levels of caspase activation were lower compared with those in the ubiquitous Tab2-deficient liver described above (see Fig. 2B). This suggests that Tab2 deficiency highly elevates liver injury also through cell types other than hepatocytes, but that hepatocyte-derived TAB2 is still at least in part responsible for liver protection under the LPS-induced stress condition.
Figure 3. Hepatocyte-specific deletion of Tab2 induces late-onset fibrosis and sensitizes the liver to LPS-induced injury.

(A) Tab2 expression in the liver, lung, brain, and heart was determined by real-time PCR (liver) and immunoblotting (liver, lung, brain and heart) in hepatocyte-specific Tab2-deficient, Alb-Cre Tab2flox/flox (Tab2HepKO), and littermate or age matched control, Tab2flox/flox (wild type), mice. Wild type mice, n = 10; Tab2HepKO mice, n = 8. Means ± SEM and P values are shown. β-actin or α-tubulin is shown as a loading control. Arrows indicate the position of TAB2. (B) Hematoxylin and eosin staining of 2-month-old controls, including wild type and Tab2 heterozygous deletion, Alb-Cre Tab2flox/+ (Tab2HepHet), and Tab2HepKO liver sections. Scale bars, 200 µm. (C) Liver sections of 10- to 12-month-old control Tab2HepHet, Tab2HepKO and Tak1-deficient (Tak1HepKO) mice. Left panels show overall structure of liver. Arrows indicate immune cell infiltration, and dashed line areas contain highly eosinophilic cells. Scale bars, 200 µm (left panels), 40 µm (right panels). (D) Sirius Red staining of 12-month-old control, Tab2HepKO, and Tak1HepKO liver sections. Scale bars, 200 µm. (E) Fibrosis marker mRNA levels were determined by real-time PCR. Wild type mice, n = 6; Tab2HepKO mice, n = 3. Means ± SEM and P values are shown. (F) 2- to 3-month-old Tab2-deficient and control mice were treated with 10 mg/kg mouse weight LPS for 24 h. Protein extracts from the liver were subjected to caspase 3 assay. Wild type mice, n = 5; Tab2HepKO mice, n = 7. Means ± SEM and P values are shown.
Figure 4. Hepatocyte-specific deletion of Tab2 sensitizes the liver to diethylnitrosamine (DEN)-induced liver injury.

(A) 2- to 4-month-old Tab2HepKO and control mice were treated with 100 mg/kg DEN or vehicle (PBS) for 24 h. Liver sections were analyzed by TUNEL staining. Scale bars, 50 µm. TUNEL-positive cells per at least 2000 total liver cells per sample were counted (a graph shown below TUNEL image panels). Controls including wild type and Tab2HepHet mice, n = 4; Tab2HepKO mice, n = 4. Means ± SEM and P values are shown. (B) ALT assay. Controls including wild type and Tab2HepHet mice, n = 7; Tab2HepKO mice, n = 5. Means ± SEM and P values are shown. (C) Expression levels of inflammatory chemokines, Cxcl2 and Ccl2, were determined by real time PCR. Controls including wild type and Tab2HepHet mice, n = 24; Tab2HepKO mice, n = 12. Means ± SEM and P values are shown. (D) Liver from 2- to 4-month-old controls, Tab1 flox/flox and Alb-Cre Tab1 flox/+ and Tab1HepKO mice were analyzed. ALT assay. Control mice, n = 6; Tab1HepKO mice, n = 11. Expression levels of inflammatory chemokines, Cxcl2 and Ccl2, mRNA levels were determined by real time PCR. Controls, mice, n = 4; Tab1HepKO mice, n = 4. Means ± SEM and P values are shown.
Hepatocyte-Specific Deletion of Tab2 Sensitizes Mice to Diethylnitrosamine (DEN)-Induced Liver Injury
To further determine the hepatocyte-specific role of TAB2 in stress protection, we utilized a strong liver carcinogen diethylnitrosamine (DEN), which is activated by cytochrome P450 within hepatocytes and induces oxidative stress [39], [40]. DEN-induced stress causes hepatocellular carcinoma in the long term, and acute liver injury in the short term [41]. Tab2HepKO and control mice were treated with DEN for 24 h. We observed highly increased TUNEL-positive cells in Tab2HepKO liver (Fig. 4A) and elevation of serum ALT (Fig. 4B), indicating profound liver injury. Furthermore, the expression of inflammatory genes, Cxcl2 and Ccl2, was elevated in Tab2HepKO liver (Fig. 4C). These results show that hepatocyte-derived TAB2 is required for liver protection from DEN-derived stress. As described earlier, Tab2 deletion alone in primary hepatocytes diminished TAK1 activation upon LPS challenge (Fig. 1C). However, other stressors may also utilize the TAB1-dependent pathway to activate the TAK1-cell protection pathway. We then examined whether Tab1-deficiency sensitizes the liver to DEN. To this end, we generated hepatocyte-specific Tab1-deficient (Tab1HepKO) mice using the same system as Tab2HepKO. Tab1HepKO mice did not exhibit any overt abnormality. Tab1HepKO mice were treated with DEN for 24 h, but did not exhibit statistically significant increases in ALT or in the expression levels of Cxcl2 and Ccl2 (Fig. 4E). Thus, TAB1 minimally, if any, contributes to liver protection, whereas TAB2 is indispensable for the protection against stress-induced liver injury.
Discussion
Tak1 deficiency in hepatocytes causes spontaneous liver injury which further engages hepatocellular carcinoma development [19], [20]. A previous study in Tak1 deficient liver used a mouse model that expresses a truncated version of TAK1, which has 38 amino acids deleted around the ATP binding site of the protein kinase domain but the other domains intact [19], [30]. Thus, TAK1 kinase activity but not TAK1 protein itself is important for hepatocyte survival. Accordingly, it is reasonable to assume that TAK1 is active to some extent in the liver under homeostatic conditions, and constitutively upregulates pro-survival factors such as cIAP and c-Jun [15], [16], and anti-oxidants such as Nrf2 and NAD(P)H:quinone oxidoreductase 1 [18], [42]. Earlier studies have established that this TAK1 pro-survival signaling pathway is indispensable for liver protection [19], [20]. In the current study, we found that a TAK1 activator protein, TAB2, is indispensable for stressor-induced TAK1 activation in hepatocytes, and that TAB2 is required for prevention of hepatocyte death in response to stressor challenge in vivo. We propose that TAK1 activity is required for prevention of hepatocyte cell death under basal conditions, and that, under stressed conditions, TAK1 is activated beyond the basal level through TAB2, which is required for liver protection against stressors as summarized in Fig. 5.
Figure 5. Model.
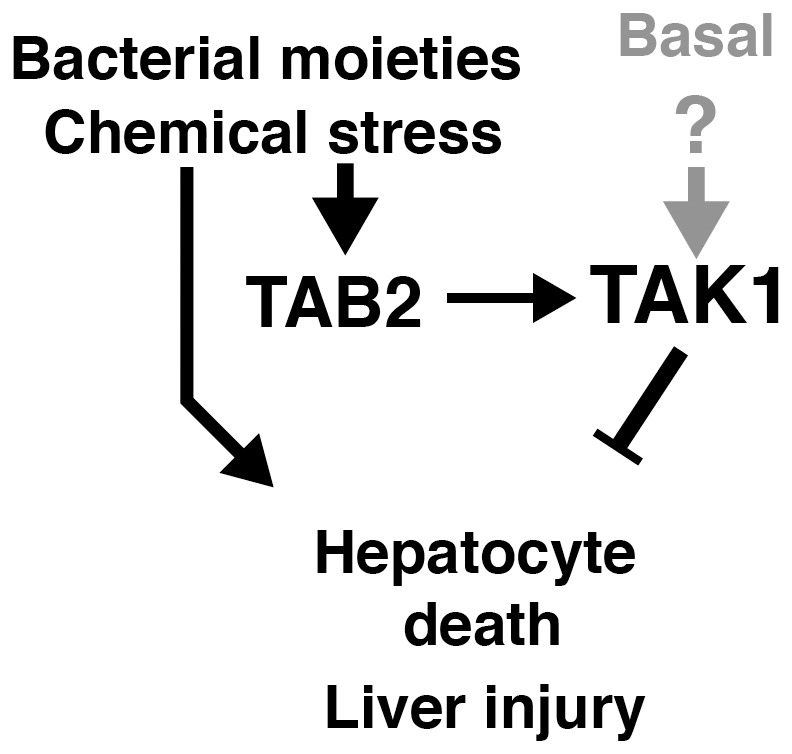
As shown in Fig. 3, hepatocyte-specific Tab2 deletion did not cause any abnormality at least for several months, whereas Tak1 deficiency induced profound liver injury and even tumors within 4 weeks of age (Fig. S2) in agreement with the earlier studies [19], [20]. Thus, TAB2 is not required for the basal TAK1 activity to protect hepatocytes under the unstressed condition. TAB2 is known to play a major role in cytokine and Toll-like receptor (TLR)-induced TAK1 activation through a polyubiquitin-dependent mechanism [24]. Hence, the cytokine- or TLR ligand-induced pathway is not essential for basal hepatic TAK1 activity. How is TAK1 activated in the absence of TAB2? Another TAK1 binding partner, TAB1, can activate TAK1 through promoting oligomerization of TAK1 [13], [21]. TAB1 has been identified to be essential for maintenance of TAK1 activity in the epidermis [28]. We show that Tab1 deficiency greatly reduces TAK1 activity in the epidermis. TAB1 may be responsible for the basal TAK1 activity in the liver, too. However, Tab1 deletion alone does not cause any abnormalities in the liver. Thus, Tab1 is not essential as is Tab2 for hepatocyte survival in vivo. TAB3 can mediate polyubiquitin-dependent activation of TAK1, but Tab3 deletion alone does not cause any abnormalities during embryogenesis or in adult mice [23]. Collectively, it is likely that TAB1 and TAB2 and possibly TAB3 redundantly function to activate TAK1 under the basal conditions. Thus, single deletion of Tab1, Tab2 or Tab3 does not cause spontaneous liver injury.
Blockade of TAK1 signaling is a powerful method to kill cells in vivo. However, since the TAK1-mediated cell survival pathway is indispensable for liver integrity, TAK1 is not an ideal target to remove undesired hepatocytes such as tumor cells. Tak1 deletion even causes hepatocellular carcinoma [19], [20]. Tab2 deficiency alone does not cause any abnormality in unstressed liver at least for several months in the mouse model. Therefore, inhibition of TAB2 would not cause acute liver injury in normal liver. Our results suggest that TAB2-dependent activation of TAK1 beyond the basal level is required for hepatocyte survival only when hepatocytes are under stressed conditions. Thus, we anticipate that inhibition of TAB2 might selectively kill stressed hepatocytes such as hepatocellular carcinoma cells. TAB2 could be a new target to kill stressed or damaged hepatocytes without affecting unstressed cells.
Supporting Information
LPS-induced NF-κB activation is reduced by Tak1 or Tab2 deficiency in primary hepatocytes.
(TIF)
Tak1 deficiency induces spontaneous liver injury within one month.
(TIF)
Acknowledgments
We thank S. Akira for Tak1-floxed and Tab2-floxed mice, and S. Elliott, L. Hester and M. Mattmuler for support.
Funding Statement
This work was supported by National Institutes of Health Grant GM068812 (to JN-T). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, et al. (2007) TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med 13: 1324–1332. [DOI] [PubMed] [Google Scholar]
- 2. Szabo G, Dolganiuc A, Mandrekar P (2006) Pattern recognition receptors: a contemporary view on liver diseases. Hepatology 44: 287–298. [DOI] [PubMed] [Google Scholar]
- 3. He G, Yu GY, Temkin V, Ogata H, Kuntzen C, et al. (2010) Hepatocyte IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 17: 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karin M (2006) Nuclear factor-κB in cancer development and progression. Nature 441: 431–436. [DOI] [PubMed] [Google Scholar]
- 5. Das M, Garlick DS, Greiner DL, Davis RJ (2011) The role of JNK in the development of hepatocellular carcinoma. Genes Dev 25: 634–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maeda S, Kamata H, Luo JL, Leffert H, Karin M (2005) IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 121: 977–990. [DOI] [PubMed] [Google Scholar]
- 7. Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF (2008) Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest 118: 3943–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sakurai T, Maeda S, Chang L, Karin M (2006) Loss of hepatic NF-κ B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci U S A 103: 10544–10551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baldwin AS (2012) Regulation of cell death and autophagy by IKK and NF-κB: critical mechanisms in immune function and cancer. Immunol Rev 246: 327–345. [DOI] [PubMed] [Google Scholar]
- 10. Li F, Sethi G (2010) Targeting transcription factor NF-κB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta 1805: 167–180. [DOI] [PubMed] [Google Scholar]
- 11. Pasparakis M (2009) Regulation of tissue homeostasis by NF-κB signalling: implications for inflammatory diseases. Nat Rev Immunol 9: 778–788. [DOI] [PubMed] [Google Scholar]
- 12. Hayden MS, Ghosh S (2008) Shared principles in NF-κB signaling. Cell 132: 344–362. [DOI] [PubMed] [Google Scholar]
- 13. Inagaki M, Omori E, Kim JY, Komatsu Y, Scott G, et al. (2008) TAK1-binding protein 1, TAB1, mediates osmotic stress-induced TAK1 activation but is dispensable for TAK1-mediated cytokine signaling. J Biol Chem 283: 33080–33086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, et al. (1999) The kinase TAK1 can activate the NIK-IκB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398: 252–256. [DOI] [PubMed] [Google Scholar]
- 15. Morioka S, Omori E, Kajino T, Kajino-Sakamoto R, Matsumoto K, et al. (2009) TAK1 kinase determines TRAIL sensitivity by modulating reactive oxygen species and cIAP. Oncogene 28: 2257–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Omori E, Morioka S, Matsumoto K, Ninomiya-Tsuji J (2008) TAK1 regulates reactive oxygen species and cell death in keratinocytes, which Is essential for skin integrity. J Biol Chem 283: 26161–26168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morioka S, Inagaki M, Komatsu Y, Mishina Y, Matsumoto K, et al. (2012) TAK1 kinase signaling regulates embryonic angiogenesis by modulating endothelial cell survival and migration. Blood 120: 3846–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kajino-Sakamoto R, Omori E, Nighot PK, Blikslager AT, Matsumoto K, et al. (2010) TGF-β-activated kinase 1 signaling maintains intestinal integrity by preventing accumulation of reactive oxygen species in the intestinal epithelium. J Immunol 185: 4729–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, et al. (2010) Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci U S A 107: 844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bettermann K, Vucur M, Haybaeck J, Koppe C, Janssen J, et al. (2010) TAK1 suppresses a NEMO-dependent but NF-κB-independent pathway to liver cancer. Cancer Cell 17: 481–496. [DOI] [PubMed] [Google Scholar]
- 21. Scholz R, Sidler CL, Thali RF, Winssinger N, Cheung PC, et al. (2010) Autoactivation of transforming growth factor β-activated kinase 1 is a sequential bimolecular process. J Biol Chem 285: 25753–25766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kishimoto K, Matsumoto K, Ninomiya-Tsuji J (2000) TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J Biol Chem 275: 7359–7364. [DOI] [PubMed] [Google Scholar]
- 23. Ori D, Kato H, Sanjo H, Tartey S, Mino T, et al. (2013) Essential roles of K63-linked polyubiquitin-binding proteins TAB2 and TAB3 in B cell activation via MAPKs. J Immunol 190: 4037–4045. [DOI] [PubMed] [Google Scholar]
- 24. Chen ZJ (2012) Ubiquitination in signaling to and activation of IKK. Immunol Rev 246: 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Broglie P, Matsumoto K, Akira S, Brautigan DL, Ninomiya-Tsuji J (2010) Transforming growth factor β-activated kinase 1 (TAK1) kinase adaptor, TAK1-binding protein 2, plays dual roles in TAK1 signaling by recruiting both an activator and an inhibitor of TAK1 kinase in tumor necrosis factor signaling pathway. J Biol Chem 285: 2333–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xia ZP, Sun L, Chen X, Pineda G, Jiang X, et al. (2009) Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 461: 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanayama A, Seth RB, Sun L, Ea CK, Hong M, et al. (2004) TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell 15: 535–548. [DOI] [PubMed] [Google Scholar]
- 28. Omori E, Inagaki M, Mishina Y, Matsumoto K, Ninomiya-Tsuji J (2012) Epithelial transforming growth factor β-activated kinase 1 (TAK1) is activated through two independent mechanisms and regulates reactive oxygen species. Proc Natl Acad Sci USA 109: 3365–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Inagaki M, Komatsu Y, Scott G, Yamada G, Ray M, et al. (2008) Generation of a conditional mutant allele for Tab1 in mouse. Genesis 46: 431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, et al. (2005) Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 6: 1087–1095. [DOI] [PubMed] [Google Scholar]
- 31. Sanjo H, Takeda K, Tsujimura T, Ninomiya-Tsuji J, Matsumoto K, et al. (2003) TAB2 is essential for prevention of apoptosis in fetal liver but not for interleukin-1 signaling. Mol Cell Biol 23: 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Badea TC, Wang Y, Nathans J (2003) A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J Neurosci 23: 2314–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, et al. (1999) Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274: 305–315. [DOI] [PubMed] [Google Scholar]
- 34. Takaesu G, Inagaki M, Takubo K, Mishina Y, Hess PR, et al. (2012) TAK1 (MAP3K7) signaling regulates hematopoietic stem cells through TNF-dependent and -independent mechanisms. PLoS ONE 7: e51073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, et al. (2000) TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell 5: 649–658. [DOI] [PubMed] [Google Scholar]
- 36. Ma Y, Temkin V, Liu H, Pope RM (2005) NF-κB protects macrophages from lipopolysaccharide-induced cell death: the role of caspase 8 and receptor-interacting protein. J Biol Chem 280: 41827–41834. [DOI] [PubMed] [Google Scholar]
- 37. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, et al. (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325: 332–336. [DOI] [PubMed] [Google Scholar]
- 38. He S, Liang Y, Shao F, Wang X (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A 108: 20054–20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwabe RF, Brenner DA (2006) Mechanisms of Liver Injury. I. TNF-α-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol 290: G583–589. [DOI] [PubMed] [Google Scholar]
- 40. Chowdhury G, Calcutt MW, Guengerich FP (2010) Oxidation of N-Nitrosoalkylamines by human cytochrome P450 2A6: sequential oxidation to aldehydes and carboxylic acids and analysis of reaction steps. J Biol Chem 285: 8031–8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, et al. (2007) Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317: 121–124. [DOI] [PubMed] [Google Scholar]
- 42. Omori E, Matsumoto K, Zhu S, Smart RC, Ninomiya-Tsuji J (2010) Ablation of TAK1 upregulates reactive oxygen species and selectively kills tumor cells. Cancer Res 70: 8417–8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
LPS-induced NF-κB activation is reduced by Tak1 or Tab2 deficiency in primary hepatocytes.
(TIF)
Tak1 deficiency induces spontaneous liver injury within one month.
(TIF)