

Abstract
Abnormalities of lipid metabolism through overexpression of fatty acid synthase (FASN), which catalyzes the formation of long-chain fatty acids, are associated with the development of inflammatory bowel disease (IBD). C75 is a synthetic α-methylene-γ-butyrolactone compound that inhibits FASN activity. We hypothesized that C75 treatment could effectively reduce the severity of experimental colitis. Male C57BL/6 mice were fed 4% dextran sodium sulfate (DSS) for 7 d. C75 (5 mg/kg body weight) or dimethyl sulfoxide (DMSO) (vehicle) was administered intraperitoneally from d 2 to 6. Clinical parameters were monitored daily. Mice were euthanized on d 8 for histological evaluation and measurements of colon length, chemokine, cytokine and inflammatory mediator expression. C75 significantly reduced body weight loss from 23% to 15% on d 8, compared with the vehicle group. The fecal bleeding, diarrhea and colon histological damage scores in the C75-treated group were significantly lower than scores in the vehicle animals. Colon shortening was significantly improved after C75 treatment. C75 protected colon tissues from DSS-induced apoptosis by inhibiting caspase-3 activity. Macrophage inflammatory protein 2, keratinocyte-derived chemokine, myeloperoxidase activity and proinflammatory cytokines (tumor necrosis factor-α, interleukin [IL]-1β and IL-6) in the colon were significantly downregulated in the C75-treated group, compared with the vehicle group. Treatment with C75 in colitis mice inhibited the elevation of FASN, cyclooxygenase-2 and inducible nitric oxide synthase expression as well as IκB degradation in colon tissues. C75 administration alleviates the severity of colon damage and inhibits the activation of inflammatory pathways in DSS-induced colitis. Thus, inhibition of FASN may represent an attractive therapeutic potential for treating IBD.
INTRODUCTION
Inflammatory bowel disease (IBD), which encompasses ulcerative colitis (UC) and Crohn’s disease (CD), consists of chronic and relapsing immune responses and inflammation in the gastrointestinal tract (1). According to the Centers for Disease Control and Prevention, IBD is one of the top five prevalent gastrointestinal disease burdens in the United States, which accounts for >700,000 physician visits, 100,000 hospitalizations and disability in 119,000 patients each year (2). Over the long term, up to 75% of patients with CD and 25% of patients with UC require surgery (3). IBD is thought to result from an inappropriate and continuing inflammatory response to the commensal microbes in a genetically susceptible host. Recent progress in the understanding of IBD pathobiology indicates an elevated production of inflammatory mediators to induce inflammation, as well as tissue injury due to the migration and infiltration of hyperactive innate and adaptive immune cells (1). Proinflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6, as well as a potent chemokine IL-8 for attracting infiltration of activated leukocytes are under the control of nuclear factor (NF)-κB and mitogen-activated protein kinase (MAPK) to cause a variety of inflammatory events during colitis (4). Oxygen-derived free radicals and reactive oxygen species (ROS) have also emerged as a common pathway of tissue injury and inflammation in IBD (5). This scenario has given rise to the hope that efforts directed toward the pharmacologic control of free radical–mediated tissue injury may have a particular application toward patients suffering from IBD (6). Taken together, suppressing the inflammatory and ROS pathways will be a rational strategy to alleviate IBD.
Fatty acids (FAs) are aliphatic acids required for the production and storage of energy in the form of ATP to maintain cellular structure, as well as in the biosynthesis of hormones and other biologically active molecules (7). Free or unesterified FAs are ubiquitous in all living tissues and are unbound to other molecules (especially albumin) (7). Recently, free FA has emerged as an important component in transmitting signals as ligands of either membrane receptors that are involved in intracellular signaling or as nuclear receptors that mediate gene regulation (8). Accumulation of FAs due to altered metabolism and/or unbalanced diet has been described to be toxic for several organs (9). In numerous cell types, cell death, cytokine secretion and activation of inflammatory processes appear to be consequences of FA accumulation (9). FAs are known to stimulate NF-κB and activator protein 1 for transcriptional activation that ultimately leads to enhanced levels of monocyte chemoattractant protein-1, vascular cell adhesion molecule-1, intercellular adhesion molecule-1 and TNF-α (10). FAs affect biological systems by stimulating the production of eicosanoids, ROS and reactive nitrogen species, as well as inducing cell death and tissue injury (11). Apart from that, a recent study reveals that saturated FAs activate toll-like receptor (TLR)-mediated upregulation of proinflammatory cytokine expression in macrophages via NF-κB and MAPK pathways (12).
Fatty acid synthase (FASN) is a lipogenic enzyme that catalyzes the condensation of acetyl-CoA and malonyl-CoA to generate long-chain FAs (13). FASN consists of two identical multi-functional polypeptides, each including seven catalytic domains (13). Because the generation of FAs by FASN is known to initiate several biochemical and immunological pathways that lead to inflammation, FASN could be an attractive target for novel antiinflammatory therapies. In support of this, overexpression of FASN was observed in patients with UC (14). C75 is a synthetic cell-permeable α-methylene-γ-butyrolactone compound that abrogates FASN activity and has been well studied for its anti-tumor activity (15,16). C75 interferes with the binding of malonyl-CoA to the β-ketoacyl synthase domain of FASN, thus inhibiting long-chain FA elongation (17). Herein, we hypothesized that C75, an FASN inhibitor, might play an important role in reducing the inflammatory consequences in IBD. On the basis of this hypothesis, we induced experimental colitis in mice by dextran sodium sulfate (DSS) and evaluated the efficacy of C75 treatment by monitoring several clinical symptoms. We then examined the effect of C75 treatment on tissue integrity, neutrophil infiltration and inflammatory responses to further elucidate the molecular mechanisms involved in attenuating the disease severity by C75.
MATERIALS AND METHODS
Experimental Model
Male C57BL/6 mice (12 wks old, 20–25 g) were obtained from Taconic (Albany, NY, USA) and randomly divided into three groups, consisting of sham, DSS and DSS with C75 treatment. To produce a DSS colitis model, mice were fed 4% DSS (molecular weight 36,000–50,000; MP Biomedical, Solon, OH, USA) in drinking water for 7 d, whereas the sham group received only normal drinking water throughout the experiment. Mice were administered 0.2 mL C75 (5 mg/kg BW) or dimethyl sulfoxide (DMSO) in saline (vehicle) by intraperitoneal injection once a day from d 2 to 6. The dose of C75 was on the basis of a previous study that has shown its effectiveness on mice (18). All experiments were performed in accordance with the guidelines for use of experimental animals by the National Institutes of Health (Bethesda, MD, USA) and were approved by the Institutional Animal Care and Use Committee of The Feinstein Institute of Medical Research (Manhasset, NY, USA).
Clinical Assessment
The physical and clinical parameters were monitored daily by visual inspection throughout the experiment. The diarrhea score (stool consistency) and fecal bleeding were recorded in a 0–3 scale (0 = normal; 1 = slightly loose stool or bloody; 2 = loose stool or bloody; and 3 = watery diarrhea or blood in whole feces). Disease activity index was determined by combining scores of stool consistency, fecal bleeding and weight loss. On d 8, mice were euthanized and the colon length was measured. Proximal, medial and distal parts of colon tissues were harvested in liquid nitrogen and stored at −80°C for further analysis. All the measurements presented in this study are from distal colon tissues.
Histopathological Examination
Colon tissues were fixed in 10% formalin and embedded in paraffin. Tissue blocks were sectioned at a thickness of 5 μm and stained with hematoxylin and eosin. Colonic epithelial damage was assigned scores in a blinded fashion, described as follows: 0 = normal; 1 = hyper-proliferation, irregular crypts and goblet cell loss; 2 = mild to moderate crypt loss (10–50%); 3 = severe crypt loss (50–90%); 4 = complete crypt loss, surface epithelium intact; 5 = small- to medium-sized ulcer (<10 crypt widths); and 6 = large ulcer (≥10 crypt widths). Infiltration with inflammatory cells was assigned scores separately for mucosa (0 = normal; 1 = mild; 2 = modest; and 3 = severe), submucosa (0 = normal; 1 = mild to modest; and 2 = severe), and muscle/serosa (0 = normal and 1 = moderate to severe). Scores for epithelial damage and inflammatory cell infiltration were added, resulting in a total scoring range of 0–12 (19).
Measurements of Cytokines, Malondialdehyde and Myeloperoxidase Activity
Colon tissue was homogenized in lysis buffer (10 mmol/L Tris-HCl, pH 7.5, 120 mmol/L NaCl, 1% NP-40, 1% sodium deoxycholate and 0.1% sodium dodecyl sulfate) with protease inhibitor (Roche Diagnostics, Indianapolis, IN, USA) by sonication. Protein concentration of the lysate was determined by the DC protein assay kit (Bio-Rad, Hercules, CA, USA). The levels of TNF-α, IL-6 and IL-1β in colon tissues were quantified by using specific mouse enzyme-linked immunosorbent assay (ELISA) kits from BD Biosciences (San Jose, CA, USA). Malondialdehyde (MDA) was measured by using 2-thiobarbituric acid reactive substances assay kit from Cayman (Ann Arbor, MI, USA). For measuring myeloperoxidase (MPO) activity, colon tissue was homogenized in KPO4 buffer containing 0.5% hexa-decyltrimethyl-ammonium bromide by sonication. After centrifuging, the supernatant was diluted in the reaction solution containing o-dianisidine hydrochloride and H2O2 in phosphate buffer. The rate of change in absorbance for 1 min was measured at 460 nm to calculate MPO activity.
Quantitative Real-Time Polymerase Chain Reaction
Total RNA was extracted from colon tissues by using a TRIzol reagent (Invitrogen/Life Technologies, Carlsbad, CA, USA) and was reverse transcribed into cDNA by using murine leukemia virus reverse transcriptase (RT) (Applied Biosystems/Life Technologies). A polymerase chain reaction (PCR) was carried out in 25 μL of a final volume containing 0.08 μmol of each forward and reverse primer, cDNA and 12.5 μL SYBR Green PCR Master Mix (Applied Biosystems/Life Technologies). Amplification was conducted in an Applied Biosystems 7300 real-time PCR machine (Applied Biosystems/Life Technologies) under the thermal profile of 50°C for 2 min and 95°C for 10 min, followed by 45 cycles of 95°C for 15 s and 60°C for 1 min. The level of mouse β-actin mRNA was used for normalization. Relative expression of mRNA was expressed as the fold-change in comparison with the sham. The primers used for this study are listed in Table 1.
Table 1.
Primer sequences used in this study.
| Name | GenBank | Forward | Reverse |
|---|---|---|---|
| MIP-2 | NM_009140 | CCCTGGTTCAGAAAATCATCCA | GCTCCTCCTTTCCAGGTCAGT |
| KC | NM_008176 | GCTGGGATTCACCTCAAGAA | ACAGGTGCCATCAGAGCAGT |
| TNF-α | X_02611 | AGACCCTCACACTCAGATCATCTTC | TTGCTACGACGTGGGCTACA |
| IL-6 | NM_031168 | CCGGAGAGGAGACTTCACAG | CAGAATTGCCATTGCACAAC |
| IL-1β | NM_008361 | CAGGATGAGGACATGAGCACC | CTCTGCAGACTCAAACTCCAC |
| COX-2 | NM_011198 | CTCAGCCAGGCAGCAAATC | ACATTCCCCACGGTTTTGAC |
| iNOS | NM_010927 | GCAGGTCGAGGACTATTTCTTTCA | GAGCACGCTGAGTACCTCATTG |
| β-actin | NM_007393 | CGTGAAAAGATGACCCAGATCA | TGGTACGACCAGAGGCATACAG |
Western Blotting
Colon tissue was homogenized in lysis buffer with protease inhibitor (Roche Diagnostics, Indianapolis, IN, USA) by sonication. Protein concentration of the lysate was determined by the DC protein assay kit. Equal amounts of protein from total lysate were fractionated on a 4–12% Bis-Tris gel (Invitrogen/Life Technologies) and transferred to nitrocellulose membranes. Membranes were blocked by phosphate-buffered saline with 0.1% casein and then incubated with anti-cleaved caspase-3 (Cell Signaling, Beverly, MA, USA), cyclooxygenase (COX)-2, inducible nitric oxide synthase (iNOS), IκB (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or β-actin (Sigma-Aldrich, St. Louis, MO, USA) antibody diluted in phosphate-buffered saline with 0.1% casein and 0.1% Tween-20. After washing, the membranes were incubated with fluorescently labeled secondary antibody. The membranes were scanned by an Odyssey imaging system (LI-COR, Lincoln, NE, USA) for detection and quantification.
Immunohistochemical Staining of FASN
Paraffin-embedded colon sections were dewaxed in xylene and rehydrated in a graded series of ethanol. Tissues were incubated in 0.92% citric acid buffer (Vector Laboratories, Burlingame, CA, USA) at 95°C for 15 min. After cooling to room temperature, the slides were incubated with 2% H2O2 in 60% methanol and blocked in 2% normal donkey serum/Tris-buffered saline, after which they were incubated with anti-FASN antibody (1:50; Cell Signaling) in 1% normal donkey serum/Tris-buffered saline with 0.02% Triton X-100 at 4°C overnight. The detection was carried with VECTASTAIN Elite ABC Kit (Vector Laboratories) and 3,3′-diaminobenzidine chromogen substrate solution. The slides were counter-stained with Mayer’s hematoxylin.
Statistical Analysis
Data were expressed as mean ± standard error of the mean (SEM) and compared by one-way analysis of variance and the Student-Newman-Keuls test. Differences in values were considered significant if P < 0.05.
RESULTS
C75 Alleviates Clinical Parameters of DSS-Induced Colitis
Body weight loss is a key indicator of the onset and severity of DSS colitis (20). Mice received 4% DSS in their drinking water and started losing weight on d 4 and reached a 23.5% loss from original weight on d 8 (Figure 1A). However, colitis mice with C75 treatment only had a 15.3% weight loss on d 8 (Figure 1A). C75 treatment also significantly improved the clinical presentations of DSS colitis by lowering fecal blood and diarrhea scores (Figures 1B, C). Disease activity index, which reflects the overall clinical condition, was decreased by 28.3% in the C75-treated group in comparison with the vehicle group (Figure 1D).
Figure 1.

Effect of C75 treatment on the severity of DSS-induced colitis. Sham or mice exposed to 4% DSS in drinking water either treated with vehicle or C75 were monitored daily to record their body weight change (A), fecal blood score (B) and diarrhea score (C). Disease activity index (D), macroscopic appearance (E) and colon length (F) were assessed at d 8. Data are expressed as mean ± SEM (n = 5 per group). *P < 0.05 versus sham, #P < 0.05 versus vehicle.
Shortening of colon length is another index to reflect the disease severity of DSS-induced colitis (21). We first examined the macroscopic appearance at d 8 and observed that colons from colitis mice were shorter, had a small cecum, were bloody, and had very little stool retained in comparison with the sham mice (Figure 1E). In contrast, colons of colitis treated with C75 had a larger cecum, were less bloody, and had a good amount of stool retained, compared with the vehicle (Figure 1E). With quantification by measuring, the colon length from colitis mice shortened by 34.6% on average, whereas C75 treatment restored it to 25.4%, compared with the sham (Figure 1F). Administration of C75 to mice given normal drinking water did not affect the macroscopic appearance and colon length in comparison with the sham (data not shown).
C75 Reduces Colon Damage and Apoptotic Marker in DSS Colitis
We next performed histopathological evaluation of colon tissues at d 8. Colons exposed to DSS had massive infiltration of inflammatory cells in mucosa and submucosa with epithelial denudation, crypt loss, edema and ulceration in comparison with the sham (Figure 2A). However, the inflammatory cell infiltration was only observed in the mucosa of the colon after C75 treatment. Apart from that, C75 treatment markedly reduced inflammation and improved the integrity of structure in the inflamed colon (Figure 2A). The quantitative measurements of histological damage scores showed significant reduction in the C75 treatment group by 60% in comparison with the vehicle group (Figure 2B). We also examined the effect of C75 treatment on apoptosis by assessing caspase-3 activity (22). Cleaved caspase-3 was well detected by Western blotting in the vehicle group, while its levels were reduced by 33.2% in the C75-treated group (Figure 2C).
Figure 2.

Effect of C75 treatment on the histological morphology of colon in DSS colitis. (A) Representative images of colon tissues stained with hematoxylin and eosin from sham, vehicle and C75-treated mice at d 8. Original magnification 40×. (B) Histological damage score of the colons as described in Materials and Methods. (C) Western blot analysis of cleaved caspase-3 expression in colon tissues collected at d 8. Expression in the sham group is designated as 1 for comparison. Data are expressed as mean ± SEM (n = 5 per group). *P < 0.05 versus sham; #P < 0.05 versus vehicle.
FASN Expression Increases in DSS Colitis
To correlate the severity of DSS colitis to FASN expression, we performed immunohistochemical staining in colon tissues. FASN expression in the colons of the sham mice was mainly localized at the top of the crypts; however, FASN was expressed in the entire crypt after DSS exposure (Figure 3A). Treatment with C75 restored the pattern of FASN expression resembling the sham, although there was still some patchy FASN expression throughout the crypt (Figure 3A). A transverse section indicated that FASN expression was increased after DSS exposure, but reduced with C75 treatment (Figure 3B).
Figure 3.

Expression of FASN in the colon of DSS colitis. Colon tissues from sham, vehicle and C75-treated groups at d 8 were subjected to immunohistochemical analysis against FASN with brown staining and counterstained with hematoxylin. Representative images of longitudinal sections (A) (100× magnification) and transverse sections (B) (200× magnification) are shown.
C75 Decreases Chemokine Levels and Neutrophil Infiltration in DSS Colitis
Chemokines, such as macrophage inflammatory protein (MIP)-2 and keratinocyte-derived chemokine (KC), attracting neutrophil transmigration across mucosal epithelia is a hallmark of several inflammatory diseases, including IBD (23,24). The mRNA levels of MIP-2 and KC in colons dramatically increased to 326- and 28-fold, respectively, after DSS exposure, whereas their levels were significantly inhibited by 78.2% and 68.0%, respectively, with C75 treatment (Figures 4A, B). Correspondingly, MPO activity, an indicator of neutrophil infiltration, was well detected in the inflamed colon, whereas it was significantly lower in the C75-treated group (Figure 4C).
Figure 4.
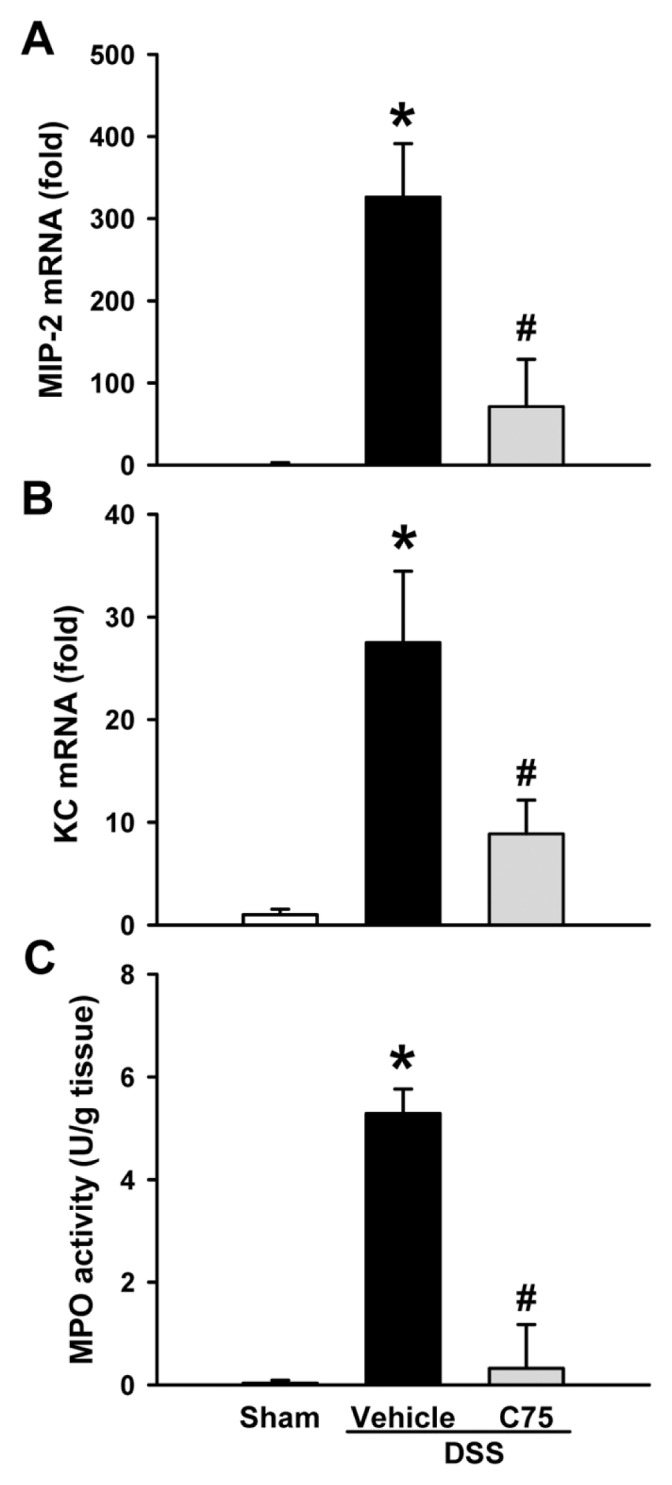
Effect of C75 treatment on chemokine expression and MPO activity in DSS colitis. The mRNA levels of MIP-2 (A) and KC (B) in colons from sham, vehicle and C75-treated groups at d 8. Expression in the sham group is designated as 1 for comparison. (C) Colon MPO activity, a marker for neutrophil infiltration, was determined spec-trophotometrically. Data are expressed as mean ± SEM (n = 5 per group). *P < 0.05 versus sham; #P < 0.05 versus vehicle.
C75 Lowered Proinflammatory Cytokine Levels in DSS Colitis
A cytokine is a principal mediator of the innate and adaptive arms of the immune responses in mucosal inflammation (25). We examined the effect of C75 treatment on the expression of TNF-α, IL-6 and IL-1β in colon tissues at d 8. The mRNA levels of TNF-α, IL-6 and IL-1β, determined by RT-PCR, were decreased by 83.2%, 68.7% and 97.1%, respectively (Figures 5A–C), as well as their protein levels, measured by ELISA, were decreased by 19.2%, 21.9% and 30.2%, respectively, in the C75-treated group, compared with the vehicle group (Figures 5D–E).
Figure 5.

Effect of C75 treatment on proinflammatory cytokine expression in DSS colitis. The mRNA levels of TNF-α (A), IL-6 (B) and IL-1β (C) in colons from sham, vehicle and C75-treated groups at d 8. Expression in the sham group is designated as 1 for comparison. Protein levels of TNF-α (D), IL-6 (E) and IL-1β (F) in colons, measured by ELISA, are shown. Data are expressed as mean ± SEM (n = 5 per group). *P < 0.05 versus sham; #P < 0.05 versus vehicle.
MDA Production and NF-κB Activation in DSS Colitis Are Attenuated by C75
ROS can degrade polyunsaturated lipids to form MDA as a direct indicator of lipid peroxidation-induced injury and oxidative stress in tissues (26). After DSS exposure, MDA levels in colon tissues increased to 1.5-fold in comparison with the sham group, while its levels were reduced by 36.5% with C75 treatment (Figure 6A). The NF-κB signaling pathway plays a key role in regulating inflammatory responses including in IBD (27,28). NF-κB is usually found in the cytoplasm bound to its inhibitory protein termed IκB. Degradation of IκB via proteasomes, triggered by inflammatory signals, can result in a translocation of NK-κB into the nucleus for the activation (29). We detected a downregulation of IκB expression in the inflamed colon, while it was partially restored in the C75-treated group (Figure 6B), indicating the inhibition of NF-κB activation by C75 treatment.
Figure 6.

Effect of C75 treatment on MDA production and IκB degradation in DSS colitis. (A) MDA levels in colons from sham, vehicle and C75-treated groups at d 8. (B) Western blot analysis of IκB expression in colons. Expression in the sham group is designated as 1 for comparison. Data are expressed as mean ± SEM (n = 5 per group). *P < 0.05 versus sham; #P < 0.05 versus vehicle.
Elevation of COX-2 and iNOS Expression in DSS Colitis Are Inhibited by C75
The inflamed colon mucosa in DSS colitis produces high amounts of prostaglandins and nitric oxide through COX-2 and iNOS pathways, respectively (30). By using RT-PCR, we observed that the mRNA levels of COX-2 and iNOS were significantly reduced by 55.6% and 66.5 %, respectively, with C75 treatment (Figures 7A, B). In the Western blot analysis, COX-2 and iNOS expression were also significantly reduced by 42.0% and 60.7%, respectively, with C75 treatment (Figures 7C, D).
Figure 7.

Effect of C75 treatment on the expression of inflammatory mediators in DSS colitis. The mRNA levels of COX-2 (A) and iNOS (B) in colons from sham, vehicle and C75-treated groups at d 8. Western blot analysis of COX-2 (C) and iNOS (D) expression in colons. Expression in the sham group is designated as 1 for comparison. Data are expressed as mean ± SEM (n = 5 per group). *P < 0.05 versus sham; #P < 0.05 versus vehicle.
DISCUSSION
IBD is a chronic relapsing form of inflammatory disease with unknown etiology. It occurs at any age, especially in the young age-groups, leading to a decreased quality of life. The patients can live an ordinary life because of advanced therapeutic approaches, even though a major portion of IBD cases are resistant to such advanced treatments and require surgery. If left untreated, IBD patients have a high possibility of developing colon carcinoma latter on (31). Steroids, nonsteroids, aminosalicylates, butyrates and anti-TNF drugs have now been clinically used with significant therapeutic efficacy to treat IBD (32). Nevertheless, these therapies for IBD are usually associated with several side effects or clinical limitations. Thus, there is still an unmet medical need for the innovated approaches for treating IBD patients. In this study, we demonstrate the therapeutic potential of C75, a FASN inhibitor, against a murine model of DSS-induced colitis in terms of attenuating clinical symptoms; improving macroscopic appearance and microscopic structure of colons of colitis; lowering the production of chemokines, proinflammatory cytokines and mediators; and inhibiting neutrophil infiltration and cellular apoptosis.
The abnormalities in FA metabolism have been reported in IBD, which can be considered as one of the etiologies for the development of this disease (33). We have demonstrated that FASN expression extends from the top of the crypt in the sham mice to the entire mucosa in the colitis mice, detected by immunohistochemistry. Intensity of the FASN expression was also seen to be increased in colitis mice when compared with sham. This observation agrees with other studies showing high FASN expression in patients with UC and colorectal neoplasia (14,34). However, by using gene expression prolife analysis, a decrease of FASN expression has been reported in ileum and colon of UC patients (35). A recent study also indicates that FASN plays an important role in maintaining the intestinal barrier function, and loss of FASN in mouse intestine can initiate intestinal inflammation (36). Therefore, a balance of controlling FASN expression is crucial for preventing the development of colitis.
Although colon damage is mainly caused by necrosis in the DSS-induced colitis, the elevated apoptosis was also observed in both the mouse DSS model and patients with UC (37–39). The apoptosis of epithelial cells results in loss of intestinal epithelial barrier integrity and exaggerated mucosal inflammatory response in IBD (40). In this study, we detected an increased activation of caspase-3 in the inflamed colon, whereas its activation is attenuated by C75. This result suggests that reduction of apoptosis by C75 may contribute to the improvement of the colitis. The detail pathways and mechanisms meditated by C75 to regulate apoptosis in colitis need further investigation.
Along the lines of the attenuation of severity of colitis mice in several clinical measurements, C75 treatment also effectively lowers immune cells in responding to the inflammation in the inflamed colon. Macrophages are the major source of MIP-2 and KC, which play a major role in neutrophil migration to sites of inflammation (41). We have demonstrated a significant reduction of MIP-2 and KC expression as well as MPO activity, an indicator of neutrophil infiltration, in the inflamed colon after C75 treatment. The main function of neutrophils recruited to the inflamed organs is to contain and eradicate invading pathogens (42). These recruited, activated neutrophils can release proteolytic enzymes and reactive oxygen species, not only for killing invading pathogens, but their excessive production can disrupt the endothelial barrier and cause extravascular tissue damage (43,44). Indeed, we have observed an increase in apoptosis, determined by activation of caspase-3, in the inflamed colon. In congruence with its effect on MPO activity, C75 also inhibits the activation of cas-pase-3 in the inflamed colon. The therapy designed to attenuate neutrophil infiltration has been an attractive strategy to treat colitis (45).
In addition to producing chemokines, macrophages are mainly responsible for the production of proinflammatory cytokines (46). Again, we have demonstrated that C75 effectively decreases the expression of TNF-α, IL-6 and IL-1β in the inflamed colon at their mRNA as well as protein levels. Furthermore, it is well known that NF-κB is the major transcriptional factor for controlling the expression of chemokines and cytokines (27). We have demonstrated that administration of C75 prevents the degradation of IκB, which leads to activation of NF-κB, in the inflamed colon. A recent study reveals that the effects of FA as inflammatory players are generated through the TLR-mediated pathways (12). Future study will determine whether C75 treatment affects the FA profile in the inflamed colon, leading to disruption of the TLR/NF-κB signaling pathway.
We have further examined other pathways that contribute to the development of colitis. Elevation of MDA levels in the inflamed colon are significantly suppressed by C75 treatment. MDA low- molecular-weight end products are formed via the degradation of lipids, especially polyunsaturated FA and arachidonic acid (47). Overexpression of FASN in the inflamed colon, as indicated in this study, can produce high amounts of long-chain FAs for being further degraded as MDA. Inhibition of FASN by C75 can limit the resources necessary for generating MDA. In addition to MDA, ROS is another contributing factor for MDA production. ROS can be produced by the activation of several inflammatory enzymes such as nicotinamide adenine dinucleotide phosphate oxidase, COX, iNOS, myeloperoxidas and lipoxygenase (48). We have demonstrated that C75 can inhibit mRNA and protein expression of COX-2 and iNOS elevated in the inflamed colon. Of note, COX-2 and iNOS are the main enzymes used to produce prostaglandins from arachidonic acid and nitric oxide, two other prominent inflammatory mediators (49).
CONCLUSION
C75 treatment downregulates the expression of inflammatory mediators, including chemokines, cytokines, ROS, COX-2 and iNOS, leading to the reduction of colon tissue damage and improvement of clinical symptoms in DSS-induced experimental colitis. Although C75 has a limitation for the clinical use because of its side effects (50), our study shows for the first time antiinflammatory activity of C75. Therefore, targeting FASN may provide a therapeutic potential for treating patients with IBD.
ACKNOWLEDGMENTS
We thank Lana M Corbo for editorial assistance. This study was supported in part by National Institutes of Health grants GM057468, GM053008 and HL076179 (to P Wang).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Inflammatory bowel disease (IBD) Atlanta (GA): Centers for Disease Control and Prevention; [updated 2011 Jul 15 cited 2013 Dec 23]. Available from: http://www.cdc.gov/ibd. [Google Scholar]
- 3.Sonnenberg A. Disability from inflammatory bowel disease among employees in West Germany. Gut. 1989;30:367–70. doi: 10.1136/gut.30.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Isaacs KL, Sartor RB, Haskill S. Cytokine messenger RNA profiles in inflammatory bowel disease mucosa detected by polymerase chain reaction amplification. Gastroenterology. 1992;103:1587–95. doi: 10.1016/0016-5085(92)91182-4. [DOI] [PubMed] [Google Scholar]
- 5.Harris ML, et al. Free radicals and other reactive oxygen metabolites in inflammatory bowel disease: cause, consequence or epiphenomenon? Pharmacol Ther. 1992;53:375–408. doi: 10.1016/0163-7258(92)90057-7. [DOI] [PubMed] [Google Scholar]
- 6.Reilly PM, Schiller HJ, Bulkley GB. Pharmacologic approach to tissue injury mediated by free radicals and other reactive oxygen metabolites. Am J Surg. 1991;161:488–503. doi: 10.1016/0002-9610(91)91120-8. [DOI] [PubMed] [Google Scholar]
- 7.Simopoulos AP. Omega-3 fatty acids in health and disease and in growth and development. Am J Clin Nutr. 1991;54:438–63. doi: 10.1093/ajcn/54.3.438. [DOI] [PubMed] [Google Scholar]
- 8.Unger RH. The physiology of cellular liporegulation. Annu Rev Physiol. 2003;65:333–47. doi: 10.1146/annurev.physiol.65.092101.142622. [DOI] [PubMed] [Google Scholar]
- 9.Savary S, et al. Fatty acids: induced lipotoxicity and inflammation. Curr Drug Metab. 2012;13:1358–70. doi: 10.2174/138920012803762729. [DOI] [PubMed] [Google Scholar]
- 10.Toborek M, Lee YW, Garrido R, Kaiser S, Hennig B. Unsaturated fatty acids selectively induce an inflammatory environment in human endothelial cells. Am J Clin Nutr. 2002;75:119–25. doi: 10.1093/ajcn/75.1.119. [DOI] [PubMed] [Google Scholar]
- 11.Martins de Lima T, et al. Mechanisms by which fatty acids regulate leucocyte function. Clin Sci (Lond) 2007;113:65–77. doi: 10.1042/CS20070006. [DOI] [PubMed] [Google Scholar]
- 12.Huang S, et al. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res. 2012;53:2002–13. doi: 10.1194/jlr.D029546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chirala SS, Wakil SJ. Structure and function of animal fatty acid synthase. Lipids. 2004;39:1045–53. doi: 10.1007/s11745-004-1329-9. [DOI] [PubMed] [Google Scholar]
- 14.Consolazio A, et al. Overexpression of fatty acid synthase in ulcerative colitis. Am J Clin Pathol. 2006;126:113–8. doi: 10.1309/pubvqndnvqkjvc8m. [DOI] [PubMed] [Google Scholar]
- 15.Kuhajda FP, et al. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci U S A. 2000;97:3450–4. doi: 10.1073/pnas.050582897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010;6:551–62. doi: 10.2217/fon.10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–77. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 18.Kim EK, et al. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. J Biol Chem. 2004;279:19970–6. doi: 10.1074/jbc.M402165200. [DOI] [PubMed] [Google Scholar]
- 19.Kim TW, et al. Involvement of lymphocytes in dextran sulfate sodium-induced experimental colitis. World J Gastroenterol. 2006;12:302–5. doi: 10.3748/wjg.v12.i2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krieglstein CF, et al. Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of super-oxide and nitric oxide. J Exp Med. 2001;194:1207–18. doi: 10.1084/jem.194.9.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohkawara T, et al. DNA vaccination targeting macrophage migration inhibitory factor prevents murine experimental colitis. Clin Exp Immunol. 2011;163:113–22. doi: 10.1111/j.1365-2249.2010.04277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chin AC, Parkos CA. Neutrophil transepithelial migration and epithelial barrier function in IBD: potential targets for inhibiting neutrophil trafficking. Ann N Y Acad Sci. 2006;1072:276–87. doi: 10.1196/annals.1326.018. [DOI] [PubMed] [Google Scholar]
- 24.Dutra RC, et al. Preventive and therapeutic euphol treatment attenuates experimental colitis in mice. PLoS One. 2011;6:e27122. doi: 10.1371/journal.pone.0027122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alex P, et al. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–52. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. Biomarkers of oxidative damage in human disease. Clin Chem. 2006;52:601–23. doi: 10.1373/clinchem.2005.061408. [DOI] [PubMed] [Google Scholar]
- 27.Karrasch T, Jobin C. NF-kappaB and the intestine: friend or foe? Inflamm Bowel Dis. 2008;14:114–24. doi: 10.1002/ibd.20243. [DOI] [PubMed] [Google Scholar]
- 28.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–43. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 30.Dudhgaonkar SP, Tandan SK, Kumar D, Raviprakash V, Kataria M. Influence of simultaneous inhibition of cyclooxygenase-2 and inducible nitric oxide synthase in experimental colitis in rats. Inflammopharmacology. 2007;15:188–95. doi: 10.1007/s10787-007-1603-3. [DOI] [PubMed] [Google Scholar]
- 31.Karvellas CJ, Fedorak RN, Hanson J, Wong CK. Increased risk of colorectal cancer in ulcerative colitis patients diagnosed after 40 years of age. Can J Gastroenterol. 2007;21:443–6. doi: 10.1155/2007/136406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Girardin M, et al. First-line therapies in inflammatory bowel disease. Digestion. 2012;86(Suppl 1):6–10. doi: 10.1159/000341951. [DOI] [PubMed] [Google Scholar]
- 33.Shores DR, Binion DG, Freeman BA, Baker PR. New insights into the role of fatty acids in the pathogenesis and resolution of inflammatory bowel disease. Inflamm Bowel Dis. 2011;17:2192–204. doi: 10.1002/ibd.21560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rashid A, et al. Elevated expression of fatty acid synthase and fatty acid synthetic activity in colorectal neoplasia. Am J Pathol. 1997;150:201–8. [PMC free article] [PubMed] [Google Scholar]
- 35.Heimerl S, et al. Alterations in intestinal fatty acid metabolism in inflammatory bowel disease. Biochim Biophys Acta. 2006;1762:341–50. doi: 10.1016/j.bbadis.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 36.Wei X, et al. Fatty acid synthase modulates intestinal barrier function through palmitoylation of mucin 2. Cell Host Microbe. 2012;11:140–52. doi: 10.1016/j.chom.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–49. [PubMed] [Google Scholar]
- 38.Martinez JA, et al. Deletion of Mtgr1 sensitizes the colonic epithelium to dextran sodium sulfate-induced colitis. Gastroenterology. 2006;131:579–88. doi: 10.1053/j.gastro.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 39.Iwamoto M, Koji T, Makiyama K, Kobayashi N, Nakane PK. Apoptosis of crypt epithelial cells in ulcerative colitis. J Pathol. 1996;180:152–9. doi: 10.1002/(SICI)1096-9896(199610)180:2<152::AID-PATH649>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 40.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 41.De Filippo K, Henderson RB, Laschinger M, Hogg N. Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J Immunol. 2008;180:4308–15. doi: 10.4049/jimmunol.180.6.4308. [DOI] [PubMed] [Google Scholar]
- 42.Haziot A, Hijiya N, Gangloff SC, Silver J, Goyert SM. Induction of a novel mechanism of accelerated bacterial clearance by lipopolysaccharide in CD14-deficient and Toll-like receptor 4-deficient mice. J Immunol. 2001;166:1075–8. doi: 10.4049/jimmunol.166.2.1075. [DOI] [PubMed] [Google Scholar]
- 43.Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001;7:1–7. doi: 10.1097/00075198-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 44.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31:S195–9. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 45.Farooq SM, et al. Therapeutic effect of blocking CXCR2 on neutrophil recruitment and dextran sodium sulfate-induced colitis. J Pharmacol Exp Ther. 2009;329:123–9. doi: 10.1124/jpet.108.145862. [DOI] [PubMed] [Google Scholar]
- 46.Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79:319–26. doi: 10.1172/JCI112815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Del Rio D, Stewart AJ, Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis. 2005;15:316–28. doi: 10.1016/j.numecd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 48.Koppula S, Kumar H, Kim IS, Choi DK. Reactive oxygen species and inhibitors of inflammatory enzymes, NADPH oxidase, and iNOS in experimental models of Parkinson’s disease. Mediators Inflamm. 2012;2012;823902 doi: 10.1155/2012/823902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vane JR, et al. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc Natl Acad Sci U S A. 1994;91:2046–50. doi: 10.1073/pnas.91.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loftus TM, et al. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science. 2000;288:2379–81. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]