

Abstract
Purpose
Approximately 5% to 8% of renal cell carcinoma (RCC) is hereditary. No guidelines exist for patient selection for RCC germline mutation testing. We evaluate how age of onset could indicate the need for germline mutation testing for detection of inherited forms of kidney cancer.
Patients and Methods
We analyzed the age distribution of RCC cases in the SEER-17 program and in our institutional hereditary kidney cancer population. The age distributions were compared by sex, race, histology, and hereditary cancer syndrome. Models were established to evaluate the specific age thresholds for genetic testing.
Results
The median age of patients with RCC in SEER-17 was 64 years, with the distribution closely approaching normalcy. Statistical differences were observed by race, sex, and subtype (P < .05). The bottom decile cutoff was ≤ 46 years of age and slightly differed by sex, race, and histology. The mean and median ages at presentation of 608 patients with hereditary kidney cancer were 39.3 years and 37 years, respectively. Although age varied by specific syndrome, 70% of these cases were found to lie at or below the bottom age decile. Modeling age-based genetic testing thresholds demonstrated that the 10th percentile maximized sensitivity and specificity.
Conclusion
Early age of onset might be a sign of hereditary RCC. Even in the absence of clinical manifestations and personal/family history, an age of onset of 46 years or younger should trigger consideration for genetic counseling/germline mutation testing and may serve as a useful cutoff when establishing genetic testing guidelines.
INTRODUCTION
Although most cancers occur sporadically, there is increasing recognition that many individuals have a hereditary predisposition. As of 2011, almost 50 cancer-susceptibility syndromes were recognized.1 Many of these represent monogenic syndromes that are related to germline alterations in tumor suppressor genes or proto-oncogenes. In the past two decades, research on cancer susceptibility has demonstrated that hereditary cancer may be more common than previously recognized. Besides monogenic syndromes, genome-wide association studies demonstrate that multiple, inherited predisposition loci can increase specific cancer risk.2 A hereditary predisposition has been described for common cancers such as breast and colon cancer as well as for less commonly encountered cancers such as osteosarcoma, adrenocortical carcinoma, and retinoblastoma.
Renal cell carcinoma (RCC) occurs in approximately 55,000 patients per year in the United States; 1% to 2% of the population develops RCC by the age of 75 years.3 Hereditary RCC may account for 5% to 8% of kidney cancers; however, this approximation may be significantly underestimated.4 A study by Gudbjartsson et al5 suggested that nearly 60% of patients with RCC have a hereditary predisposition. Several hereditary RCC syndromes have been characterized, including von Hippel-Lindau (VHL), hereditary papillary renal cell carcinoma (HPRC), Birt-Hogg-Dubé (BHD), hereditary leiomyomatosis and RCC (HLRCC), succinate dehydrogenase kidney cancer (SDH-RCC), tuberous sclerosis complex (TSC), Cowden syndrome, and microphthalmia-associated transcription factor (MITF), that have been shown to be associated with germline mutations in VHL, MET, FLCN, FH, SDHB/C/D, TSC1/2, PTEN, or MITF, respectively.4,6 Although these syndromes have similarities, they vary in histology, aggressiveness, penetrance, and associated clinical manifestations.
It is the role of the clinician to recognize patients who may benefit from germline or somatic mutation testing and to institute appropriate evaluation and management. Individuals may present with a known familial diagnosis of a hereditary syndrome, thus simplifying evaluation. However, as a result of occult family history, lack of recognition of subtle clinical features, incomplete penetrance, de novo germline mutations, and an incomplete family history (either because of the patient or provider), patients may present with a previously unrecognized hereditary syndrome. Patients with RCC who present with associated clinical manifestations such as a cerebellar hemangioblastoma (VHL), cutaneous fibrofolliculomas (BHD), or leiomyomas (HLRCC) are easily identified as candidates for germline mutation testing. However, many hereditary RCC syndromes have a subtle presentation, and their diagnosis depends on an astute clinician whose concern leads to consideration for germline mutation testing.
When evaluating patients with RCC, certain findings should trigger clinical concern for a hereditary syndrome, including family history of RCC, bilateral/multifocal renal masses, associated clinical manifestations of known RCC hereditary cancer syndromes, and certain specific renal tumor histologies (eg, hybrid/oncocytic RCC in patients with BHD). Most forms of cancer have been considered a disease of old age. RCC also has a similar pattern, with higher incidence in older individuals and a median age of onset in the seventh decade of life.7 In contrast, many hereditary forms of cancer develop at a younger age than observed in the generation population. Therefore, early age of onset is recommended as a general criterion for consideration for genetic testing.8 For specific cancers such as breast and colorectal cancer, genetic testing guidelines exist and include specific age cutoffs for the selection of individuals for consideration for germline mutation testing.2,9
Studies of a number of RCC syndromes indicate a significant propensity for early onset.10,11 Currently, no established RCC guidelines are available to aid in the selection of appropriate candidates for germline mutation testing. To establish an age threshold for genetic evaluation in RCC, we evaluated the age distribution of RCC in the hereditary and general populations. To do so, we used the SEER-17 program as well as our institutional experience with hereditary cancer syndromes.
PATIENTS AND METHODS
To evaluate the age of RCC onset in the US population, the SEER-17 database was reviewed. From 1990 to 2008, all patients listed as having cancer of the kidney were selected. Cases with International Classification of Diseases (ICD) –0-3 histology codes representing RCC (8140, 8260, 8270, 8290, 8310, 8312, 8316, and 8317 through 8320) were chosen for additional review. Individual records were queried for age, histology, sex, and race. The age distribution, normalcy, and skewness were assessed overall, and by sex, race, and subtype (clear cell, 8310, 8320, and 8316; papillary, 8320; chromophobe, 8270 and 8317; and collecting duct, 8319). We compared the median age between sex (male, female), race (white, black, Asian, and Native American), and subtype (clear cell, papillary, chromophobe, and collecting duct). The overall age distribution deciles were calculated, and the cutoff for the bottom 10th percentile was noted for sex, race, and subtype. For each histology, box plots were used to demonstrate the RCC age distribution. Differences between groups were calculated using t test and analysis of variance.
To better understand the age of onset for hereditary RCC, we reviewed the age distribution of patients identified with hereditary RCC at our institution. Individuals with suspected hereditary kidney cancer syndromes continue to be enrolled onto protocols at the Urologic Oncology Branch of the National Cancer Institute (NCI; Clinical Manifestations and Molecular Bases of Heritable Urologic Malignant Disorders trial). As part of the clinical protocol, affected/at-risk patients undergo clinical evaluation and meet with a kidney cancer genetic counselor for consideration for Clinical Laboratory Improvement Amendments–certified genetic testing.
We queried the protocol database for clinical, genetic, and pathologic information to select for patients with the diagnosis of hereditary RCC. Patients affected with VHL, BHD, HPRC, HLRCC, or succinate dehydrogenase subunit B (SDHB) were included in the hereditary cohort. To be considered, patients had to have RCC and a clinical diagnosis of VHL, HPRC, BHD, or SDHB-RCC, and/or a personal or family germline mutation in the VHL, FLCN, MET, FH, or SDHB genes. The age of onset of RCC was recorded on the basis of the first presentation of a solid kidney tumor in imaging studies performed at the NCI; for patients initially treated by an outside facility, when detailed imaging information was not available, the age of onset was chosen by either the date of their first identified solid tumor or renal surgery. For patients with hereditary RCC, we evaluated the age distribution and determined the median, mean, standard deviation (SD), and range overall and by specific syndrome. The age of RCC onset in SEER-17 was compared with age of onset in patients with hereditary RCC. For each syndrome, box plots demonstrated the age of onset distribution. We compared the hereditary age distribution to that of the SEER-17 population using two-tailed t testing with statistical significance considered as P < .05.
We modeled how an age threshold alone would aid selection for germline mutation testing and enrich detection of hereditary RCC. We chose the 25th, 10th, and 2.5th percentile of SEER-17's RCC age distribution to determine its influence on the number needed to test (NNT) to find one case of hereditary RCC. This value is dependent on a currently unknown incidence of hereditary RCC. Despite the fact that many cases seem to have a strong familial component, many do not represent a known monogenic syndrome that can be identified with genetic testing. To perform modeling using an unknown incidence of a monogenic, hereditary RCC syndrome, we chose an estimate of 0.5% to 2.0%, approximately 25% to 50% of modest estimates of the incidence of hereditary RCC.4,12
On the basis of different testing thresholds, we modeled the NNT by incidence estimates. To determine how different age thresholds would affect the sensitivity and specificity of detecting hereditary RCC, we set up a model that was based on a 1% incidence of hereditary, monogenic RCC. Using a similar estimated 55,000 RCC cases and the defined age distributions for SEER and hereditary RCC, we evaluated different age thresholds for genetic testing (50th, 25th, 10th, and 2.5th percentile). Choosing an incidence of monogenic, hereditary RCC to be 1%, we set up a receiver operating characteristic (ROC) curve (sensitivity versus 100%−specificity) to evaluate potential testing thresholds. For all models, we estimated 55,000 cases of RCC per year.9
RESULTS
A total of 106,224 SEER kidney cancer records were reviewed. The mean and median ages of RCC were 63.4 and 64 years, respectively (SD, 13.4). The distribution was relatively normal, with skewness and kurtosis of −0.223 and −0.272, respectively (Fig 1). Estimates of the overall RCC age in the 25th, 10th, and 2.5th percentiles were found to be 54, 46, and 36 years. The mean ages of men and women with RCC were 62.9 and 64.3 years, respectively (P < .001). The mean ages of clear cell, papillary, chromophobe, and collecting duct were 61.9, 62.5, 60.1, and 61.8 years, respectively (P < .001). Racial differences were apparent; white and Asian patients (age 63.9 and 62.6 years, respectively) had a slightly older age of onset than black and Native American patients (age 60.7 and 60.3 years, respectively; P < .001). Although ages statistically differed by race, sex, and histology, these factors did not greatly alter the age percentiles. Therefore, we did not consider these clinically relevant for determining a genetic testing threshold.
Fig 1.

Distribution of the age of onset of kidney cancer in SEER-17 from 1990 to 2008 for 106,224 patients. The distribution approaches a normal curve; skewness and kurtosis are −0.223 and −0.272, respectively.
The NCI protocol database identified 608 patients with hereditary kidney cancer who met the defined criteria. Of these 608 patients, 387 patients were identified with VHL, 127 with BHD, 25 with HPRC, 56 with HLRCC, and 13 with SDHB (Table 1). Overall, the median and mean ages of the patients with hereditary RCC were 37 and 39.2 years, respectively (SD, 13.4), significantly lower than in the SEER population (P < .001; Fig 2) and compared with all RCC histologies (Figs 3A and 3B). The median and mean ages of onset for each hereditary syndrome were, respectively, 35 and 35.4 years for VHL, 50 and 50.3 years for BHD, 41 and 42.1 years for HPRC, 37 and 41 years for HLRCC, and 32 and 34.3 years for SDHB (Table 1). The median age and 25th/75th percentile were well below those of the SEER population (Fig 4).
Table 1.
Age of Onset by Hereditary Syndrome of NCI Urologic Oncology Branch Patients With Kidney Cancer
| Age, Years | Hereditary Syndrome |
||||
|---|---|---|---|---|---|
| VHL (n = 387) | BHD (n = 127) | HLRCC (n = 56) | HPRC (n = 25) | SDHB (n = 14) | |
| Median | 35 | 50 | 37 | 41 | 30 |
| Mean | 35.39 | 50.29 | 40.95 | 42.12 | 33.00 |
| Range | 16-79 | 23-74 | 10-77 | 19-66 | 15-61 |
| SD | 11.42 | 11.46 | 14.99 | 12.09 | 14.91 |
Abbreviations: BHD, Birt-Hogg-Dubé syndrome; HLRCC, hereditary leiomyomatosis and renal cell carcinoma; HPRC, hereditary papillary renal cell carcinoma; NCI, National Cancer Institute; SD, standard deviation; SDHB, succinate dehydrogenase B renal cell carcinoma; VHL, von Hippel-Lindau syndrome.
Fig 2.

A normalized distribution of the age of onset of kidney cancer in SEER-17 compared with National Cancer Institute patients with hereditary kidney cancer (blue, hereditary cases; gold, SEER cases).
Fig 3.


Renal cell carcinoma histology and National Cancer Institute patients with hereditary kidney cancer. (A) Box plot analysis and (B) normalized distribution analysis. UOB, Urologic Oncology Branch.
Fig 4.

Box plot of age of onset for National Cancer Institute Urologic Oncology Branch hereditary renal cell carcinoma (RCC) syndromes. BHD, Birt-Hogg-Dubé syndrome; HLRCC, hereditary leiomyomatosis and RCC; HPRC, hereditary papillary RCC; SDHB, succinate dehydrogenase B renal cell carcinoma; VHL, von Hippel-Lindau syndrome.
We evaluated whether a specific age threshold alone would enrich for patients with hereditary RCC by arbitrarily choosing the 25th, 10th, and 2.5th percentiles of the general population, which represented individuals up to the ages of 54, 46, and 36 years respectively, and this demonstrated enrichment for 85%, 70%, and 46% of the hereditary RCC cases, respectively. Knowing the number of RCC cases in the United States and how these percentiles would affect the distribution of hereditary and overall cases, statistical modeling was performed. As previously mentioned, we estimated the incidence of monogenic, hereditary RCC to range from 0.5% to 2.0%. An ROC model was generated on the basis of the sensitivity and specificity for age-based testing thresholds in the SEER distribution. The sensitivity for the 2.5th, 10th, and 25th percentiles would be 46%, 70%, and 85%, respectively, and specificity would be 98%, 90%, and 75%, respectively. The ROC model demonstrated that the 10th percentile threshold would balance the importance of both sensitivity and specificity (Fig 5A). Using age alone as a selection criterion for genetic counseling, the NNT would greatly decrease, with a higher incidence of hereditary RCC (Fig 5B). For the 10th percentile age threshold, the NNT would range from seven to 28 individuals. For the 2.5th and 25th percentiles, the NNT would range from three to seven and from 14 to 59 individuals, respectively.
Fig 5.
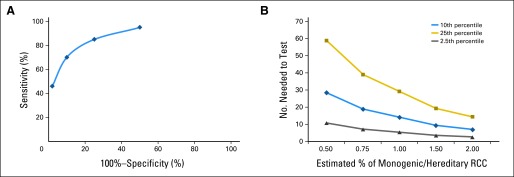
Statistical models demonstrating the ability of age threshold alone to enrich selection for renal cell carcinoma (RCC) genetic counseling germline mutation testing. (A) Receiver operating characteristic curve of sensitivity versus 100%−specificity, considering age threshold testing at the 2.5th, 10th, 25th, and 50th percentiles. For this analysis, a 1% incidence of hereditary RCC was used. (B) Number needed to test approximated on the basis of various estimates of the incidence of hereditary, monogenic RCC syndrome.
DISCUSSION
Detection of hereditary forms of RCC relies on the clinician to appropriately recognize individuals with potentially inherited forms of cancer. The importance of understanding the genetic basis of an individual's predisposition for RCC has been well established.4,6 Management strategies in those with specific hereditary RCC syndromes have been refined during the last two to three decades. On the basis of the experience at the NCI and other centers specializing in hereditary RCC, specific recommendations in regard to the role of nephron-sparing surgery, the method of partial nephrectomy (enucleation or margin), the timing of intervention, and renal mass surveillance have been defined for individual syndromes.13–15
The diagnosis of many hereditary RCC syndromes can be challenging for a number of reasons. Some features have incomplete penetrance; some disorders, such as HLRCC, have sex-specific manifestations, such as uterine leiomyomas; and others, such as HPRC, do not have extrarenal manifestations.16–19 Additionally, bilateral or multifocal renal tumors may only become apparent with follow-up because of their metachronous nature of presentation.
We performed an age distribution study of RCC in the general population (SEER-17) and reviewed our experience with hereditary RCC syndromes. These results clearly demonstrate that hereditary forms of RCC present at a much earlier age than sporadic, nonhereditary forms of RCC. We found that the median age of presentation for hereditary RCC is 27 years younger than that observed in the general population.
When germline mutation testing is considered in younger individuals, it would be useful to identify and follow an age threshold in addition to the family history and characteristic clinical manifestations. Among the individual syndromes, the median age of onset for kidney tumors in VHL, HLRCC, HPRC, and SDHB-RCC is lower than the 10th percentile of the general RCC population. Although BHD occurred at an older age than the other syndromes, the median age of onset was significantly lower than in the general RCC population and was less than the 20th percentile of age. To better understand how specific age thresholds would affect these characteristics, we constructed several models that were based on the age distribution in SEER and our patients with hereditary kidney cancer. Although initiating genetic counseling at the 2.5th and 10th age percentile brought the NNT to single digits, our ROC curve demonstrates that being too selective with age (the 2.5th percentile) diminishes the detection sensitivity. On the basis of our models, we propose that a useful threshold lies around the 10th percentile (46 years of age or younger), given that this cut point maximizes sensitivity and specificity while limiting the NNT.
On the basis of our findings, we recommend that all patients age 46 years or younger with RCC be referred for genetic counseling. Gene panel testing is an important breakthrough that is now emerging and may allow testing for all genes associated with hereditary RCC. Panel testing and the emergence of whole-exome sequencing may make interpretation of genetic testing more difficult and potentially alter our understanding of the phenotypes of existing syndromes. Understanding the difference between genetic variants and mutations can be complicated; therefore, counseling should be performed by an experienced team that is able to advise patients on the current clinical recommendations.
Although this work focused on germline mutation testing, somatic mutation testing can be useful in early-onset kidney cancer. Translocation RCC involves somatic fusion translocations and was first recognized in a fusion of the PRCC gene on chromosome 1 to the TFE3 gene on the X chromosome.20 TFE3, TFEB, and MITF are part of the microphthalmia-associated transcription family of transcription factors, and each is associated with RCC.21 MITF mutations may be found in the germline of families with RCC and melanoma, whereas TFE3 and TFEB are somatic fusion translocations. Translocation RCC may affect 15% of patients with kidney cancer younger than age 45 years,22 and 20% to 45% of children and young adults who have kidney cancer.23,24 Occasionally, pathologists can recognize this histology, but the diagnosis is reliably made by dual-color fluorescent in situ hybridization.25,26 Consequently, fluorescent in situ hybridization should therefore be considered as an adjunct to germline mutation testing for patients with early-onset kidney cancer in the appropriate clinical context, such as when pathology is suggestive of translocation RCC or after negative genetic testing.
Although our data provide important information to aid in the establishment of genetic testing guidelines, multiple limitations must be mentioned. Despite the fact that our study involved the largest cohort of patients with hereditary RCC to date, our experience with less frequent syndromes such as SDHB and HPRC is limited. Given that specific germline mutations may affect age of onset, the inclusion of several members of the same family may have altered the age distribution of the less common syndromes.27 Combining the patients with VHL, BHD, HLRCC, HPRC, and SDHB together as a hereditary RCC cohort to generate a common age distribution could be limited by the fact that these syndromes vary in age of onset, and the relative frequency of each could influence our data. However, when restricting analysis of patients with VHL (our most frequent syndrome) and SEER clear cell RCC, the 10th percentile still maximized sensitivity and specificity (data not shown). Another limitation is the retrospective nature of this cohort that was accrued over nearly three decades. Knowledge of which patients lacked clinical characteristics of a syndrome would add value to our proposed threshold; however, this information was not available. Additional studies are ongoing that involve the prospective analysis of age-based screening, which will allow validation of our recommended age threshold.
When evaluating our age threshold models, one must realize that our approach is an oversimplification of the intricacies of genetic counseling for a group of syndromes that only share RCC as a common feature. We ignored the differences in the median age distribution for sex, race, and histology (range of 1.4, 2.4, and 3.6 years, respectively), choosing to simplify our models. However, these differences, although statistically apparent, likely have minimal clinical relevance. Our models are based on estimates of the true incidence of hereditary RCC, which has not been clearly determined, nor has it been determined what percentage of these cases is linked to a testable, monogenic syndrome. Additionally, our NNT relies on the identification of one individual found on genetic testing to have a hereditary RCC syndrome. Whereas some syndromes such as VHL, HPRC, BHD, and HLRCC have greater than 90% sensitivity of detection, current genetic testing practices are not always as accurate, especially with syndromes such as TSC.28 Notwithstanding these limitations, these models demonstrate a proof of concept and, to our knowledge, serve as the first attempt to identify an evidence-based age threshold for RCC genetic testing.
We provide a detailed analysis of the age distribution of kidney cancer in the United States. The age of onset for hereditary RCC syndromes is much younger than that observed in the general population, with the median age lying near or below the bottom age decile of nonhereditary forms of RCC. Genetic testing remains an integral part of the management of patients with suspected hereditary RCC. Although family history, bilateral/multifocal renal tumors, associated clinical manifestations, and tumor histology are important for the selection of patients for germline mutation testing, early age of onset is a major indicator of a possible hereditary kidney cancer syndrome. When clinicians encounter individuals with early-onset kidney cancer (age 46 years or younger), they should strongly consider referral for genetic counseling/germline mutation testing.
Glossary Terms
- germline mutation:
An inherited variation in the lineage of germ cells. Germline mutations can be passed on to offspring.
- MET:
The receptor for hepatocyte growth factor receptor. MET is a transmembrane receptor tyrosine kinase. The primary single chain precursor protein is post-translationally cleaved to produce the alpha and beta subunits; the mature receptor is composed of these subunits linked via disulfide bonds. Various mutations in the MET gene have been associated with papillary renal carcinoma.
- von Hippel-Lindau (VHL):
A tumor suppressor gene. Patients with von Hippel-Landau (VHL) syndrome have mutations, allelic deletions, or silencing of the VHL gene. The phosphorylated or active VHL protein is a substrate recognition component of an ubiquitin-ligase complex that targets the hypoxia-inducible factor for proteolytic degradation. Loss of gene function permits the hypoxia-inducible factor to activate survival and proliferative cellular pathways.
Footnotes
Supported by Grants No. ZIA1BC011028-05, ZIA BC011038-05, ZIA BC011043-05, ZID BC011089-05, and ZIE BC 011023-05 from the National Institutes of Health.
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The author(s) indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: Brian Shuch, W. Marston Linehan
Financial support: W. Marston Linehan
Administrative support: Brian Shuch, W. Marston Linehan
Provision of study materials or patients: Brian Shuch, W. Marston Linehan
Collection and assembly of data: Brian Shuch, W. Marston Linehan
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Riley BD, Culver JO, Skrzynia C, et al. Essential elements of genetic cancer risk assessment, counseling, and testing: Updated recommendations of the National Society of Genetic Counselors. J Genet Couns. 2012;21:151–161. doi: 10.1007/s10897-011-9462-x. [DOI] [PubMed] [Google Scholar]
- 2.Purdue MP, Johansson M, Zelenika D, et al. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat Genet. 2011;43:60–65. doi: 10.1038/ng.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 4.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: A metabolic disease. Nat Rev Urol. 2010;7:277–285. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gudbjartsson T, Jónasdóttir TJ, Thoroddsen A, et al. A population-based familial aggregation analysis indicates genetic contribution in a majority of renal cell carcinomas. Int J Cancer. 2002;100:476–479. doi: 10.1002/ijc.10513. [DOI] [PubMed] [Google Scholar]
- 6.Linehan WM. Genetic basis of kidney cancer: Role of genomics for the development of disease-based therapeutics. Genome Res. 2012;22:2089–2100. doi: 10.1101/gr.131110.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallen EM, Pruthi RS, Joyce GF, et al. Kidney cancer. J Urol. 2007;177:2006–2018. doi: 10.1016/j.juro.2007.01.126. [DOI] [PubMed] [Google Scholar]
- 8.Trepanier A, Ahrens M, McKinnon W, et al. Genetic cancer risk assessment and counseling: Recommendations of the National Society of Genetic Counselors. J Genet Couns. 2004;13:83–114. doi: 10.1023/B:JOGC.0000018821.48330.77. [DOI] [PubMed] [Google Scholar]
- 9.Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 10.Linehan WM, Zbar B. Focus on kidney cancer. Cancer Cell. 2004;6:223–228. doi: 10.1016/j.ccr.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Ricketts CJ, Shuch B, Vocke CD, et al. Succinate dehydrogenase kidney cancer: An aggressive example of the Warburg effect in cancer. J Urol. 2012;188:2063–2071. doi: 10.1016/j.juro.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linehan WM, Bratslavsky G, Pinto PA, et al. Molecular diagnosis and therapy of kidney cancer. Annu Rev Med. 2010;61:329–343. doi: 10.1146/annurev.med.042808.171650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grubb RL, 3rd, Franks ME, Toro J, et al. Hereditary leiomyomatosis and renal cell cancer: A syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007;177:2074–2080. doi: 10.1016/j.juro.2007.01.155. [DOI] [PubMed] [Google Scholar]
- 14.Pavlovich CP, Grubb RL, 3rd, Hurley K, et al. Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol. 2005;173:1482–1486. doi: 10.1097/01.ju.0000154629.45832.30. [DOI] [PubMed] [Google Scholar]
- 15.Walther MM, Choyke PL, Weiss G, et al. Parenchymal sparing surgery in patients with hereditary renal cell carcinoma. J Urol. 1995;153:913–916. [PubMed] [Google Scholar]
- 16.Lubensky IA, Schmidt L, Zhuang Z, et al. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999;155:517–526. doi: 10.1016/S0002-9440(10)65147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt LS, Warren MB, Nickerson ML, et al. Birt-Hogg-Dubé syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001;69:876–882. doi: 10.1086/323744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duffey BG, Choyke PL, Glenn G, et al. The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease. J Urol. 2004;172:63–65. doi: 10.1097/01.ju.0000132127.79974.3f. [DOI] [PubMed] [Google Scholar]
- 19.Wei MH, Toure O, Glenn GM, et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet. 2006;43:18–27. doi: 10.1136/jmg.2005.033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sidhar SK, Clark J, Gill S, et al. The t(X;1) (p11.2;q21.2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Hum Mol Genet. 1996;5:1333–1338. doi: 10.1093/hmg/5.9.1333. [DOI] [PubMed] [Google Scholar]
- 21.Bertolotto C, Lesueur F, Giuliano S, et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011;480:94–98. doi: 10.1038/nature10539. [DOI] [PubMed] [Google Scholar]
- 22.Komai Y, Fujiwara M, Fujii Y, et al. Adult Xp11 translocation renal cell carcinoma diagnosed by cytogenetics and immunohistochemistry. Clin Cancer Res. 2009;15:1170–1176. doi: 10.1158/1078-0432.CCR-08-1183. [DOI] [PubMed] [Google Scholar]
- 23.Wu A, Kunju LP, Cheng L, et al. Renal cell carcinoma in children and young adults: Analysis of clinicopathological, immunohistochemical and molecular characteristics with an emphasis on the spectrum of Xp11.2 translocation-associated and unusual clear cell subtypes. Histopathology. 2008;53:533–544. doi: 10.1111/j.1365-2559.2008.03151.x. [DOI] [PubMed] [Google Scholar]
- 24.Bruder E, Passera O, Harms D, et al. Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am J Surg Pathol. 2004;28:1117–1132. doi: 10.1097/01.pas.0000131558.32412.40. [DOI] [PubMed] [Google Scholar]
- 25.Argani P, Hicks J, De Marzo AM, et al. Xp11 translocation renal cell carcinoma (RCC): Extended immunohistochemical profile emphasizing novel RCC markers. Am J Surg Pathol. 2010;34:1295–1303. doi: 10.1097/PAS.0b013e3181e8ce5b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong M, De Angelo P, Osborne L, et al. Dual-color, break-apart FISH assay on paraffin-embedded tissues as an adjunct to diagnosis of Xp11 translocation renal cell carcinoma and alveolar soft part sarcoma. Am J Surg Pathol. 2010;34:757–766. doi: 10.1097/PAS.0b013e3181dd577e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt LS, Nickerson ML, Angeloni D, et al. Early onset hereditary papillary renal carcinoma: Germline missense mutations in the tyrosine kinase domain of the met proto-oncogene. J Urol. 2004;172:1256–1261. doi: 10.1097/01.ju.0000139583.63354.e0. [DOI] [PubMed] [Google Scholar]
- 28.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]