

Abstract
A 48-year-old man was admitted for workup of stroke-like symptoms and generalised tonic–clonic seizures. History and examination revealed that the patient had background diagnoses of type 2 diabetes mellitus, epilepsy and had suffered a temporal lobe infarct 3 years ago. The unusual presentation and physical findings, along with subsequent MRI findings led to a diagnosis of mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). MELAS is a mitochondrial disorder typified by the aforementioned symptoms, and is typically diagnosed in the first two decades of life.
Background
Vascular infarcts are uncommon in young patients and accurate history taking, examination and appropriate biochemical and imaging investigations should be paramount in elucidating possible underlying syndromes or genetic aetiologies in such patients presenting with stroke-like syndromes. In this case, a young adult was diagnosed with a vascular infarct 3 years prior to current presentation. In fact, an underlying inherited mitochondrial disorder was the aetiology of his neurological symptoms. An awareness of mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) presenting in adult populations could lead to more timely and accurate diagnoses of this condition.
Case presentation
A 48-year-old right-handed man was admitted with a 3-day history of brief generalised tonic–clonic seizures, a 3-month history of weight loss with increasing difficulty in eating and a 1-week history of word finding difficulties and speech apraxia with reduced fine motor skills. On examination, he had bilateral lower limb and significant right upper limb and orolabial dyspraxia. Most notable was his difficulty in conversing, displaying the expressive and receptive dysphasia. He had an ataxic gait and coordination was impaired in the upper and lower limbs. Power, tone and sensation were normal throughout and plantars were downgoing. He had no fever and had stable vital signs.
He had a diagnosis of type 2 diabetes mellitus, epilepsy and a history of a left temporal lobe infarct 3 years ago.
The patient denied any family history of neurological or endocrine disorders.
Investigations
Basic blood biochemical markers were within normal range. Non-contrast CT of the brain was performed given the patients presenting neurological symptoms. This showed bilateral basal ganglia calcification and global parenchymal volume loss involving the cortical sulci and cerebellar fissures, disproportionate of the patient's age (figure 1). MRI of the brain precontrast and postcontrast was performed. This again revealed extensive global and symmetric parenchymal volume loss. In addition, focal cortical abnormality was seen in both insular regions bilaterally, greater on the left side, with slight associated gyral swelling and corresponding diffusion hyperintensity. Subtle cortical hypointensity on the corresponding ADC map reflected acute cortical ischaemic insults. Further focal abnormalities were seen within the left parietal cortex and right cerebellar hemisphere with corresponding diffusion hyperintensities (figures 2–4). There was no abnormal contrast enhancement. Given the patient's symptomatology and MRI findings, MR spectroscopy (MRS) was performed. This showed a lactate doublet peak with relative decrease in N-acetylaspartate when sampling was performed over the areas of cortical abnormality, indicating increased anaerobic glycolysis and loss of neuronal integrity, respectively (figure 5). The patient's venous lactate level was sampled and was elevated at 2.4 mmol/L (0.9–1.7), peaking at 3 mmol/L during admission. Muscle biopsy was performed to aid diagnosis (figure 6). Genetic analysis revealed m.3243A<G mitochondrial mutation, associated with MELAS.
Figure 1.
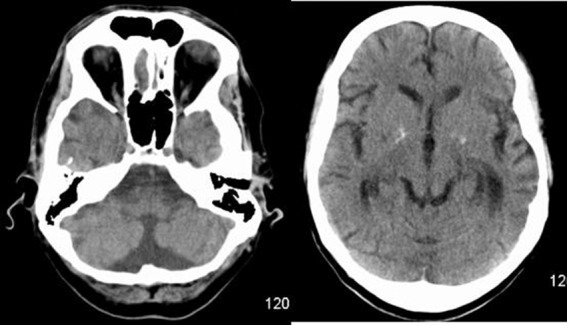
Standard non-contrast CT of the brain showing bilateral basal ganglia calcifications and global cerebral and cerebellar parenchymal volume loss.
Figure 2.
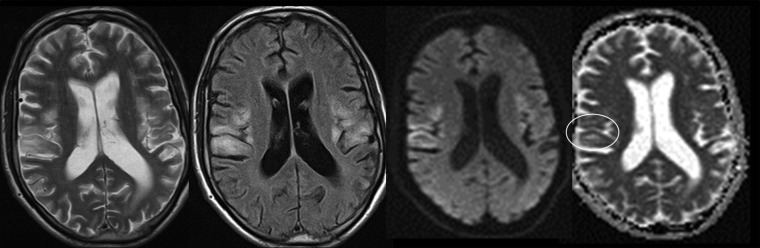
T2, fluid attenuation inversion recovery and diffusion-weighted MRI of the brain showing bilateral cortical-based signal abnormality with associated oedema, with corresponding diffusion hyperintensity. ADC map reveals subtle correlating cortical low signal, in keeping with diffusion restriction within some of the lesions.
Figure 3.
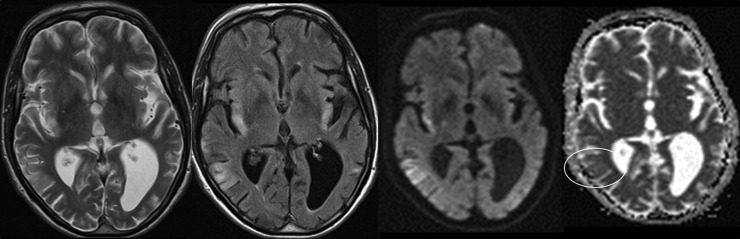
Further bilateral insular cortex T2, fluid attenuation inversion recovery and diffusion hyperintensities with subtle corresponding cortical ADC low signal.
Figure 4.
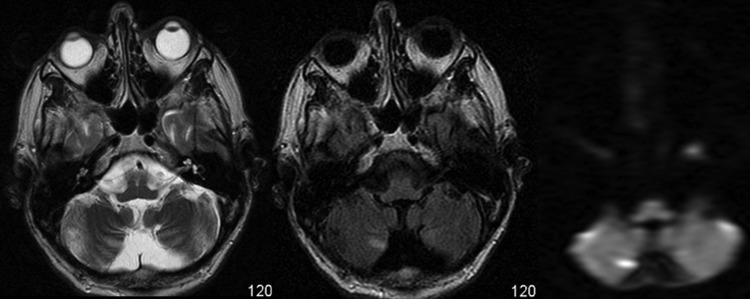
T2, fluid attenuation inversion recovery and diffusion-weighted MRI sequences showing concomitant right inferior cerebellar hemisphere signal abnormality with corresponding diffusion hyperintensity.
Figure 5.

MR spectroscopy showing a lactate doublet peak (white), a relative reduction in N-acetylaspartate when voxel sampling was performed over the foci of cortical abnormality.
Figure 6.
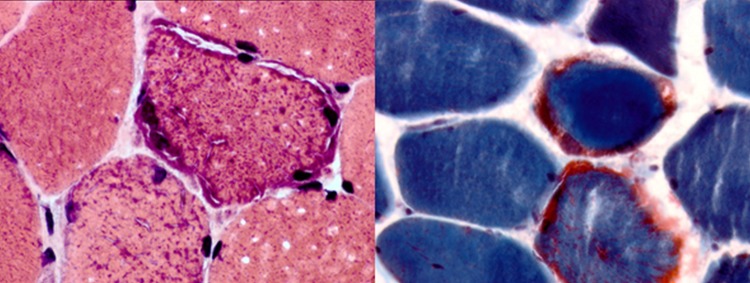
Muscle biopsy staining showing the presence of ragged red fibres, pathognomonic of mitochondrial myopathy.
Treatment
Treatment options for MELAS are limited and largely focus on supportive therapy. Clinical trials have failed to show efficacy in the use of various therapies in the management of acute flares.1 2 The patient was aggressively hydrated and supported nutritionally with enteral feeding supplementation. The patient was treated with L-arginine, though its efficacy is uncertain and his antiseizure and diabetic medications were optimised. The patient underwent ophthalmological and cardiac evaluation to assess for multisystem involvement. In the long term, education regarding the avoidance of fasting was highlighted. Genetic counselling was also offered.
Outcome and follow-up
The patient's speech and gait improved greatly over the course of his 3-week admission. The event did, however, significantly impact on his activities of daily living and was therefore referred to a specialist neurological rehabilitation centre for further care where there was almost complete recovery of neurological function.
Discussion
MELAS is a disabling, progressive multisystem mitochondrial disease that is characterised by early onset.3 4 Non-ischaemic stroke-like episodes are the defining features though the clinical heterogeneity of the disorder commonly results in difficulty in diagnosis. Neuronal hyperexcitability and increased capillary permeability result in focal hyperaemia and typical oedematous cortical-based brain lesions.5 Initially reported in a paediatric population, MELAS was originally felt to present in the first decade or two of life though an adult onset form is now recognised.6 While vascular infarcts are uncommon in children and suspicion of an underlying metabolic condition is raised more readily, adult onset MELAS can go undiagnosed if appropriate investigations are not performed. MELAS is, therefore, an important syndrome to consider in any patient presenting with unexplained stroke disorders. It is the most common maternally inherited mitochondrial disease and while treatment options are limited, supportive therapy and genetic counselling are important component of patient management. Cortical and subcortical lesions on MRI that cross vascular territories should raise the possibility of a mitochondrial disorder.7 The transient nature of the stroke-like episodes is reflected in fleeting abnormalities on neuroimaging. MRS offers an important diagnostic adjunct,8 along with serum and CSF lactate during acute episodes and respiratory enzyme defects in skeletal muscle biopsy samples. The utilisation of MRS, in addition to conventional MRI in the workup of young patients with stroke-like episodes, offers a non-invasive yet highly pathognomonic test for mitochondrial disorders. Knowledge of this condition should lead to appropriate radiological and biochemical investigations and ultimately invasive muscle biopsy and genetic analysis in order to avoid its misdiagnosis particularly in adult patients.
Learning points.
Stroke-like symptoms in young adults warrant careful consideration of non-vascular aetiologies.
Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke (MELAS) can present late in the adulthood, and is an uncommon multisystem mitochondrial disorder, which should be considered in any patient presenting with unusual stroke-like symptoms.
MRI findings of cortical and subcortical lesions that cross vascular territories in a patient with stroke-like symptoms should raise suspicion for a mitochondrial disorder, leading to consideration of MR spectroscopy.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.El-Hattaba A, Emrickb L, Craigenb W, et al. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol Genet Metab 2012;107:247–52 [DOI] [PubMed] [Google Scholar]
- 2.Scaglia F, Northrop JL. The mitochondrial myopathy encephalopathy, lactic acidosis with stroke-like episodes (MELAS) syndrome: a review of treatment options. CNS Drugs 2006;20:443–64 [DOI] [PubMed] [Google Scholar]
- 3.Montagna P, Gallassi R, Medori R, et al. MELAS syndrome: characteristic migrainous and epileptic features and maternal transmission. Neurology 1988;38:751–4 [DOI] [PubMed] [Google Scholar]
- 4.Pavlakis SG, Phillips PC, DiMauro S, et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol 1984;16:481–8 [DOI] [PubMed] [Google Scholar]
- 5.Lizuka T, Sakai F, Suzuki N, et al. Neuronal hyperexcitability in stroke-like episodes of MELAS syndrome. Neurology 2002;59:816–24 [DOI] [PubMed] [Google Scholar]
- 6.Goodfellow JA, Dani K, Stewart W, et al. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes: an important cause of stroke in young people. Postgrad Med J 2012;88:326–34 [DOI] [PubMed] [Google Scholar]
- 7.Castillo M, Kwock L, Green C. MELAS syndrome: imaging and proton MR spectroscopic findings. AJNR Am J Neuroradiol 1995;16:233–9 [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z, Zheng D, X Wang J, et al. Apparent diffusion coefficients of metabolites in patients with MELAS using diffusion-weighted MR spectroscopy. AJNR Am J Neuroradiol 2011;32:898–902 [DOI] [PMC free article] [PubMed] [Google Scholar]