

Abstract
Altered DNA methylation occurs ubiquitously in human cancer from the earliest measurable stages. A cogent approach to understanding the mechanism and timing of altered DNA methylation is to analyze it in the context of carcinogenesis by a defined agent. Epstein-Barr virus (EBV) is a human oncogenic herpesvirus associated with lymphoma and nasopharyngeal carcinoma, but also used commonly in the laboratory to immortalize human B-cells in culture. Here we have performed whole-genome bisulfite sequencing of normal B-cells, activated B-cells, and EBV-immortalized B-cells from the same three individuals, in order to identify the impact of transformation on the methylome. Surprisingly, large-scale hypomethylated blocks comprising two-thirds of the genome were induced by EBV immortalization but not by B-cell activation per se. These regions largely corresponded to hypomethylated blocks that we have observed in human cancer, and they were associated with gene-expression hypervariability, similar to human cancer, and consistent with a model of epigenomic change promoting tumor cell heterogeneity. We also describe small-scale changes in DNA methylation near CpG islands. These results suggest that methylation disruption is an early and critical step in malignant transformation.
The original discovery of altered DNA methylation in cancer involved widespread loss of DNA methylation (Feinberg and Vogelstein 1983). Recently, whole-genome bisulfite sequencing (WGBS) by us and others has shown that approximately one-half of the tumor genome is hypomethylated, involving one-third of single-copy genes (Hansen et al. 2011; Berman et al. 2012). Furthermore, this hypomethylation includes large blocks corresponding to large organized chromatin lysine (K)-modified regions associated with the nuclear lamina, called LOCKs, blocks, or LADs (Guelen et al. 2008; Wen et al. 2009; Hawkins et al. 2010).
The timing and role of altered DNA methylation in cancer has not been fully worked out, although some changes like hypomethylation occur at the earliest discernible time points in human tumor formation (Goelz et al. 1985; Teschendorff et al. 2012). One way to approach the issue of epigenetic timing mechanistically is to relate epigenetic changes to known causal agents. One such agent is Epstein-Barr virus (EBV), associated with Burkitt's lymphoma, nasopharyngeal carcinoma, post-transplant lymphoproliferative disease, and to a large extent, Hodgkin's disease (Rickinson and Kieff 2007).
Epstein-Barr virus (EBV) is a tumorigenic human herpesvirus that promotes proliferation and inhibits apoptosis in infected cells. The association of EBV with cancer was initially discovered in Burkitt's lymphoma, and a causative link with the disease was suggested by the finding that EBV infection immortalized B-lymphocytes in vitro, generating continuously proliferating lymphoblastoid cell lines (LCL) (Pope et al. 1973). After the primary infection, EBV persists in memory B-cells in an inert latent state (Kieff and Rickinson 2007). In addition to this state, designated as latency type 0, there are three additional latency types, called types I, II, and III, characterized by the differential expression of latent viral proteins (Thorley-Lawson 2001; Young and Rickinson 2004). Although cells with an LCL (type III) phenotype can be found in infectious mononucleosis, their proliferation is controlled by the immune system. Although type III latency viral products (especially EBNA-2 and LMP-1) are essential to induce and maintain B-cell activation and proliferation, and several cellular pathways and genes targeted by these proteins have been described (Kieff and Rickinson 2007), the process of EBV-induced immortalization is still not well understood. Several observations, however, suggested that the epigenetic reprogramming of the host genome by viral products may play a central role in the process of immortalization (Niller et al. 2012).
An adequate characterization of EBV-induced alterations in the host methylome is lacking, since most publications analyzed the effects of EBV infection on only a few selected genes (Tsai et al. 2002; Paschos et al. 2009), and prior genome-wide DNA methylation studies have not used biological measurement and/or analytical methods that would permit detection of the major finding in this study.
Here we performed whole-genome bisulfite sequencing on quiescent, CD40/IL4-activated, and matched EBV transformed B-cells in order to characterize their methylome at high resolution. Including CD40/IL4-activated cells in our study design enabled us to identify DNA methylation changes specific to the process of B-cell immortalization by EBV, in contrast to earlier studies. In addition, the comprehensive methylome profile we ascertained by WGBS allowed us to discover the presence of long hypomethylated blocks following EBV transformation.
Results
In order to address the epigenetic effects of EBV transformation independent of activation effects per se or confounding genetic differences between subjects, we chose to study matched B-cells from three healthy volunteers. Furthermore, in contrast to previous studies (Grafodatskaya et al. 2010; Sun et al. 2010; Caliskan et al. 2011; Sugawara et al. 2011; Aberg et al. 2012), we directly compared EBV immortalized cells to activated B-cells. Our reasoning was that since EBV infection and antigen stimulation induce similar gene pathways in quiescent B-cells (Calender et al. 1987; O'Nions and Allday 2004), we compared DNA methylation levels between EBV-transformed and CD40L/IL4-activated cells to identify differences specific to the process of transformation (Fig. 1). We also compared activated B-cells to quiescent B-cells (Fig. 1). Since we did not know in advance where the most significant differences in methylation might occur, we performed whole-genome bisulfite sequencing (WGBS). This method also allows us to detect large-scale changes to the methylome, such as large hypomethylated blocks.
Figure 1.

DNA methylation and gene expression changes following EBV transformation and CD40L/IL4 activation. The number of differentially methylated regions (DMRs), differentially expressed (DE) genes, and bases covered by hypomethylated blocks is listed for each condition. Numbers after the slash are at a family-wise error rate of <5% using permutation testing. Differences specific to the process of transformation (dashed line) were identified by comparing EBV immortalized B-cells to CD40L/IL4 activated B-cells.
We generated sequencing data to a depth of 8–12 times across samples and analyzed it using BSmooth (Hansen et al. 2012), an algorithm designed for determining DNA methylation in low-coverage WGBS data. Our previous work demonstrated that BSmooth reliably estimates methylation levels at single base-pair resolution by borrowing information from nearby CpGs. BSmooth also allows for the identification of small and large-scale changes in DNA methylation, properly accounting for biological replicates. Using BSmooth we were earlier able to discover large-scale blocks of hypomethylation in colon cancer (Hansen et al. 2011), which was later confirmed in an independent study (Berman et al. 2012).
We performed quality control, mapping, and smoothing of our WGBS data using the BSmooth pipeline (Methods; Supplemental Fig. 1; Supplemental Tables 1,2). After filtering out reads with methylation bias and low-mapping quality (see Methods), we obtained on average 180 million reads (±26 million reads) that contained at least one CpG site (Supplemental Table 2). This amount of sequencing provided us with at least one sequencing read for 24.3 million CpG sites per sample (±1 million CpG sites) (Supplemental Table 2). Bisulfite conversion rates were estimated to be 96%–99% across samples using lambda phage DNA as a spike-in control (Methods; Supplemental Table 2).
We observed extensive differences in the genome-wide distribution of DNA methylation between EBV-transformed and activated B-cells from each of the three individuals. Remarkably, there was a dramatic change in large regions of the genome, with 10,565 large-scale blocks of hypomethylation encompassing 2.18 Gb of the genome, of which 485 were longer than 1 Mb (Fig. 2A; Supplemental Data 1). As a control, we permuted the data labels and reran the analysis a total of nine times. We use these permutations to compute a family-wise error rate (corrected for multiple testing) for each of these blocks. This error rate describes how often we see another block of similar length and effect size anywhere in the genome and in any of the permutations. Due to the small number of permutations, this error rate has a very coarse resolution, and we choose a stringent cutoff of 5%. This cutoff translates to a requirement that we cannot see an equally good block in any of the permutations anywhere in the genome. Since we believe this to be a very stringent cutoff, perhaps too stringent, we report results before and after permutation testing. Surprisingly, we found a total of 3888 blocks encompassing 1.96 Gb at a family-wise error rate of <5%. This confirms the large amount of difference between the two conditions.
Figure 2.

Large hypomethylated genomic blocks in EBV-immortalized B-cells. (A) Smoothed methylation values from bisulfite sequencing data for quiescent (dark blue), activated (light blue), and EBV immortalized (red) B-cells, top panel. The smoothed methylation values estimate average DNA methylation on the kilobase scale. Hypomethylated EBV blocks are demarcated in pink shading. The bottom panel shows smoothed DNA methylation values for normal colon (purple) and colon tumor (orange) samples, from Hansen et al. (2011). (B) Genome-wide distribution of DNA methylation. The large block domains appear as a large bump around 0.6. (C) Simulations show that block locations co-occur. For each of the three EBV transformed samples, we find sample-specific blocks by comparing the sample in question to all three activated samples. For each set of sample-specific blocks, we computed the distance from the observed start position of each sample-specific block to the closest start position in the other two sets. The boxplot on the left shows the distribution of these distances, pooled across all six comparisons. The boxplot on the right shows the expected distribution of distances under the null hypothesis that the block start positions do not agree. The smaller values seen in the left boxplot demonstrates that the start positions of the sample-specific blocks co-occur much more frequently than expected by chance. (D) Enrichment of hypervariable genes in EBV-transformed cell lines, inside EBV blocks. The x-axis denotes a standard deviation cutoff, above which genes are considered hypervariable. The y-axis is the log2 odds ratio of enrichment of these hypervariable genes inside EBV blocks. The gray shaded area is a 95% confidence interval, and values above 0 mark enrichment.
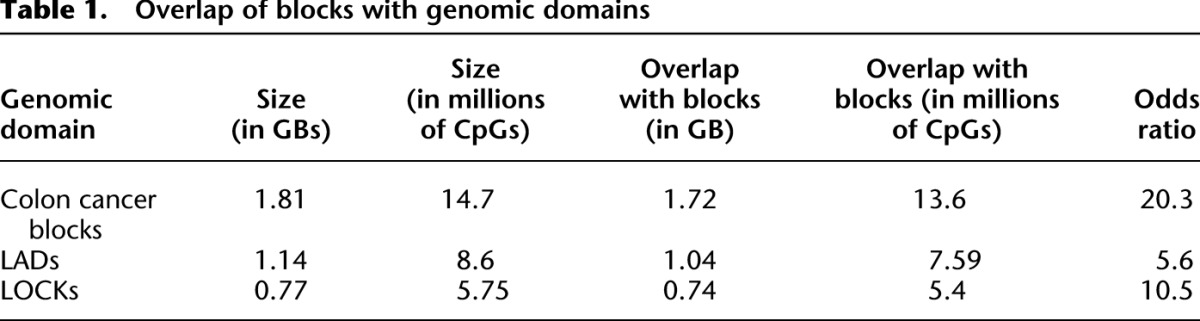
While BSmooth is capable of estimating methylation levels at single-base resolution, the smoothed methylation values in Figure 2A estimate methylation levels at the kilobase scale. These blocks are relatively gene poor and contain roughly one-third of the annotated UCSC gene promoters despite encompassing roughly two-thirds of the genome. The methylation level inside these hypomethylated blocks is generally above 50% (Supplemental Fig. 2). To ensure that copy-number variation did not confound our results we estimated genome-wide CNV levels using our bisulfite-converted sequencing reads, and did not find any large-scale copy-number changes. We also compared the position of EBV blocks with other large-scale genomic domains, specifically LADs (Guelen et al. 2008) and LOCKs (Wen et al. 2012), and found a significant overlap with both of these domains (Table 1) (P < 2.2 × 10−16). Note that these epigenetic marks are tissue specific and that we used data derived from lung fibroblasts for LADs (Guelen et al. 2008) and pulmonary fibroblasts for LOCKS (Wen et al. 2009). Hence, we may be underestimating the true correlation between hypomethylated blocks and these domains. We also compared the EBV blocks with epigenetics marks from the ENCODE Project obtained on the Tier 1 cell line GM12878, which is a lymphoblastoid cell line. We used all available ChIP-seq tracks on this cell line, for a total of 192 tracks, including all transcription factors and histone marks assays. We found enrichment of H3K27me3 (a repressive mark) inside the EBV blocks (odds-ratio = 3.48, P < 2.2 × 10−16 and present in 97% of all blocks) and depletion of all transcription factors. We also examined block boundaries (defined as 10 kb on either side of a block). Again we found enrichment of H3K27me3 (odds-ratio = 2.16, P < 2.2 × 10−16 and present in 72% of all boundaries), but also a number of other marks involved in either gene regulation or chromatin stability during mitosis. BATF (odds-ratio = 1.55, P < 2.2 × 10−16, present in 15% of block boundaries) encodes a transcription factor that complexes with IRF4 in targeting genes during immune activation (Glasmacher et al. 2012). H4K20me1 (odds-ratio = 1.36, P < 2.2 × 10−16, present in 48% of block boundaries) is thought to regulate S-phase progression and genome stability (Jorgensen et al. 2013) and is a WNT-signaling mediator (Li et al. 2011). EZH2 (odds-ratio = 1.59, P < 2.2 × 10−16, present in 66% of block boundaries) encodes a canonical histone methyltransferase for H3K27me3 and is both overexpressed and mutated in lymphomas and other neoplasms (Chase and Cross 2011). The protein encoded by RAD21 (odds-ratio = 1.43, P < 2 × 10−16, present in 30% of block boundaries) associates with mitotic chromatin stability (Deardorff et al. 2012). The protein encoded by BCL11A (odds-ratio = 1.44, P < 2 × 10−16, present in 11% of block boundaries) has been shown to interact with a complex of transcriptional corepressors (RCOR1/KDM1A) in modulating hemoglobin switching and fetal hemoglobin silencing (Xu et al. 2013). Note that since ENCODE only profiled EBV-transformed cells, we cannot observe whether these marks change as a consequence of transformation.
Table 1.
Overlap of blocks with genomic domains
We then searched for large-scale DNA methylation differences between activated and quiescent cells and found hypomethylated blocks encompassing 60.7 Mb of the genome with the largest block being just under 95 kb (Supplemental Data 1). Of these, <1%, i.e., 0.6 Mb, were hypermethylated, which we think is likely noise and do not report. Using a permutation approach as above (see Methods), we found that none of these putative activation blocks had a family-wise error rate of <5%. The close agreement between activated and quiescent cells is apparent in Figure 2A, where the kilobase-scale methylation estimates for the activated cells very closely track the estimates for the quiescent cells. Similarly, the genome-wide distribution of DNA methylation showed little or no differences between activated and quiescent cells, but a dramatic shift in distribution was observed between activated and EBV-transformed cells (Fig. 2B). We conclude that the formation of hypomethylated blocks occur specifically in the immortalization step and is not associated with activation.
To further pinpoint the timing of the hypomethylation and to investigate the possibility that an increased number of cell divisions could lead to hypomethylation, we examined additional samples at 16 d and 3 wk post EBV infection (one sample each time point) and CD40 activation (two samples at each time point), as 3 wk is the longest that CD40 activation can be maintained in culture. We repeated the analysis with these new samples, comparing each of the four new conditions to the three CD40-activated samples described above (which were measured at day 6). For the activated samples, at day 16 we found 10,433 blocks encompassing 267 Mb, of which one block/0.03 Mb had a family-wise error rate of <5% using a permutation procedure. At 3 wk post-activation we found 11,195 blocks/242 Mb, of which three blocks/0.22 Mb had a family-wise error rate of <5%. In conclusion, we see no evidence of extensive large-scale hypomethylation at up to 3 wk post-activation. For the EBV infected cells we also observed, at most, very small differences at these two time points. Specifically, at day 16 we found 8984 blocks/165 Mb reducing to 194 blocks/3.89 Mb at a family-wise error rate of <5%, and at wk 3 we found 9418 blocks/208 Mb reducing to 13 blocks/1.45 Mb at a family-wise error rate of <5%. We conclude that the large-scale hypomethylation described above occurs after 3 wk post-infection, but before 6 wk.
We compared the EBV blocks to the large blocks of hypomethylation that we previously discovered in colon cancer (Hansen et al. 2011). The degree of overlap between hypomethylated EBV and cancer blocks was a striking 1.72 Gb. The consistency between the block boundaries between EBV and colon cancer was also remarkably high (Fig. 2A). An analysis of individual specific blocks confirms this to be significant (P < 0.001) (Fig. 2C; Methods).
By the same token, there was a 25% difference in location of the two sets of blocks, with 462 Mb-hypomethylated and 96 Mb-hypermethylated blocks unique to EBV-transformed cells. Thus, the specific blocks are not identical across all cancer mechanisms. These differences between hypomethylated blocks in EBV transformed cells and colon cancer appeared meaningful, based on a comparison of our data to gene-expression barcode (Zilliox and Irizarry 2007; McCall et al. 2011) and publicly available microarray data (Runne et al. 2007; Gyorffy et al. 2009) (see Methods) between EBV transformed cell lines and colon cancer. Specifically, genes inside EBV blocks, but not in colon-cancer blocks, were more likely to be expressed in normal colon and be transcriptionally silent in lymphocytes (OR = 3.5, P < 2.2 × 10−16). The converse is true for genes inside colon-cancer blocks but not in EBV blocks (OR = 4.0, P < 2.2 × 10−16). This correlation suggests that EBV and colon-cancer blocks have biological implications, and our findings are not due to chance.
In Hansen et al. (2011) we found hypomethylated blocks to be enriched for genes with hypervariable expression in colon cancer, which could drive tumor cell heterogeneity. To investigate whether the same mechanism might be in play during EBV immortalization, we examined the methylation variability in our current data and noted that the hypomethylated blocks were also notably more variable in methylation in the EBV immortalized cells than in activated B-cells (Fig. 2A). When we compared the between-sample variation in methylation for both cancer and EBV transformed cells, we found EBV hypomethylated blocks to be much more consistent, with cancer samples showing an increased variance in 98% of common blocks (t-stat, P < 2.2 × 10−16) (Fig. 2A).
In order to relate the hypervariable methylation of blocks after EBV immortalization to gene-expression variability, we needed to examine large numbers of samples for gene expression in order to generate such a metric. We reasoned that since the HapMap project was based on EBV-immortalized cell lines, we could use publicly available data on gene expression from 257 EBV-transformed HapMap samples (Choy et al. 2008) to address this question. We normalized the array data using the gene-expression barcode and discarded unexpressed genes. The remaining genes were then sorted based on whether they were located inside or outside of hypomethylated blocks, and the standard deviation in expression was calculated. This analysis revealed EBV hypomethylated blocks to be enriched for highly variable genes, no matter which standard deviation cutoff was used to define high variability (Fig. 2D).
Surprisingly, the genes exhibiting the highest degree of hypervariability in hypomethylated blocks are genes encoding immunoglobulin variable domains including IGHV3-7, IGHV3-9, IGHV3-21, IGHV3-23, and IGKV4-1. A functional annotation analysis of genes with hypervariable expression in the blocks (most relevant functionally) shows that the most enriched category is immune response genes (P = 1.2 × 10−9). The presence of these genes in hypomethylated blocks is intriguing and suggests that it is feasible that hypomethylated blocks have properties that enable inactive genes to be induced, perhaps in a coordinated manner, when required.
In addition to large-scale blocks, we also identified small DMRs associated with transformation. Note that these latter results would be akin to the studies done in a more limited way on microarrays by other investigators (Grafodatskaya et al. 2010; Sun et al. 2010; Caliskan et al. 2011; Sugawara et al. 2011; Aberg et al. 2012), although the current results are more comprehensive, based on WGBS. These small DMRs were typically 250–500 bp in length, with the longest being 2.5 kb, and encompassed roughly 1 Mb of the genome. In total, we identified 2970 small DMRs, of which 1502 were hypermethylated and 1468 were hypomethylated following EBV transformation (Supplemental Data 2). Using permutation testing, as for the block analysis above, all of these DMRs have a family-wise error rate of <5%. In total, 644 of these DMRs overlapped and another 588 DMRs were within 2 kb of an annotated UCSC gene promoter. We then looked for methylation differences between quiescent and activated B-cells and identified 1014 small DMRs associated with activation (Supplemental Data 2). These small DMRs covered 273 kb, and were unevenly spread between hypermethylation (293) and hypomethylation (721). However, only one of these DMRs has a family-wise error rate of <5%. In total, 235 of these DMRs overlapped and another 191 DMRs were within 2 kb of an annotated UCSC gene promoter. Using a GO analysis, genes with a hypermethylated small DMR within 2 kb of their promoter region were found to be enriched for genes associated with translation (P = 7.81 × 10−8), chromatin reorganization (P = 4.34 × 10−3), and RNA catabolism (P = 6.37 × 10−3). No enrichment was found for any functional categories for genes near hypermethylated small DMRs. We found that genes harboring a small DMR within 2 kb of their promoter were not enriched for genes with hypervariable expression, using the same analytical strategy we used to associate hypervariable genes with hypomethylated blocks.
As above, we also examined the time points of 16 d and 3 wk post-activation and EBV infection using two activation samples and one EBV-infected sample measured at the two time points. As for the block analysis, we initially identify some number of small DMRs, but almost none are present after permutation testing. Specifically, comparing samples at 16 d post-activation to samples at day 6 after activation, we found 1788 small DMRs/432 kb, but none of these have a family-wise error rate of <5%. At 3 wk post-activation we found 2870 small DMRs/746 kb, but only three of these DMRs have a family-wise error rate of <5%. We conclude that after permutation testing there remains little evidence of small differentially methylated regions between activation at day 6 and 16 d and 3 wk post-activation. At 16 d post-EBV infection we find 938 small DMRs/246 kb of which seven DMRs have a family-wise error rate of <5%, and at 3 wk post-EBV infection we find 1641 DMRs/406 kb, of which five DMRs have a family-wise error rate of <5%.
To validate the small DMRs, we performed bisulfite pyrosequencing on a number of small DMRs. Based on the estimated mean difference between EBV-transformed cells and activated cells, we picked seven high-ranked DMRs (ranks between 16 and 186 out of 2970) and two DMRs much lower ranked (ranks 2328 and 2586 out of 2970). In the seven high-ranking DMRs, bisulfite pyrosequencing demonstrated a large decrease in DNA methylation of EBV transformed cells compared with quiescent and activated B-cells, which is in agreement with our whole-genome bisulfite sequencing results (Supplemental Figs. 3,4). Differential methylation was not observed in the two low-ranking DMRs.
To determine the relationship between DNA methylation and functional properties, we next measured gene expression using Affymetrix microarrays on the same nine samples used for WGBS. Gene-expression barcodes were used to normalize the array data. Comparing EBV transformed and activated cells, we identified 1769 genes to be differentially expressed above background with a fold change of two or greater (Supplemental Data 3). A total of 959 of these genes were up-regulated and 810 were down-regulated. We identified genes with promoters within 2 kb or overlapping a small DMR and found, as expected, an inverse relationship between DNA methylation and gene expression with a correlation of −0.36 (P < 2.2 × 10−16) (Supplemental Figs. 5, 6). Comparing activated and quiescent cells, we found 4502 genes to be differentially expressed above background with a fold change of two or greater (Supplemental Data 3). These genes include markers of activation (FCER2) and proliferation (CCND2, CCNE1, CCNE2), highlighting the need for using activated cells as controls when studying EBV transformation. We also found the lamin genes LMNB1, LMNB2, and LMNA to be more than fourfold up-regulated between quiescent and activated cells, but unchanged between EBV transformed and activated cells. We performed TaqMan qPCR, and measured relative expression levels for the CCND2, CCNE1, CCNE2, FCER2, LMNA, LMNB1, and LMNB2 genes (Supplemental Fig. 7; Methods). Consistent with our microarray results, we found the expression of the cyclin and the FCER2 genes, which are activation markers, to be up-regulated in activated cells compared with quiescent cells. We also found these genes to be up-regulated in EBV transformed cells. In addition, we found overexpression of LMNA, LMNB1, and LMNB2 genes to be up-regulated in both EBV transformed and activated cells compared with quiescent cells. Similar to the microarray results, the expression levels between the EBV transformed and activated cells did not differ significantly in these three genes.
Discussion
In summary, we show here that EBV immortalization of B-lymphocytes causes widespread demethylation of the genome, affecting 2.18 Gb and including one-third of genes. The study adds mechanistic weight to an emerging and growing story of large domains providing a higher-order organization of the genome that are functionally altered in development and disease. While not entirely overlapping functionally or physically, there is nevertheless strong correspondence between lamin-associated domains, large regions with heterochromatin-associated lysine methylation, alternately called LOCKs or blocks and characterized by H3K9me2 and H3K27me3, partially methylated domains in fibroblasts, and hypomethylated blocks in human colorectal cancer and likely other malignancies. These domains change during iPSC reprogramming, comparing ES and differentiated cells, and between cancer and normal (Reddy and Feinberg 2012). Moreover, these hypomethylated blocks, and the genes contained within them, overwhelmingly correspond to those seen in cancer, with an overlap of 1.72 GB. It is remarkable that the location of hypomethylated blocks between EBV-transformed lymphocytes and colon tumors correlate highly with each other despite the fact that lymphocytes and colon cells are very different in phenotype.
It is striking that the hypomethylated blocks we observe are specific to the immortalization process itself and not to B-cell activation by the oncogenic virus. Previous studies did not use the matched control of activated B-cells, and therefore many of the differences they saw between EBV-immortalization and control cells was likely related to activation, but not the key step of immortalization. Indeed, our own data show extensive differential expression and methylation as a result of activation, with 4502 differentially expressed genes and 1014 small DMRs. Essentially no blocks appear from activation per se. One previous study (Aberg et al. 2012) did use a whole-genome tiling array that in theory could see the same hypomethylated blocks we report here. However, those investigators performed quantile normalization, which by design removes global methylation differences between samples, and thus the finding we present here. None of the previous studies used whole-genome bisulfite sequencing.
Finally, the present study suggests that block hypomethylation is an early event in human cancer, consistent with its observation in premalignant adenomas as well as colorectal carcinomas (Hansen et al. 2011; Berman et al. 2012). What is the functional importance of these changes? While much work needs to be done to understand its role, an important clue comes from examination of gene expression. It is noteworthy that both hypomethylated blocks in EBV transformed cell lines and colon tumors are enriched for genes that exhibit hypervariable gene expression, consistent with a role in establishing tumor cell heterogeneity early in malignant transformation.
Methods
Collection, activation, and EBV immortalization of B-cells
Blood samples were collected from three healthy donors at the Karolinska Hospital (Stockholm) following institutional guidelines for human subjects research (study #02-277). B-cells were isolated by positive selection using CD19 Dynabeads PanB magnetic beads (Invitrogen). An aliquot of the purified primary B-cells was frozen and used as the quiescent cells in our study. A second aliquot of the purified B-cells was activated with CD40 Ligand (CD40L) and Interleukin-4 (IL4) utilizing a previously described procedure (Kis et al. 2005). The activated B-cells were kept in culture for up to 3 wk by twice weekly replacement of media with addition of fresh CD40L and IL4. A third aliquot of B-cells was incubated with B95-8 cell supernatant for 1.5 h at 37°C in order to infect them with EBV. Aliquots of infected cells were collected at different time points for analysis: day 16, week 3, and week 6 (to ensure that EBV immortalization had occurred).
Whole-genome bisulfite sequencing
Bisulfite sequencing libraries were constructed using the Illumina TruSeq DNA Library Preparation Kit protocol with the following modifications. Thirty nanograms of unmethylated lambda DNA were added to 3 μg of genomic DNA prior to shearing in order to monitor the efficiency of the bisulfite conversion. The sheared DNA ends were then repaired using 1× NEB Buffer2, 400 nm each of dATP, dGTP, and dTTP (dCTP was not included), 15 units of T4 DNA polymerase (NEB), 5 units of Klenow DNA polymerase (NEB), and 50 units of T4 Polynucleotide kinase (NEB). In the bisulfite conversion step, 24 μL of formamide was added to an equal volume of DNA and incubated at 95°C for 5 min. Subsequently, 100 μL of Zymo Gold bisulfite conversion reagent (Zymo) was added, and the mixture was incubated for 8 h in 50°C. Samples were then desulphonated and purified using spin columns following the Zymo EZ DNA Methylation-Gold Kit protocol. The bisulfite converted library was amplified in 1× PCR buffer, 0.2 mM dNTP, 5 μL of the TruSeq PCR Primer Cocktail, 5 units of Taq (Denville), and 0.25 units of PfuTurbo DNA polymerase (Stratagene). The DNA was subjected to 10 cycles of PCR.
Bisulfite pyrosequencing
Four hundred nanograms of genomic DNA was bisulfite converted using the EZ DNA Methylation-Gold Kit. Nested PCR was performed using the primers listed in Supplemental Table 3. The annealing temperature used for all PCR reactions was 50°C. The resulting PCR reactions were used directly for pyrosequencing (Tost and Gut 2007) in a Pyromark 96 ID instrument (Qiagen). The sequencing primers used for pyrosequencing are listed in Supplemental Table 4.
Gene expression
Total RNA was extracted from the B-cells using the Qiagen RNeasy Mini Kit. Two hundred fifty nanograms of total RNA were then hybridized onto Affymetrix GeneChip Human Genome U133 Plus 2.0 arrays.
qPCR
Gene expression assays were performed using TaqMan predesigned assays purchased from Life Technologies, Inc. The catalog numbers for the assays are Hs00233627_m1 for FCER2 (CD23), Hs00153380_m1 for CCND2 (cyclin D2), Hs01026536_m1 for CCNE1 (cyclin E1), Hs00180319_m1 for CCNE2 (cyclin E2), Hs01059210_m1 for LMNB1 (Lamin B1), Hs00383326_m1 for LMNB2 (Lamin B2), and Hs00153462_m1 for LMNA (Lamin A). Expression between samples was normalized using the beta glucuronidase (GUSB) gene (Hs00939627_m1). The reactions were carried out in an ABI Prism 7900HT real time PCR machine following the manufacturer's recommended protocol.
Mapping and quality control of WGBS reads
We ran the BSmooth (Hansen et al. 2012) bisulfite alignment pipeline (version 0.4.5-beta) on the 100-by-100 bp HiSeq 2000 paired end sequencing reads obtained for each sample using Bowtie 2 version 2.0.0-beta8 (Langmead and Salzberg 2012) and the GRCh37 build of the human genome including sex chromosomes, nonchromosomal sequences, and mitochondrial sequence, as well as the genome for lambda phage (accession NC_001416.1) and for Epstein-Barr virus (accession AJ507799.2). Supplemental Table 1 summarizes alignment results. We were unable to determine bisulfite conversion rates for samples quiescent 2 and EBV immortalized 3 since we had too few reads originating from the lambda genome, likely because lambda DNA added to these samples was degraded.
We then used BSmooth to extract read-level measurements, and we filtered out unreliable read-level measurements in three ways. First, we removed read-level measurements from alignments with mapping quality less than 10, indicating that the read aligner could not place the read in its place of origin with high confidence. Second, we removed read-level measurements where the read nucleotide's base quality was less than 20, indicating that the sequencing software had relatively low confidence in the base call. Finally, we removed read-level measurements from the 5′ most 10 nucleotides of both mates, based on inspecting the “M-bias” plot (Hansen et al. 2012) (Supplemental Fig. 1). After filtering, we used BSmooth to sort read-level measurements by genome coordinate and compile them into a table summarizing methylation evidence at each CpG in the reference genome. Supplemental Table 2 summarizes the read-level measurements obtained and how they were filtered.
Smoothing WGBS data and identification of small DMRs and large-scale blocks
We used BSmooth to identify small DMRs and large hypomethylated blocks as previously described (Hansen et al. 2011; Hansen et al. 2012). We analyzed CpGs that had at least a coverage of 2 in two of the three main samples for each condition (quiescent, activated, transformed). We used the same cutoffs as previously (Hansen et al. 2011), specifically a t-statistics cutoff of −4.6,4.6 for small DMRs and −2,2 for blocks. We estimated variance assuming that samples in all conditions had equal variance. Small DMRs were ranked by absolute mean difference, and blocks were ranked by length. Unlike earlier work, we did not postprocess inferred blocks to break them if a block contains an unmethylated CpG island, following the suggestion of Berman et al. (2012).
Permutation testing
Depending on the comparison, we had either nine null permutations (for three vs. three comparisons and for two vs. three comparisons) or three null permutations (one vs. three comparisons). The relevant method was applied to the new sample labels and blocks and/or DMRs were identified using the methods described above, resulting in a set of null blocks/DMRs for each permutation. We then asked, for each original block/DMR, in how many permutations did we see a null block/DMR anywhere in the genome with the same or better characteristics as the original block/DMR. Since we are comparing each original block/DMR against anything found anywhere in the genome, we are correcting for multiple testing. The characteristics were length (in base pairs) and signed mean methylation difference for blocks and size (in CpGs), and signed mean methylation difference for small DMRs. This resulted in a number between 0 and the number of permutations, for each original block/DMR, and an original block/DMR was conservatively found to have a family-wise error rate of <5% if this number was zero.
Publicly available data
Large-scale hypomethylated blocks in cancer (Hansen et al. 2011), LADs (Guelen et al. 2008), and LOCKs (Hansen et al. 2012; Wen et al. 2012) were retrieved from public sources. Gene expression data for HapMap samples were obtained from GEO GSE11582 (Choy et al. 2008), using only samples that were processed at the Broad Institute and were labeled “technical replicate 1” for a total of 257 samples. Gene expression data for colon cancer was obtained from GEO GSE4183 (Gyorffy et al. 2009), for a total of eight normal samples and 15 cancer samples. Gene expression data for lymphocytes was obtained from GEO GSE8762 (Runne et al. 2007) for a total of 10 samples.
Overlap with ENCODE data and other large domains
We obtained 192 ENCODE tracks for the GM12878 cell line from The ENCODE Project Consortium (2012) data hub at UCSC. Each track consists of a number of genomic intervals, and we divided the genome into four compartments based on the relationship between EBV blocks and the track in question (inside both, outside both, etc.). We only used EBV blocks with a family-wise error rate of <5%. Because hypomethylation only occurs at CpGs, and because we only have enough data on a subset of CpGs (which we call “covered”) we counted the number of covered CpGs inside each of the four compartments, forming a 2 × 2 table of CpGs. We calculated an odds-ratio for this table and used Fisher's exact test to compute a P-value. The same analysis was performed for other large domains described in the manuscript, such as LOCKs, LADs, and colon-cancer blocks.
Analysis of gene expression microarray data
All gene-expression array data was normalized using frozen RMA (McCall et al. 2010), and we used the gene-expression barcode (Zilliox and Irizarry 2007; McCall et al. 2011) to decide whether a gene was expressed in a given condition by requiring an average Z-score > 5 (from the gene-expression barcode).
To decide which genes were differentially expressed between the quiescent, activated, and transformed state, we used limma (Smyth 2004) from Bioconductor (Gentleman et al. 2004) and utilized an empirical Bayes shrinkage method to estimate a gene-specific variance. In order for a gene to be differentially expressed between two conditions, it had to be expressed (using the gene-expression barcode) in at least one of the two conditions, it had to have an estimated absolute log2 fold change > 1, and it had to have a Benjamini-Hochberg adjusted P-value < 5%.
Copy-number analysis
We performed copy-number analysis as previously described (Hansen et al. 2011).
Co-occurrence of sample-specific blocks
We evaluated the co-occurrence of sample-specific blocks as described previously (Hansen et al. 2011). Briefly, we found sample-specific blocks by in turn comparing each of the EBV transformed samples to the three activated samples (BSmooth requires multiple samples in the reference group). For each chromosome (excluding Y) we computed the observed distribution of distances between consecutive start locations of the sample-specific blocks. For each chromosome, we simulated 1000 sets of start positions of blocks on that chromosome, according to the observed distribution. We constrained the simulated start positions to CpG locations, accounting for the nonrandom distribution of CpGs across the genome. Next, we (in turn) picked one of the three individuals as reference, and computed, for each start position, the distance to the nearest start position of a block for each of the two other sets of sample-specific individuals. This serves as the observed pairwise distances. Next, we computed the same distances from the reference to each of the 1000 simulated sets of blocks, yielding 1000 simulated expected pairwise distances with the specific sample as a reference. Since this was done using in turn each of the three EBV samples as reference, this yielded 3 × 2 = 6 different sets of pairwise distances and 3 × 1000 = 3000 expected pairwise distances. Figure 2C shows the distribution of the observed and expected distribution, pooled across all chromosomes.
GO analysis
Small DMRs were mapped to known genes by establishing whether the small DMR was within 2 kb of a promoter region of a gene. As promoter regions, we used a 2-kb interval around the transcription start site of UCSC known genes. The known gene identifiers were converted to official gene symbols using the kgXref table from the UCSC Genome Browser website. Duplicate gene names were removed so that each gene was only counted a single time during the analysis. Hyper- and hypomethylated genes were then separated into two different files, and analysis was performed separately for each list using the DAVID Functional Annotation Tool (Huang da et al. 2009a,b). The analysis was performed using the GOTERM_BP_FAT annotation, which is the summarized version of Biological Processes in gene ontology. All tests were corrected for multiple testing using the Benjamini procedure implemented in DAVID. For the functional annotation analysis of hypervariable genes in blocks, we used all genes on the 133A array as background.
Data access
Whole-genome bisulfite sequencing and gene expression data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE49629.
Acknowledgment
This work was supported by NIH Grant CA54358 to A.P.F. In addition, K.D.H. was partially supported by NIH Grant GH005520.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.157743.113.
Freely available online through the Genome Research Open Access option.
References
- Aberg K, Khachane AN, Rudolf G, Nerella S, Fugman DA, Tischfield JA, van den Oord EJ 2012. Methylome-wide comparison of human genomic DNA extracted from whole blood and from EBV-transformed lymphocyte cell lines. Eur J Hum Genet 20: 953–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, et al. 2012. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 44: 40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calender A, Billaud M, Aubry JP, Banchereau J, Vuillaume M, Lenoir GM 1987. Epstein-Barr virus (EBV) induces expression of B-cell activation markers on in vitro infection of EBV-negative B-lymphoma cells. Proc Natl Acad Sci 84: 8060–8064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliskan M, Cusanovich DA, Ober C, Gilad Y 2011. The effects of EBV transformation on gene expression levels and methylation profiles. Hum Mol Genet 20: 1643–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase A, Cross NC 2011. Aberrations of EZH2 in cancer. Clin Cancer Res 17: 2613–2618 [DOI] [PubMed] [Google Scholar]
- Choy E, Yelensky R, Bonakdar S, Plenge RM, Saxena R, De Jager PL, Shaw SY, Wolfish CS, Slavik JM, Cotsapas C, et al. 2008. Genetic analysis of human traits in vitro: Drug response and gene expression in lymphoblastoid cell lines. PLoS Genet 4: e1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff MA, Wilde JJ, Albrecht M, Dickinson E, Tennstedt S, Braunholz D, Monnich M, Yan Y, Xu W, Gil-Rodriguez MC, et al. 2012. RAD21 mutations cause a human cohesinopathy. Am J Hum Genet 90: 1014–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B 1983. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301: 89–92 [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. 2004. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasmacher E, Agrawal S, Chang AB, Murphy TL, Zeng W, Vander Lugt B, Khan AA, Ciofani M, Spooner CJ, Rutz S, et al. 2012. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 338: 975–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP 1985. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science 228: 187–190 [DOI] [PubMed] [Google Scholar]
- Grafodatskaya D, Choufani S, Ferreira JC, Butcher DT, Lou Y, Zhao C, Scherer SW, Weksberg R 2010. EBV transformation and cell culturing destabilizes DNA methylation in human lymphoblastoid cell lines. Genomics 95: 73–83 [DOI] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. 2008. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948–951 [DOI] [PubMed] [Google Scholar]
- Gyorffy B, Molnar B, Lage H, Szallasi Z, Eklund AC 2009. Evaluation of microarray preprocessing algorithms based on concordance with RT-PCR in clinical samples. PLoS ONE 4: e5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. 2011. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 43: 768–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Langmead B, Irizarry RA 2012. BSmooth: From whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol 13: R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. 2010. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6: 479–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA 2009a. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Jorgensen S, Schotta G, Sorensen CS 2013. Histone H4 lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Res 41: 2797–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieff E, Rickinson AB 2007. Epstein-Barr virus and its replication. In Fields virology (ed. Knipe DM, Howley PM), pp. 2603–2654. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- Kis LL, Nishikawa J, Takahara M, Nagy N, Matskova L, Takada K, Elmberger PG, Ohlsson A, Klein G, Klein E 2005. In vitro EBV-infected subline of KMH2, derived from Hodgkin lymphoma, expresses only EBNA-1, while CD40 ligand and IL-4 induce LMP-1 but not EBNA-2. Int J Cancer 113: 937–945 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Nie F, Wang S, Li L 2011. Histone H4 Lys 20 monomethylation by histone methylase SET8 mediates Wnt target gene activation. Proc Natl Acad Sci 108: 3116–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MN, Bolstad BM, Irizarry RA 2010. Frozen robust multiarray analysis (fRMA). Biostatistics 11: 242–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MN, Uppal K, Jaffee HA, Zilliox MJ, Irizarry RA 2011. The Gene Expression Barcode: Leveraging public data repositories to begin cataloging the human and murine transcriptomes. Nucleic Acids Res 39: D1011–D1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niller HH, Banati F, Ay E, Minarovits J 2012. Epigenetic changes in virus-associated neoplasms. In Patho-epigenetics of disease (ed. Minarovits J, Niller HH), pp. 179–225. Springer, New York [Google Scholar]
- O'Nions J, Allday MJ 2004. Proliferation and differentiation in isogenic populations of peripheral B cells activated by Epstein-Barr virus or T cell-derived mitogens. J Gen Virol 85: 881–895 [DOI] [PubMed] [Google Scholar]
- Paschos K, Smith P, Anderton E, Middeldorp JM, White RE, Allday MJ 2009. Epstein-Barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog 5: e1000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope JH, Scott W, Moss DJ 1973. Human lymphoid cell transformation by Epstein-Barr virus. Nat New Biol 246: 140–141 [DOI] [PubMed] [Google Scholar]
- Reddy KL, Feinberg AP 2012. Higher order chromatin organization in cancer. Semin Cancer Biol 109–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickinson AB, Kieff E 2007. Epstein-Barr virus. In Fields virology (ed. Knipe DM, Howley PM), pp. 2655–2700. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- Runne H, Kuhn A, Wild EJ, Pratyaksha W, Kristiansen M, Isaacs JD, Regulier E, Delorenzi M, Tabrizi SJ, Luthi-Carter R 2007. Analysis of potential transcriptomic biomarkers for Huntington's disease in peripheral blood. Proc Natl Acad Sci 104: 14424–14429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3. [DOI] [PubMed] [Google Scholar]
- Sugawara H, Iwamoto K, Bundo M, Ueda J, Ishigooka J, Kato T 2011. Comprehensive DNA methylation analysis of human peripheral blood leukocytes and lymphoblastoid cell lines. Epigenetics 6: 508–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YV, Turner ST, Smith JA, Hammond PI, Lazarus A, Van De Rostyne JL, Cunningham JM, Kardia SL 2010. Comparison of the DNA methylation profiles of human peripheral blood cells and transformed B-lymphocytes. Hum Genet 127: 651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Jones A, Fiegl H, Sargent A, Zhuang JJ, Kitchener HC, Widschwendter M 2012. Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med 4: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorley-Lawson DA 2001. Epstein-Barr virus: Exploiting the immune system. Nat Rev Immunol 1: 75–82 [DOI] [PubMed] [Google Scholar]
- Tost J, Gut IG 2007. DNA methylation analysis by pyrosequencing. Nat Protoc 2: 2265–2275 [DOI] [PubMed] [Google Scholar]
- Tsai CN, Tsai CL, Tse KP, Chang HY, Chang YS 2002. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc Natl Acad Sci 99: 10084–10089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP 2009. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet 41: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wu H, Loh YH, Briem E, Daley GQ, Feinberg AP 2012. Euchromatin islands in large heterochromatin domains are enriched for CTCF binding and differentially DNA-methylated regions. BMC Genomics 13: 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bauer DE, Kerenyi MA, Vo TD, Hou S, Hsu YJ, Yao H, Trowbridge JJ, Mandel G, Orkin SH 2013. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci 110: 6518–6523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LS, Rickinson AB 2004. Epstein-Barr virus: 40 years on. Nat Rev Cancer 4: 757–768 [DOI] [PubMed] [Google Scholar]
- Zilliox MJ, Irizarry RA 2007. A gene expression bar code for microarray data. Nat Methods 4: 911–913 [DOI] [PMC free article] [PubMed] [Google Scholar]