

Abstract
Genomic instability is a hallmark of human cancers, including the 5% caused by human papillomavirus (HPV). Here we report a striking association between HPV integration and adjacent host genomic structural variation in human cancer cell lines and primary tumors. Whole-genome sequencing revealed HPV integrants flanking and bridging extensive host genomic amplifications and rearrangements, including deletions, inversions, and chromosomal translocations. We present a model of “looping” by which HPV integrant-mediated DNA replication and recombination may result in viral–host DNA concatemers, frequently disrupting genes involved in oncogenesis and amplifying HPV oncogenes E6 and E7. Our high-resolution results shed new light on a catastrophic process, distinct from chromothripsis and other mutational processes, by which HPV directly promotes genomic instability.
Human papillomavirus (HPV) causes ∼610,000 cancers worldwide each year (Forman et al. 2012). Landmark studies established that HPV is the principal cause of virtually all cervical cancers (Walboomers et al. 1999) and subsets of other anogenital and head and neck cancers (Gillison et al. 2000; IARC Working Group on the Evaluation of Carcinogenic Risks to Humans 2007). The incidence of HPV-associated head and neck squamous cell carcinoma (HNSCC) in particular is increasing at an alarming rate (Chaturvedi et al. 2011). The transforming ability of oncogenic HPV types has been attributed to two viral oncoproteins, E6 and E7, which inactivate p53 and members of the pRb family (Moody and Laimins 2010), respectively. E6 and E7 expression is essential to the viral life cycle, as HPV coopts the cellular DNA replication machinery. Although their expression is sufficient for cellular immortalization in vitro (Hawley-Nelson et al. 1989; Munger et al. 1989), secondary genetic events are necessary for cancer development in vivo (Moody and Laimins 2010).
Early upon infection, HPV genomes replicate as extra-chromosomal elements in the nucleus. Viral integration frequency increases with severity of cervical precancers, and viral integrants are present in the majority of cervical cancers (Wentzensen et al. 2004; Xu et al. 2013). Integration imparts both a selective growth advantage and genomic instability of the infected cell (Pett et al. 2004) through enhanced expression and stabilization of viral oncogene transcripts (Jeon and Lambert 1995). Increased transcription has been attributed to frequent disruption of the viral repressor HPV E2 (Thierry and Yaniv 1987). HPV integrants in cervical cancers have been statistically associated with regional structural abnormalities (Peter et al. 2010), but the relationship of the virus to such variants, their detailed genomic structures, and their functional significance remain largely unknown.
To study the impact of HPV integration on host genomic structure and function, we performed whole-genome sequencing (WGS), RNA-seq, spectral karyotyping (SKY), fluorescence in situ hybridization (FISH), and other molecular assays. We chose 10 HPV-positive and -negative human cancer cell lines because they provided relatively unlimited and experimentally tractable genome samples representing cervical and head and neck cancers. Our comprehensive, genome-wide analysis, performed at single-nucleotide resolution, identified a striking and recurrent association between HPV integrants and adjacent genomic amplifications and rearrangements. WGS also confirmed that similar HPV-associated structural variations occur frequently in primary, HPV-positive head and neck cancers.
Results
We conducted WGS of 2 × 100-bp paired-end libraries from 10 cancer cell lines (two cervical and eight head and neck) and two HPV16/18-positive primary HNSCC (tonsillar and oral cavity), using the HiSeq 2000 platform (Illumina Genome Network). The cell lines were established from five HPV16-positive (HMS001, UM-SCC-104, UD-SCC-2, UM-SCC-47, UPCI:SCC090) and three HPV-negative (CAL 27, D562, SCC-25) HNSCC samples, and two HPV16-positive (SiHa, CaSki) cervical cancers (Table 1).
Table 1.
Summary of 12 cancer samples analyzed in this study: clinical origin, HPV viral status, and references

Variability in HPV genomes and transcriptomes
Significant numbers of WGS reads aligned to the reference viral genome in HPV-positive (Fig. 1A) but not HPV-negative cases. Viral copy number estimates ranged from zero to 830 per cell, using sequence depths of coverage in autosomes as a guide. Comparable viral copy number estimates were obtained by quantitative PCR (qPCR) assays of the viral E6 gene, normalized against a control (Table 1).
Figure 1.
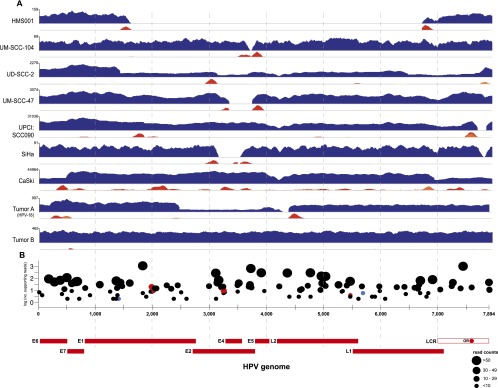
The HPV virome in human cancers. WGS reads were aligned to the HPV16 (x-axis coordinates, bottom) or HPV18 (Tumor A only) reference genomes. (A) In each row, histograms for various cancer samples indicate copy numbers of viral sequences (blue) and counts of discordant paired-end reads supporting insertional (red), intraviral (yellow), or both (orange) breakpoints. The scale (y-axis) of each plot was normalized to maximum read counts (upper left, each row). HPV breakpoints are defined further in Supplemental Figure 1 and Supplemental Table 3. (B) Verification of insertional breakpoints. Counts of discordant paired-end reads supporting each breakpoint (displayed both on the y-axis and by dot sizes), identified in any of the samples studied, are shown according to position in HPV16 genome (x-axis). (Black dots) Confirmed by PCR and Sanger sequencing; (red) failed PCR; (blue) not tested; (LCR) long control region; (OR) origin of replication. See also Supplemental Figure 2 and Supplemental Tables 3 and 4.
Numerous sample-specific single-nucleotide polymorphisms (SNPs; n = 24–119) were identified in homozygosity in viral sequences, confirming a clonal relationship between virus and tumor (Supplemental Tables 1, 2; Gillison et al. 2000). The depth of sequence coverage across the ∼8-kb viral genome showed considerable variability (Fig. 1A). Uniform viral coverage was found in Tumor B, consistent with a predominance of viral episomes. In contrast, the other eight HPV-positive samples had nonuniform coverage or were missing portions of the viral genome, prompting us to search for insertional breakpoints, defined as the junctions between viral and host genome sequences. Breakpoint calls were based on two or more discordant paired-end reads aligned to the human reference genome within a 500-bp interval. A total of 111 unique insertional breakpoints were identified in the nine HPV-positive cases; their frequencies ranged from two to 47 per sample. Targeted PCR amplification and Sanger sequencing were used to confirm the vast majority (95%; 105 of 111) (Supplemental Table 3).
Collectively, the insertional breakpoints mapped broadly across the HPV genome (Fig. 1B). In several cases (e.g., SiHa, HMS001, UM-SCC-104, and UM-SCC-47 cells), they flanked missing viral segments, indicating loss at integration. Portions of the E2 gene were missing in four of nine cases (Fig. 1A; Choo et al. 1987). Rearrangements within the viral genome also contributed to nonuniform viral coverage in UD-SCC-2, UPCI:SCC090, CaSki, and Tumor A (Supplemental Fig. 1). Despite wide-ranging viral genomic fragmentation, viral fragments containing E6 and E7 oncogenes were retained in all samples.
Since detection of HPV E6 and E7 transcripts remains the gold standard for defining cancers caused by HPV (Gillison and Shah 2003), we assessed viral gene expression using RNA-seq in all cases except SiHa (evaluated only by RACE). All virus-positive cases displayed strong but variable expression of HPV transcripts (ranging from 11.1 to 533.8 fragments per kilobase of transcript per million reads mapped, fpkm) (Supplemental Fig. 2). Canonical, spliced viral transcripts originating from the early p97 promoter, with coding potential for E6*I and E7, were found in all cases.
HPV and the cellular genome
WGS data revealed millions of SNPs and insertion/deletion (indel) polymorphisms in each host cell genome sample (Supplemental Fig. 3). We confirmed an inverse association between HPV status and inactivating mutations in TP53 and CDKN2A (Fisher's exact, P = 0.004) (Gillison et al. 2000; Westra et al. 2008). Mutations, deletions, or chromosomal translocations also were detected in genes frequently mutated in HNSCC, including PTEN, PIK3CA, PIK3API, NOTCH1, TP63, SYNE1, SYNE2, and CASP8 (Agrawal et al. 2011; Stransky et al. 2011). In UM-SCC-104 cells, homozygous deletions in NOTCH1 resulted in its predicted frameshift and premature truncation (Supplemental Table 4). Recurrent chromosomal gains and losses included 5p (gain) and 3p26 (loss) (Heselmeyer et al. 1997) found in six of seven HPV-positive HNSCC samples (Supplemental Figs. 4, 5).
Focal clustering of HPV integrants
Collectively, the HPV integrants mapped broadly across the human genome; we obtained no evidence for recurrent integration in chromosomal hotspots. However, we observed HPV insertional breakpoints to cluster at specific chromosomal regions in different cell lines (Fig. 2; Supplemental Fig. 5). Genomic distances between viral integrants in a “cluster,” defined by three or more unique insertional breakpoints, spanned up to 3 Mb (Tumor A). As examples, HPV integrant clusters mapped to chromosome Xq21 in UD-SCC-2 cells, chromosome 3p11 in UM-SCC-47, and chromosomes 3p12, 6p21, and 9q22 in UPCI:SCC090 cells (Fig. 2).
Figure 2.

HPV integration sites are clustered. HPV insertional breakpoints (dots), identified from discordant paired-end WGS reads, were mapped to human chromosomes (karyotypes, center). FISH and SKY images of chromosomes from various cancer cell lines (background colors, see key) are shown to the left and right of the karyotypes. Each set of FISH/SKY images includes (left) spectral karyotypes, (middle) chromosome banding patterns with HPV insertion sites (pink or teal fluorescent signals), and (right) pseudo-colored cartoons depicting chromosomal composition including translocations. The color key indicates the sample source. See also Supplemental Figures 5 and 6.
To examine HPV integrant clusters further, we performed both fluorescence in situ hybridization (FISH) using HPV16-specific probes and spectral karyotyping (SKY) on all cell lines. Several observations were made. Chromosomal locations of the majority of integrant clusters identified by WGS were confirmed (Fig. 2; Supplemental Fig. 6), but low-copy-number integrants were not detected (e.g., HMS001 and UM-SCC-104). HPV FISH signals were found in identical cytogenetic regions of duplicate chromosomal copies, but with variable intensities, suggesting that integration preceded chromosomal duplication and subsequent divergence in viral copy numbers. Chromosomal paints and HPV FISH signals routinely overlapped, consistent with alternating viral and host genomic sequences. Combined FISH and SKY analysis also frequently identified HPV integrants adjacent to chromosomal translocations. Although these juxtapositions also were observed from WGS data, in several instances, the physical distance between HPV insertions and translocations exceeded 1 Mb.
HPV insertions flank copy number variations (CNVs)
HPV insertional breakpoints detected by WGS were observed to be less numerous than viral copy numbers in some cases (Table 1). We hypothesized that this discrepancy was due to amplification of viral integrants and flanking genomic sequences, leading to redundant, identical breakpoints (Fig. 3). Therefore, we examined WGS coverage across chromosomal regions containing HPV integrants. We observed extensive, focal genomic CNVs at these integration sites in eight of nine HPV-positive cases. Viral integrants (Fig. 3, red histograms) resided directly adjacent to or within sharply demarcated fluctuations in depth of WGS coverage. HPV integrants were found at regions of amplification (blue histograms), ranging from an ∼1.5-fold increase in HMS001 to an ∼58-fold increase in UPCI:SCC090 cells (Supplemental Figs. 7, 8; Supplemental Table 5). Viral integrants also flanked regions wherein deletions occurred (Fig. 3, yellow), ranging from 487 bp in HMS001 (Fig. 4B) up to 234 kb in chromosome 3 of UPCI:SCC090 (data not shown). Only UM-SCC-104 cells lacked CNVs flanking a lone HPV integrant (Fig. 3G). We confirmed regions of amplification by quantitative PCR assays (Supplemental Fig. 8) and regions of deletion by spanning PCR and Sanger sequencing (data not shown).
Figure 3.

Focal CNVs and breakpoints adjacent to HPV insertions. Histograms showing the depth of WGS coverage (y-axis) of well-aligned (top; yellow, blue, and green) and breakpoint sequence reads (bottom; red and gray), mapped to the reference human genome (hg19) at indicated chromosomal loci (x-axis, gene schematics, coordinates in megabases). (Top) Focal CNVs and (bottom) HPV insertional (red) or host–host (gray) breakpoints are shown for cancer samples (A) SiHa; (B) HMS001; (C) UM-SCC-47; (D) UD-SCC2; (E) UPCI:SCC090, chromosome 9; (F) UPCI:SCC090, chromosome 6; and (G) UM-SCC-104. (Top, y-axis labels) The depth of WGS coverage is shown for cancer samples (blue); normal control (yellow); overlapping, shared coverage regions (green). (Bottom) Maximal breakpoint read counts (black) and breakpoint ID numbers (HPV bk, red).
Figure 4.

Focal amplifications and rearrangements explained by “looping” model. Schematics of HPV target sites before (top) and after (middle) viral integration in (A) SiHa and (B) HMS001 cells. We defined genomic segments (marked with capital letters and grayscale fills) based on HPV insertional (red lines) or host–host breakpoints (gray lines). (Top, white rectangles and horizontal lines) Gene exon structures; (arcing dotted lines, arrows: red) connections between HPV insertional breakpoints; (black) host–host breakpoints joining discontinuous reference genome sequences; (light blue histograms) depth of WGS coverage depicting CNVs (see Fig. 3). (Middle: direction of large red arrows) Relative orientation of sense strand of HPV reference genome, not drawn to scale; (white numbers and gaps) viral breakpoint ID numbers and rearrangements; (blue bars) confirmatory PCR amplicons and Sanger sequencing. (Bottom) Inferred, transient looping models to explain formation of CNVs and observed connections between breakpoints. (C) A generalized, stepwise looping model depicting HPV-associated structural variation at YFG (your favorite gene, top) as a target, inferred target nicking and integration of linear HPV genome (red), transient formation of circular DNA containing viral sequences, rolling circle amplification of this template (gray circular arrows), and formation of concatemers harboring identical viral–host and host–host breakpoints. The resulting observed focal amplifications and rearrangements, adjacent to HPV integrants, disrupt expression of YFG (bottom).
Statistical analysis confirmed a strong association between HPV insertional breakpoints and genomic structural variation in eight of nine cases, including chromosomal translocations, deletions, inversions, and/or intrachromosomal rearrangements (Bonferroni adjusted binomial test, all P < 10−9) (Supplemental Fig. 7). Moreover, such enrichment occurred at focal hyperamplification sites in six of nine samples (local ploidy >8N, Bonferroni adjusted binomial test, all P < 10−10). This statistical association between HPV integrants and focal hyperamplifications (>8N) was not observed in samples harboring low-copy-number or predominantly unintegrated viral genomes (i.e., UM-SCC-104, HMS001, and Tumor B).
Resolving focal genomic amplifications and rearrangements
We sought to elucidate the actual linear DNA structures flanking HPV insertional sites. Guided by the breakpoints and CNVs called from WGS data (Fig. 3), we performed targeted PCR, Sanger sequencing, and chromosome walking to develop connectivity maps (Figs. 4–6).
Figure 5.

Connectivity maps of complex, HPV-associated genomic structural variation in HNSCC cell lines. Schematics of genomic target loci before (top) and after (bottom) HPV integration in HNSCC cell lines (A) UM-SCC-47 and (B) UPCI:SCC090, chromosome 6. Target gene schematic and connectivity map features are as described in the Figure 4 legend. HPV breakpoint numbers are listed in Supplemental Table 3. These schematics including the viral insertions are not drawn to scale. A short inversion in the viral sequence flanking breakpoint 3 is not shown. (Parentheses) Genomic segments with indicated fold amplification (x N) calculated from WGS data; (upside-down letters) inverted segments. See Supplemental Figure 9 for connectivity maps for both UD-SCC-2 and UPCI:SCC090, chromosome 9.
Figure 6.

Focal structural variations are associated with HPV integrants in primary tumors. Schematics depict HPV-associated genomic structural variation in primary tumors of the (A–C) tongue (i.e., Tumor A) and (D–F) tonsil (i.e., Tumor B). (A,D) WGS coverage histogram features are as described in Figure 3 legend. (B,D,E) Target gene schematic and connectivity map features are as described in Figure 4 legend. (C) Looping model for HPV-associated structural variation in Tumor A. See Figure 4 legend. (E) Chromosomal translocation structure depicting multiple connections between segments on chromosomes 11 and 8, associated with HPV integrants (red arrows). (F) Scatterplots showing WGS coverage in 50-kb blocks on chromosomes 11 and 8, to estimate (y-axis, left) chromosomal ploidy and (right) ratios of observed local coverage versus mean autosomal coverage. (Gray vertical dots) Centromere; (red arrows) HPV insertion sites.
A recurrent pattern of focal amplifications and rearrangements adjacent to HPV integration sites was seen in six of seven HPV-positive cell lines (Figs. 4, 5). In SiHa cells, two insertional breakpoints directly flanked a twice-amplified, 300-kb segment (Figs. 3A, 4A, red lines). Targeted Sanger sequencing confirmed these two breakpoints to be connected by 7 kb of HPV16 genomic sequence, despite being mapped ∼300 kb apart in the reference genome (Fig. 4A, red lines). This HPV-mediated rearrangement occurred twice, concatemerizing large, flanking genomic segments head-to-tail and resulting in two HPV integrants bounded by identical breakpoints. In one of the duplicated segments, a secondary rearrangement resulted in an ∼72-kb internal deletion.
In HMS001 cells, two viral insertional breakpoints delineated an HPV16 integrant that spanned a small genomic deletion (Fig. 4B). Adjacent host genome fragments and viral sequences were duplicated in tandem, resulting in two identical HPV integrants and an intrachromosomal rearrangement.
These relatively simple, resolved linear genome structures adjacent to HPV integration sites prompted us to construct a “looping” model to explain their occurrence (Fig. 4C). We hypothesized that HPV integrants could bridge noncontiguous genomic sequences, perhaps by connecting nicked sites. We also surmised that replication of transient, circularized genomic segments would result in focal amplification of breakpoints involving viral and flanking host genomic sequences. Subsequently, distal free ends of amplified strands could recombine with noncontiguous sequences and undergo repair, resulting in concatemerized linear structures bounded by recurrent, identical breakpoints (Fig. 4C).
Remarkably consistent patterns of concatemerized host–virus sequences were identified in all but one of the HPV-positive cell lines (Figs. 3–6). The resolved, linear DNA structures at HPV integrant clusters in cell lines UM-SCC-47 and UD-SCC-2, and chromosomes 6 and 9 of UPCI:SCC090 were considerably more complex than in SiHa and HMS001 cells (Figs. 4, 5; Supplemental Fig. 9). Nevertheless, genomic amplifications and rearrangements connected by identical HPV integrants (red lines) and/or host–host recombinant breakpoints (gray lines) were found in each sample. HPV-associated genomic deletions (UD-SCC-2) and inversions (UM-SCC-47, UD-SCC-2, UPCI:SCC090) also were identified (Fig. 5; Supplemental Fig. 9). Notably, the viral E6 and E7 oncogenes were amplified within the viral–host concatemers in most samples studied.
For example, at the TP63 locus in UM-SCC-47 cells (Figs. 3C, 5A), we observed five insertional breakpoints to demarcate four unique HPV integrants. Consistent with our looping model, a viral integrant bridged discontinuous host sequences (Fig. 5A, segments B and F) that were amplified, resulting in a total of ∼25 HPV integrants in a concatenated array.
In UD-SCC-2 cells, HPV-mediated genomic amplifications flanked a >100-kb deletion (Fig. 3D; Supplemental Fig. 9A). Certain exons of the DIAPH2 gene were amplified (e.g., segments K–S), while others were deleted (segment J).
In UPCI:SCC090 cells, 15 HPV insertional breakpoints were clustered on chromosome 9 (Fig. 3E; Supplemental Fig. 9), and five were clustered on chromosome 6 (Figs. 3F, 5B). Several integrants on chromosome 9 shared an identical 5′ breakpoint but were connected to unique 3′ breakpoints (Supplemental Fig. 9B). The shared 5′ breakpoints again were consistent with our looping model (Fig. 4C), whereby the 3′ end of a primary HPV integrant subsequently could recombine at multiple upstream genomic sites. We also observed >90-fold amplification of host DNA bridged by another HPV integrant. These results confirmed that looping could occur at more than one chromosome in the same cell. Similar amplifications, deletions, and inversions were present in numerous HPV integrant clusters in CaSki cells (Supplemental Figs. 5, 6).
To exclude the possibility that the observed associations between HPV integrants and flanking genomic variants (Figs. 3–5) were merely artifacts of cell culture, we also evaluated two primary human tumors by WGS (Fig. 6; Table 1; Supplemental Table 1). In Tumor A, an insertional breakpoint cluster on chromosome 5 included HPV-18-associated deletions, inversions, and amplifications (Fig. 6A–C). The involved locus spanned ∼2 Mb in length and disrupted genomic structures of several genes including ACTBL2 and a candidate oncogene, PDE4D (Lin et al. 2013).
In Tumor B, which harbored predominantly episomal HPV, WGS demonstrated that a small fragment of HPV16 integrated directly into the midst of a chromosomal translocation, juxtaposed between chromosome 11q13 (directly involving CCND1, encoding cyclin D1) and chromosome 8p11 (Fig. 6D–F). We found several insertional and host–host breakpoints spanning >2-Mb segments on chromosome 11 and almost 5 Mb on chromosome 8, supporting an interwoven series of connections between the chromosomes that again is consistent with our looping model. Based on WGS sequence coverage and estimated chromosomal ploidies, we infer that HPV sequences connected segments of two chromosomes, resulting in an apparent dicentric translocation t(11;8) together with monosomic chromosomes 8 and 11 (Fig. 6E,F).
Importantly, preliminary analysis of WGS data from 21 additional HPV-positive HNSCC primary tumors indicated that approximately half possessed CNVs and rearrangements flanked directly by HPV insertional breakpoints (data not shown).
HPV integrants disrupt cellular genes by multiple mechanisms
The possibility that HPV integrants could be enriched within or near cellular genes was evaluated in the nine HPV-positive cell lines and tumors. Confirmed breakpoints mapped 5′ (12.6%) or 3′ (15.3%) to genes, within exons (2.7%), within introns (39.6%), or in intergenic loci (29.7%), using NCBI RefSeq genes as a reference. We observed a modest enrichment of HPV integrants within 50 kb of RefSeq genes (binomial test, P-value = 0.063), within genomic fragile sites (binomial test, P-value = 0.02), or within DNase I hypersensitivity sites (binomial test, P-value = 0.003) when all integrants were considered. However, these associations were no longer significant after accounting for interdependence of integrant clusters (Supplemental Table 6).
Gene expression analysis by RNA-seq and 5′ and 3′ RACE experiments revealed expression of virus–host fusion transcripts in all cell lines and Tumor A. Transcripts initiated from the viral early region promoters (p97, p670) were fused directly with or were alternatively spliced with genomic templates. In SiHa, UPCI:SCC090, and UD-SCC-2 cells, viral fusion transcripts were expressed from more than one integrant within a cluster. Importantly, transcript structures frequently confirmed genomic rearrangements found by WGS and Sanger sequencing (Fig. 7).
Figure 7.

Focal HPV-associated genomic instability disrupts gene expression by diverse mechanisms. Disruption of DIAPH2 in UD-SCC-2 cells (A–E) and of TP63 in UM-SCC-47 cells (F–I) by HPV-associated genomic variation. (A) Mapped RNA-seq read histograms show 5′ expression of DIAPH2 transcript (RefSeq NM006729) in control CAL 27 (top) but not in UD-SCC-2 cells (bottom). Maximal read counts, chromosomal coordinates, and exon numbers are indicated. Truncated 3′ transcripts are overexpressed by fourfold in UD-SCC-2 cells. (B) Resolved linear structure of DIAPH2 locus after HPV integration, displaying (top and inset, middle; red arrows) direction of HPV integrants, not drawn to scale; (red and white numbers) HPV coordinates. (Bottom) Mapped viral–host fusion transcripts identified by 3′ RACE and RT-PCR. (C) Northern blot probed against DIAPH2 3′ cDNA demonstrates aberrant transcripts (arrows) in comparison to CAL 27 and UPCI:SCC090 controls. (D) Western blot showing that the protein translated from DIAPH2, i.e., diaphanous-related formin 2, is markedly reduced compared with control (top); GAPDH loading control (bottom). (E) Northern blot probed against HPV16 E7 demonstrating fusion transcripts in UD-SCC-2 and UM-SCC-47. (F) Histograms of mapped RNA-seq reads show expression of the 5′ end of TP63 in control CAL 27 but not in UM-SCC-47 cells. (G) Resolved linear structure of TP63 locus after HPV integration (top), not drawn to scale. Viral–host fusion transcripts detected by 3′ RACE (bottom) demonstrate HPV-associated gene disruption. (H) Northern blot probed against the 3′ end of TP63, showing truncated transcripts in UM-SCC-47 cells. (I) Western blot showing aberrant expression of 25-kDa p63 carboxy-terminal protein in UM-SCC-47 in comparison to 67-kDa conventional protein in UPCI:SCC090 and SCC-25 cells; (bottom) beta-actin loading control. See also Supplemental Figures 10 and 11.
Many insertional breakpoints flanked or disrupted genes involved in carcinogenesis (Supplemental Table 7). For example, an HPV integrant in UD-SCC-2 cells deleted and rearranged portions of DIAPH2 (Fig. 7A–E), which encodes a protein involved in fidelity of sister-chromatid separation (DeWard et al. 2010; Cheng et al. 2011). Viral–host fusion transcripts flanking the deletion were expressed strongly, while native DIAPH2 transcripts were not. The protein encoded by DIAPH2, i.e., diaphanous-related formin 2, was absent from the UD-SCC-2 cell line, as shown by Western blot (Fig. 7D).
In a different form of gene disruption, HPV-mediated gene amplification in UM-SCC-47 cells resulted in aberrant expression of TP63, a key regulator of epithelial differentiation (Fig. 7F–I; Bergholz and Xiao 2012). Several distinct viral–host fusion transcripts and a novel, truncated 25-kDa protein at the carboxyl terminus of TP63 were expressed. The truncated protein includes the TID domain, a dominant-negative regulator of the pro-apoptotic protein TAp63 (Serber et al. 2002).
Additional examples of gene disruption were found (Supplemental Figs. 10, 11). These included amplification of the oncogenes FOXE1 (Katoh et al. 2013) and PIM1 (Narlik-Grassow et al. 2013) in UPCI:SCC090 cells, resulting from HPV-associated looped structures. We also found “gene breaking” (Wheelan et al. 2005) by an HPV integrant in SLC47A2, a solute carrier, in UM-SCC-104 cells, caused by premature termination of transcripts at the HPV E5 polyadenylation site.
Discussion
Genomic instability is a critical determinant of virus-induced carcinogenesis, and indeed of virtually all human cancers (Hanahan and Weinberg 2000). In the case of HPV, such instability has been attributed largely to functions of the viral E6 and E7 oncogenes (Moody and Laimins 2010). Here, at single-nucleotide resolution, we mapped HPV integrants in human cancer genomes and showed that they directly flank genomic structural variations, including focal amplifications, rearrangements, deletions, and/or translocations. These genomic alterations frequently disrupted the expression and structure of neighboring genes involved in oncogenesis, and resulted in amplification and expression of E6 and E7. Our results shed new light on these potentially critical, catastrophic steps in the progression from an initial viral infection to development of an HPV-associated cancer.
Previous studies, lacking high-resolution WGS data, reported HPV breakpoints preferentially in the viral E2 gene (Thierry and Yaniv 1987). Recently a new method based on targeted amplification of particular HPV sequences together with flanking genomic sequences was described (Xu et al. 2013). However, although we also observed recurrent disruptions of E2, viral breakpoints were collectively distributed more broadly across the viral genome (Fig. 1). Despite frequent fragmentation of HPV integrants, the E6 and E7 viral oncogenes were retained and expressed as viral–host fusion transcripts in all virus-positive samples (Fig. 1; Supplemental Fig. 2; Lace et al. 2011; Khoury et al. 2013).
Prior data, based on SNP arrays, suggested that approximately one-third of HPV integrants in cervical cancers were statistically associated with regional CNVs (Peter et al. 2010). In contrast, our identification of a recurrent pattern of extensive, focal CNVs and/or other forms of structural variation immediately adjacent to HPV integrants, all mapped at single-nucleotide resolution, highlighted the powerful capabilities of WGS in the current era of personalized, genomic medicine (Figs. 3–6). Use of cell lines facilitated FISH and SKY experiments (Fig. 2), which corroborated these viral insertion sites and their proximity near sites of structural variation. Both targeted DNA sequencing and viral–host fusion transcript structures also confirmed such structural variations including concatemerization of viral and host sequences (Figs. 4–7).
Our inclusion of primary tumors here verified that HPV-associated genomic variation is not a mere artifact of cell culture. Moreover, our WGS analysis of numerous additional primary HPV-positive HNSCC specimens independently confirmed that HPV-associated amplifications and rearrangements were present in approximately half of them (data not shown). Moreover, the recently sequenced HeLa genome also revealed multiple HPV-18 integrants (with one HPV-32) clustered in an ∼10-kb region on chromosome 8, again flanked by focal structural variants (Adey et al. 2013; Landry et al. 2013).
The recurrent patterns of focal genomic instability adjacent to HPV integrants, observed across diverse cancer samples arising in different tissues, led us to infer a possible looping model to explain their formation (Fig. 4C). Organized concatemers composed of viral and host sequences, adjoined by identical HPV-breakpoint sequences, may have accumulated as a result of DNA replication originating from the viral origin of replication. This model is compatible with previous experiments in which viral E1 and E2 proteins were overexpressed, inducing replication of extant HPV integrant sequences in HeLa and SiHa cells (Kadaja et al. 2007; Peter et al. 2010). However, the relatively low-resolution Southern blots and SNP arrays employed in those studies did not fully elucidate the resulting structural variants.
Host DNA polymerases and transcription factors are involved in HPV replication, which can occur either bi-directionally or as a rolling circle (Flores and Lambert 1997). Our model is particularly compatible with the latter. Transient formation of looped, circular structures composed of HPV integrants and adjacent chromosomal sequences would facilitate their replication as a rolling circle (Fig. 4C). Subsequent repair of linear structures consisting of virus–host concatemers would lead to the resulting, observed intrachromosomal amplifications and rearrangements (breakpoints) (Fig. 2). We note that virtually all identified looped structures contained the HPV origin of replication (OR) (Fig. 1). In the viral life cycle, HPV replication is initiated by E2 binding and recruitment of the E1 helicase to the OR (McBride 2008). It also depends on activation of the DNA damage response by ATM (Gillespie et al. 2012) and ATR (Reinson et al. 2013). Therefore, looped structures and associated rearrangements could be by-products of aberrant HPV integrant replication.
Looping as a proposed mechanism for focal genomic instability bears some similarities with other mechanisms, but also is distinct from each. In chromothripsis, seen in 2%–3% of human cancers, the observed chromosomal fragmentation is focal but disorganized. Chromothripsis breakpoints appear juxtaposed in a random pattern (Crasta et al. 2012; Forment et al. 2012; Molenaar et al. 2012; Rausch et al. 2012; Korbel and Campbell 2013) in contrast with the organized concatemers described here. However, both mechanisms may be replication-dependent (Liu et al. 2011). Hallmarks of chromothripsis recently were identified in HeLa cells, but not at the HPV integration sites (Landry et al. 2013). In breakage–fusion–bridge cycles, palindromic sequences are thought to trigger focal amplification and regional genomic instability (Tanaka and Yao 2009; Hillmer et al. 2011). However, we did not observe palindromes at intrachromosomal or HPV insertional breakpoints. L1 retrotransposition can induce forms of structural variation including deletions, inversions, and chromosomal translocations, but focal amplifications have not been associated with it (Symer et al. 2002). The formation of other breakpoints in cancers may occur via nonhomologous end joining (Chiang et al. 2012; Malhotra et al. 2013). The small numbers of HPV insertional breakpoints detected by WGS, even in the setting of extraordinarily high HPV copy numbers, argue effectively against disorganized or “random” breakpoints, as might be expected in aneuploidy, nonhomologous end joining or chromothripsis.
A temporal relationship between HPV integration, enhanced expression of E7, and the subsequent development of quantitative and structural chromosomal abnormalities has been observed in cell culture (Pett et al. 2004). Increased E7 expression confers a selective growth advantage (Jeon and Lambert 1995); thus we conclude that cells bearing HPV integrant clusters, with amplified viral genomes and transcriptionally active viral promoters, would be clonally selected. Deregulated expression of HPV E6 and E7 oncoproteins is a sine qua non of HPV-associated malignancies.
Prior evidence supporting HPV as an insertional mutagen has been scant. In a few reports, viral integration was associated with loss of heterozygosity of tumor suppressor genes (Reuter et al. 1998; Schmitz et al. 2012). HPV integrants near MYC have been associated with increased Myc expression, possibly due to viral enhancer sequences introduced nearby (Peter et al. 2006). Here we identified multiple means by which HPV integrants can disrupt neighboring genes, including gene rearrangements (DIAPH2 complex rearrangements in UD-SCC-2), introduction of aberrant promoters (TP63 in UM-SCC-47), and gene amplification (PIM1, FOXE1, C9orf156 in UPCI:SCC090). Even in UM-SCC-104 cells, where a lone HPV integrant was not associated with CNV (Fig. 3G), we nevertheless observed gene breaking (Supplemental Fig. 11; Wheelan et al. 2005). Such gene disruption by HPV integrants is likely of biological significance, given the established functions of affected genes. For example, we observed homozygous deletion of DIAPH2 in UD-SCC-2 cells. Mutations of this gene promote chromosomal instability via misalignment of sister chromatids during metaphase (Cheng et al. 2011). In UM-SCC-47 cells, the HPV integrant cluster caused aberrant expression of TP63, a critical regulator of epithelial differentiation. The highly expressed, truncated form of p63 protein contained a domain known to inhibit the pro-apoptotic protein, TAp63 (Serber et al. 2002).
We note some limitations to our analysis. A majority of samples were cultured cells, chosen intentionally to allow more extensive experimentation (e.g., Figs. 2, 7) than would be possible with primary tumors. However, since exclusively episomal HPV may be lost in the derivation of cell lines from tumors, our data may overestimate the proportion of cancers bearing HPV integrants and looping events. Because the insert size of our WGS libraries was ∼300 nucleotides (nt), we performed extensive additional PCR, cloning, and sequence analysis to confirm connections within looped structures. Although we are confident in the existence of these structures, in complex cases spanning kilobases or megabases in sequence, the exact order of recombined genomic segments could not be resolved unambiguously using conventional paired-end libraries. Although we currently are conducting long-range sequencing, even these new technologies will be unable to resolve repetitive structures on a megabase scale. We cannot entirely exclude alternative sources of variability in copy numbers or insertional breakpoints, such as the simultaneous presence of different subclones in cell populations. Also, we were unable to map insertional breakpoints unambiguously at certain repetitive elements. To test the hypothesis that insertional mutagenesis and looping by HPV may contribute causally to cancer formation, by generating driver mutations rather than passenger or bystander mutations, currently we are engineering genetic knock-ins and knockouts in the cell lines studied.
The looping model proposed here provides a framework to understand how HPV integrants could become flanked by neighboring CNVs in human cancers. Intriguing recent data showed that hepatitis B virus (HBV) integrants in hepatocellular carcinoma similarly can be associated with regional CNVs, viral–host fusion transcripts and alterations in host genome expression (Jiang et al. 2012; Sung et al. 2012; Toh et al. 2013), analogous to our findings on HPV integrants. However, although CNVs were identified near HBV integrants in ∼25% of cases, they and other structural variants were not fully resolved. Moreover, in contrast to HPV, recurrent “hotspots” of HBV integration in the host genome (e.g., at TERT) were observed. It will be interesting to determine whether other oncogenic viruses also are associated with similar genomic alterations. Our findings support an emerging hypothesis whereby the aberrant firing of origins of replication could contribute plausibly to genomic instability in human cancers in the absence of viral infection (Evertts and Coller 2012). Studies are ongoing to extend this looping model to additional HPV-associated primary tumors (e.g., anogenital cancers), and to evaluate the significance of these genomic alterations for HPV-associated cancer progression, targeted therapeutics, and patient outcomes.
Methods
Human cancer cell lines and primary tumors
SiHa (Friedl et al. 1970), CaSki (Pattillo et al. 1977), CAL 27 (Gioanni et al. 1988), D562 (Peterson et al. 1971), and SCC-25 (Rheinwald and Beckett 1981) were acquired from the American Type Culture Collection (Manassas, VA). UM-SCC-47 (Lansford et al. 1999) and UM-SCC-104 (Tang et al. 2012) were kindly provided by Thomas Carey, University of Michigan; UPCI:SCC090 (White et al. 2007) were from Susanne M. Gollin, University of Pittsburgh; UD-SCC-2 (Ballo et al. 1999) were from Henning Bier, University of Dusseldorf; and HMS001 cells were from James Rocco, Harvard Medical School. Please see Table 1 and Supplemental Table 1.
CAL 27, D562, SCC-25, SiHa, and CaSki cells were cultured as recommended by ATCC. UM-SCC-104, UM-SCC-47, UD-SCC-2, and UPCI:SCC090 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 2 mM L-glutamine, 1% nonessential amino acids, 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum (FBS; Gibco). HMS001 cells were cultured in DMEM:F12 (Gibco) medium supplemented with 10% FBS and 1× penicillin/streptomycin/glutamine (Mediatech). All cell lines were grown at 37°C in a humidified atmosphere containing 5% CO2. Cells were harvested at 90% confluence. Genomic DNA was extracted using a Puregene DNA purification kit (Gentra Systems). RNA was extracted in TRIzol (Life Technologies).
De-identified, fresh-frozen HNSCC samples were obtained from a tumor bank at The Ohio State University. Specimen banking was approved by an Institutional Review Board for research purposes, and written consent was obtained from patients. OCT-embedded frozen sections were stained with hemotoxylin and eosin to guide microdissection, ensuring >80% tumor. Please see Table 1 and Supplemental Table 1. Genomic DNA was purified by proteinase K digestion, phenol/chloroform/isoamyl alcohol extraction, and ethanol precipitation, and was quantified using Quant-iT PicoGreen dsDNA reagent (Invitrogen). RNA from primary tumors was extracted using the Ambion RNAqueous-4PCR kit.
Whole-genome sequencing and analysis
Whole-genome sequencing (WGS, 2 × 100-bp paired-end reads) was performed using the HiSeq 2000 platform via the Illumina Genome Network (IGN; http://www.illumina.com). Raw sequence reads were aligned to the human genome reference assembly (hg19) using IGN ELAND. Small variants such as SNPs and small indels were detected by IGN CASAVA. Those variants previously observed in the 1000 Genomes Project Consortium (release phase 1) (The 1000 Genomes Project Consortium 2010) and present in the dbSNP database at minor allele frequency >1% were excluded (Sherry et al. 2001). Predicted functional consequences of somatic variants, annotated using variant_effect_predictor (McLaren et al. 2010) as per the Ensembl release 70 database, were classified into four tiers of predicted functional significance (tier1: variants with coding change and essential splice sites; tier2: conserved or regulatory region; tier3: non-repeat region; tier4: other).
To detect variants in both human and HPV genomes, we created a proxy reference assembly combining the human (hg19) and HPV genomes (called hg19+HPV; GenBank accession numbers: HPV16 [NC_001526.2]; HPV18 [NC_001357.1]; HPV31 [HQ537687.1]; HPV33 [HQ537707.1]; and HPV35 [M74117.1]). We used BWA (Li and Durbin 2010) to align WGS sequence reads against hg19+HPV. Mpileup function from SAMtools (Li et al. 2009) was used to detect SNPs and small INDELs in HPV genomes. HPV variants were classified by comparison of sequences to HPV16 lineage-specific SNPs as reported by the IARC HPV Variant Study Group (Cornet et al. 2012).
To estimate HPV copy numbers per sample, mean viral genome coverage was divided by autosomal coverage (Table 1). A modified Hydra workflow (Quinlan and Hall 2010) was used to detect structural variants. Discordant read pairs were extracted by alignments against the reference assembly using GSNAP (version 2012-07-20) (Wu and Nacu 2010). To detect copy number alterations, CNAnorm (Gusnanto et al. 2012) was used to evaluate read depths of WGS data in 50-kb windows for each sample. Combined read depth data from 20 normal individuals of both genders were used as a control. For those samples with corresponding cytogenetic data, baselines for copy number detection were adjusted to average ploidy.
Alignments supporting candidate chromosomal translocations and HPV breakpoints were further confirmed by BLAT (Kent 2002). We removed SV candidates with less than four supporting read pairs. SVs were categorized into known and novel variants by comparison with the Database of Genomic Variants, DGV (Iafrate et al. 2004). De novo assembly was performed using Velvet to identify structural variants (SV) and junctions (Zerbino et al. 2009).
To visualize sequence alignments, we used the Broad Institute's Integrative Genome Viewer (IGV) (Thorvaldsdottir et al. 2013). Circos plots (Krzywinski et al. 2009) were generated to visualize variant distributions genome-wide.
PCR amplification and Sanger sequencing
To confirm insertional breakpoints, perform chromosome walking, and model resolved linear structures of the host cell genome, we designed custom primers to amplify regions of interest by PCR, using standard methods (e.g., see blue bars in “evidence tracks” in Figs. 4–6). PCR products were sequenced directly using conventional Sanger sequencing. If unsuccessful, PCR products were cloned using the TOPO TA cloning kit (Life Technologies) and sequenced from both directions using the M13F forward and M13 reverse primers at the OSUCCC Nucleic Acids Core Facility or using a commercial sequencing service (Genewiz).
Analysis of the relationship between HPV breakpoints, human RefSeq genes, and structural variation
Genes neighboring HPV breakpoints were identified using the NCBI RefSeq database. We examined the overlap between HPV breakpoints and reported fragile sites within the human genome (n = 104) in the NCBI Gene database (mean size = 7.5 Mb, range 0.7–28 Mb) and for DNase I hypersensitivity sites using 51 cell types from the UCSC Genome Browser ENCODE (The ENCODE Project Consortium 2011). In these analyses, we compared our observed distribution of HPV breakpoints to a simulated set of 1 million random breakpoints (using randomly chosen human genomic coordinates) as a control. Comparisons were performed using a binomial test.
The relationship between HPV breakpoints and predicted DNA copy number change was evaluated using CNAnorm (Gusnanto et al. 2012). For this analysis, the human genome was divided into 50-kb bins. Hyperamplification was defined as regions displaying copy numbers (ploidy) of N > 8, and breakpoint regions were defined as within ±250 kb from an HPV breakpoint. Associations were evaluated using a binomial test. P-values were adjusted by Bonferroni multiple testing correction. A similar analysis was conducted to evaluate associations between HPV breakpoints and detected structural variants (chromosomal translocation, deletion, inversion, intrachromosomal translocation) using Hydra. All statistical analyses (including those described above) were performed using the R/Bioconductor packages (http://www.bioconductor.org).
FISH (fluorescence in situ hybridization) and spectral karyotyping (SKY) analysis
To evaluate chromosomal structures and counts in the various cell lines, we used spectral karyotyping (SKY) and fluorescence in situ hybridizations (FISH) (Schrock et al. 1996). Protocols can be accessed at http://www.riedlab.nci.nih.gov/index.php/protocols. In brief, metaphase chromosome suspensions were prepared first by treating cells with a hypotonic solution (0.075 M KCl), followed by fixation using methanol:acetic acid (3:1 [v/v]) and then dropping them onto slides, using a Thermotron chamber to control humidity. Slides were aged for ∼1 wk at 37°C prior to hybridization. Chromosome preparations were hybridized for 72 h with SKY probes (prepared in-house).
SKY analysis of samples was performed using a Leica DMRXE microscope (Leica), DAPI and SKY filters (Chroma), a Xenon lamp, and a Spectracube (Applied Spectral Imaging). Spectrum-based classification and analysis of the fluorescent SKY images were achieved using SkyView software (Applied Spectral Imaging).
For FISH analysis, slides were evaluated using a Leica DMRXE microscope, fitted with a mercury lamp and a CCD camera. Leica QFISH software was used to acquire cell images.
For each cell line, ∼15–20 metaphase spreads were acquired for SKY and FISH analysis and scored for numerical and structural chromosomal aberrations according to established human chromosome nomenclature rules from ISCN (International Standing Committee on Human Cytogenetic Nomenclature 2009).
RNA sequencing (RNA-seq)
One microgram of total RNA was used for library preparation. Either 51-bp paired-end or 250-bp paired-end libraries were constructed from 1 μg of total RNA samples, using Illumina's stranded and unstranded library protocols. A TruSeq RNA sample preparation kit (version 2; Illumina) was used to construct strand-specific RNA-seq libraries. Library construction included fragmentation, first-strand cDNA synthesis, second-strand cDNA synthesis, end repairing, 3′-end adenylation, adaptor ligation, and PCR amplification with barcoded oligos. Briefly, total RNA was fragmented by heating. First-strand cDNA synthesis was performed (Life Technologies). Reaction products were precipitated with ethanol and ammonium acetate, incubated for 2 h at −80°C and centrifuged at 20,000g for 30 min at 4°C. Pellets were washed with ice-cold 70% ethanol, air-dried for 10 min, and then resuspended in second-strand synthesis buffer (New England Biolabs). After mixing, samples were incubated for 2.5 h at 16°C for second-strand synthesis. To preserve strand information in certain libraries, we added dUTP to the second-strand synthesis reaction. Samples were then cleaned using Agencourt AMPure XP beads (Beckman Coulter), ends were repaired, and 3′ adenylation was performed. Adaptor ligation was performed as per the kit manual. Then, in some cases in which strand-specific information was to be preserved, prior to PCR amplification, the incorporated uracils in the coding strand were degraded to eliminate the second strand of cDNA by adding USER enzyme (New England Biolabs) for 1 h at 37°C before being placed on ice.
The resulting libraries were amplified by PCR and purified using Agencourt AMPure XP beads (Beckman Coulter). Library quality and concentrations were measured using high-sensitivity chips on an Agilent 2100 Bioanalyzer and a Qubit 2.0 (Life Technologies). Based on measured concentrations, barcoded RNA-seq libraries were pooled and loaded onto an Illumina MiSeq instrument (Genomics Facility, Cincinnati Children's Hospital and Medical Center).
We aligned 51-bp paired-end reads against our hg19+HPV reference assembly using TopHat (version 1.4.1) (Trapnell et al. 2009), and 250-bp paired-end reads were aligned using GSNAP (Wu and Nacu 2010). Expression levels for known transcripts were determined (Ensembl release 67 human transcripts) using Cufflinks (version 1.3.0) (Trapnell et al. 2012). To detect HPV fusion transcripts, we used GSNAP to align reads and Hydra to detect chimeric HPV–host read pairs (Quinlan and Hall 2010). We used Cummerbund to visualize gene expression levels (Trapnell et al. 2012).
Rapid amplification of cDNA ends (RACE)
RNA ligase-mediated RACE analysis was conducted using the Ambion FirstChoice RLM-RACE kit (Life Technologies). HPV or gene-specific forward and reverse primers were used to amplify 5′ and 3′ cDNA ends (Klaes et al. 1999). Resulting PCR products were cloned using the TOPO TA cloning kit (Life Technologies) and sequenced in both directions using the M13F forward and M13 reverse primers.
Additional methods
Quantitative PCR (qPCR) assays were performed to quantify focal amplification at genomic regions detected by WGS (Supplemental Fig. 8). Northern blot hybridization was used to assay transcripts from TP63, DIAPH2, HPV16 E7, HPV16 E5, and RPLP0, a loading control (Fig. 7). Western blotting was performed to assess expression of TP63 and diaphanous-related formin 2 proteins (Fig. 7). Additional details for quantitative (real-time) PCR, Northern blot hybridization, Western blot hybridization, and peptide competition experiments are provided in the Supplemental Methods.
Data access
WGS data have been deposited at the European Genome-phenome Archive (EGA; http://www.ebi.ac.uk/ega/), hosted by the European Bioinformatics Institute (EBI), European Molecular Biology Laboratory (EMBL), under accession number EGAS00001000599.
Acknowledgments
We thank Dr. Douglas Lowy (National Cancer Institute) for insightful comments on the manuscript; Anthony S. Baker for expert help preparing graphical figures; Dr. Yanqiang Wang (OSUCCC) for help with Northern blots; Sylwia Wojcik for help with initial characterization of insertional breakpoints; and other members of the Gillison and Symer laboratories for helpful discussions. We thank Drs. Thomas Carey (University of Michigan), Susanne M. Gollin (University of Pittsburgh), and Henning Bier (University of Dusseldorf) for their kind gifts of cell lines. This study was funded by The Ohio State University Comprehensive Cancer Center (D.E.S., M.L.G.), the Ohio Supercomputer Center (PAS0425; K.A., D.E.S.), an Ohio Cancer Research Associate grant (GRT00024299; K.A.), the Oral Cancer Foundation (M.L.G.), and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (T.R.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.164806.113.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J 2013. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500: 207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et al. 2011. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333: 1154–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballo H, Koldovsky P, Hoffmann T, Balz V, Hildebrandt B, Gerharz CD, Bier H 1999. Establishment and characterization of four cell lines derived from human head and neck squamous cell carcinomas for an autologous tumor-fibroblast in vitro model. Anticancer Res 19: 3827–3836 [PubMed] [Google Scholar]
- Bergholz J, Xiao ZX 2012. Role of p63 in development, tumorigenesis and cancer progression. Cancer Microenviron 5: 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi A, Engels E, Pfeiffer R, Hernandez B, Xiao W, Kim E, Jiang B, Goodman M, Sibug-Saber M, Cozen W, et al. 2011. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol 29: 4294–4301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Zhang J, Ahmad S, Rozier L, Yu H, Deng H, Mao Y 2011. Aurora B regulates formin mDia3 in achieving metaphase chromosome alignment. Dev Cell 20: 342–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, Mills RE, Kirby A, Lindgren AM, Rudiger SR et al. 2012. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet 44: 390–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo KB, Pan CC, Han SH 1987. Integration of human papillomavirus type 16 into cellular DNA of cervical carcinoma: Preferential deletion of the E2 gene and invariable retention of the long control region and the E6/E7 open reading frames. Virology 161: 259–261 [DOI] [PubMed] [Google Scholar]
- Cornet I, Gheit T, Franceschi S, Vignat J, Burk RD, Sylla BS, Tommasino M, Clifford GM 2012. Human papillomavirus type 16 genetic variants: Phylogeny and classification based on E6 and LCR. J Virol 86: 6855–6861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D 2012. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482: 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWard AD, Eisenmann KM, Matheson SF, Alberts AS 2010. The role of formins in human disease. Biochim Biophys Acta 1803: 226–233 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2011. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9: e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evertts AG, Coller HA 2012. Back to the origin: Reconsidering replication, transcription, epigenetics, and cell cycle control. Genes Cancer 3: 678–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Lambert PF 1997. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J Virol 71: 7167–7179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman D, de Martel C, Lacey CJ, Soerjomataram I, Lortet-Tieulent J, Bruni L, Vignat J, Ferlay J, Bray F, Plummer M, et al. 2012. Global burden of human papillomavirus and related diseases. Vaccine (Suppl 5) 30: F12–F23 [DOI] [PubMed] [Google Scholar]
- Forment JV, Kaidi A, Jackson SP 2012. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat Rev Cancer 12: 663–670 [DOI] [PubMed] [Google Scholar]
- Friedl F, Kimura I, Osato T, Ito Y 1970. Studies on a new human cell line (SiHa) derived from carcinoma of uterus. I. Its establishment and morphology. Proc Soc Exp Biol Med 135: 543–545 [DOI] [PubMed] [Google Scholar]
- Gillespie KA, Mehta KP, Laimins LA, Moody CA 2012. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol 86: 9520–9526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillison ML, Shah KV 2003. Chapter 9: Role of mucosal human papillomavirus in nongenital cancers. J Natl Cancer Inst Monogr 2003: 57–65 [DOI] [PubMed] [Google Scholar]
- Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, et al. 2000. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst 92: 709–720 [DOI] [PubMed] [Google Scholar]
- Gioanni J, Fischel JL, Lambert JC, Demard F, Mazeau C, Zanghellini E, Ettore F, Formento P, Chauvel P, Lalanne CM, et al. 1988. Two new human tumor cell lines derived from squamous cell carcinomas of the tongue: Establishment, characterization and response to cytotoxic treatment. Eur J Cancer Clin Oncol 24: 1445–1455 [DOI] [PubMed] [Google Scholar]
- Gusnanto A, Wood HM, Pawitan Y, Rabbitts P, Berri S 2012. Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics 28: 40–47 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA 2000. The hallmarks of cancer. Cell 100: 57–70 [DOI] [PubMed] [Google Scholar]
- Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT 1989. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J 8: 3905–3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heselmeyer K, Macville M, Schrock E, Blegen H, Hellstrom AC, Shah K, Auer G, Ried T 1997. Advanced-stage cervical carcinomas are defined by a recurrent pattern of chromosomal aberrations revealing high genetic instability and a consistent gain of chromosome arm 3q. Genes Chromosomes Cancer 19: 233–240 [PubMed] [Google Scholar]
- Hillmer AM, Yao F, Inaki K, Lee WH, Ariyaratne PN, Teo AS, Woo XY, Zhang Z, Zhao H, Ukil L, et al. 2011. Comprehensive long-span paired-end-tag mapping reveals characteristic patterns of structural variations in epithelial cancer genomes. Genome Res 21: 665–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C 2004. Detection of large-scale variation in the human genome. Nat Genet 36: 949–951 [DOI] [PubMed] [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans 2007. Human papillomaviruses. IARC Monogr Eval Carcinog Risks Hum 90: 1–636 [PMC free article] [PubMed] [Google Scholar]
- International Standing Committee on Human Cytogenetic Nomenclature. 2009. ICSN 2009: An international system for human cytogenetic nomenclature. Karger Medical Scientific, Basel, Switzerland. [Google Scholar]
- Jeon S, Lambert PF 1995. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: Implications for cervical carcinogenesis. Proc Natl Acad Sci 92: 1654–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, et al. 2012. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res 22: 593–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaja M, Sumerina A, Verst T, Ojarand M, Ustav E, Ustav M 2007. Genomic instability of the host cell induced by the human papillomavirus replication machinery. EMBO J 26: 2180–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M, Igarashi M, Fukuda H, Nakagama H 2013. Cancer genetics and genomics of human FOX family genes. Cancer Lett. 328: 198–206 [DOI] [PubMed] [Google Scholar]
- Kent WJ 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury JD, Tannir NM, Williams MD, Chen Y, Yao H, Zhang J, Thompson EJ, TCGA Network, Meric-Bernstam F, Medeiros LJ, et al. 2013. Landscape of DNA virus associations across human malignant cancers: Analysis of 3,775 cases using RNA-Seq. J Virol 87: 8916–8926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaes R, Woerner SM, Ridder R, Wentzensen N, Duerst M, Schneider A, Lotz B, Melsheimer P, von Knebel Doeberitz M 1999. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res 59: 6132–6136 [PubMed] [Google Scholar]
- Korbel JO, Campbell PJ 2013. Criteria for inference of chromothripsis in cancer genomes. Cell 152: 1226–1236 [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA 2009. Circos: An information aesthetic for comparative genomics. Genome Res 19: 1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lace MJ, Anson JR, Klussmann JP, Wang DH, Smith EM, Haugen TH, Turek LP 2011. Human papillomavirus type 16 (HPV-16) genomes integrated in head and neck cancers and in HPV-16-immortalized human keratinocyte clones express chimeric virus-cell mRNAs similar to those found in cervical cancers. J Virol 85: 1645–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry JJ, Pyl PT, Rausch T, Zichner T, Tekkedil MM, Stutz AM, Jauch A, Aiyar RS, Pau G, Delhomme N, et al. 2013. The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda) 3: 1213–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansford C, Grenman R, Bier H, Somers K, Kim S, Whiteside T, Clayman G, Welkoborsky H, Carey T 1999. Head and neck cancers. Kluwer Academic Publishers, London [Google Scholar]
- Li H, Durbin R 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DC, Xu L, Ding LW, Sharma A, Liu LZ, Yang H, Tan P, Vadgama J, Karlan BY, Lester J, et al. 2013. Genomic and functional characterizations of phosphodiesterase subtype 4D in human cancers. Proc Natl Acad Sci 110: 6109–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SC, Dhar SU, Kolodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. 2011. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146: 889–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, Layer RM, Quinlan AR, Hall IM 2013. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res 23: 762–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AA 2008. Replication and partitioning of papillomavirus genomes. Adv Virus Res 72: 155–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F 2010. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26: 2069–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, et al. 2012. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483: 589–593 [DOI] [PubMed] [Google Scholar]
- Moody CA, Laimins LA 2010. Human papillomavirus oncoproteins: Pathways to transformation. Nat Rev Cancer 10: 550–560 [DOI] [PubMed] [Google Scholar]
- Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R 1989. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol 63: 4417–4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlik-Grassow M, Blanco-Aparicio C, Carnero A 2013. The PIM family of serine/threonine kinases in cancer. Med Res Rev doi: 10.1002/med.21284 [DOI] [PubMed] [Google Scholar]
- Pattillo RA, Hussa RO, Story MT, Ruckert AC, Shalaby MR, Mattingly RF 1977. Tumor antigen and human chorionic gonadotropin in CaSki cells: A new epidermoid cervical cancer cell line. Science 196: 1456–1458 [DOI] [PubMed] [Google Scholar]
- Peter M, Rosty C, Couturier J, Radvanyi F, Teshima H, Sastre-Garau X 2006. MYC activation associated with the integration of HPV DNA at the MYC locus in genital tumors. Oncogene 25: 5985–5993 [DOI] [PubMed] [Google Scholar]
- Peter M, Stransky N, Couturier J, Hupe P, Barillot E, de Cremoux P, Cottu P, Radvanyi F, Sastre-Garau X 2010. Frequent genomic structural alterations at HPV insertion sites in cervical carcinoma. J Pathol 221: 320–330 [DOI] [PubMed] [Google Scholar]
- Peterson WD Jr, Stulberg CS, Simpson WF 1971. A permanent heteroploid human cell line with type B glucose-6-phosphate dehydrogenase. Proc Soc Exp Biol Med 136: 1187–1191 [DOI] [PubMed] [Google Scholar]
- Pett MR, Alazawi WO, Roberts I, Dowen S, Smith DI, Stanley MA, Coleman N 2004. Acquisition of high-level chromosomal instability is associated with integration of human papillomavirus type 16 in cervical keratinocytes. Cancer Res 64: 1359–1368 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, et al. 2012. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148: 59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinson T, Toots M, Kadaja M, Pipitch R, Allik M, Ustav E, Ustav M 2013. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J Virol 87: 951–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter S, Bartelmann M, Vogt M, Geisen C, Napierski I, Kahn T, Delius H, Lichter P, Weitz S, Korn B, et al. 1998. APM-1, a novel human gene, identified by aberrant co-transcription with papillomavirus oncogenes in a cervical carcinoma cell line, encodes a BTB/POZ-zinc finger protein with growth inhibitory activity. EMBO J 17: 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinwald JG, Beckett MA 1981. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res 41: 1657–1663 [PubMed] [Google Scholar]
- Schmitz M, Driesch C, Beer-Grondke K, Jansen L, Runnebaum IB, Durst M 2012. Loss of gene function as a consequence of human papillomavirus DNA integration. Int J Cancer 131: E593–E602 [DOI] [PubMed] [Google Scholar]
- Schrock E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith MA, Ning Y, Ledbetter DH, Bar-Am I, Soenksen D, et al. 1996. Multicolor spectral karyotyping of human chromosomes. Science 273: 494–497 [DOI] [PubMed] [Google Scholar]
- Serber Z, Lai HC, Yang A, Ou HD, Sigal MS, Kelly AE, Darimont BD, Duijf PH, Van Bokhoven H, McKeon F, et al. 2002. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol Cell Biol 22: 8601–8611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K 2001. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res 29: 308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, et al. 2011. The mutational landscape of head and neck squamous cell carcinoma. Science 333: 1157–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, et al. 2012. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet 44: 765–769 [DOI] [PubMed] [Google Scholar]
- Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD 2002. Human L1 retrotransposition is associated with genetic instability in vivo. Cell 110: 327–338 [DOI] [PubMed] [Google Scholar]
- Tanaka H, Yao MC 2009. Palindromic gene amplification—an evolutionarily conserved role for DNA inverted repeats in the genome. Nat Rev Cancer 9: 216–224 [DOI] [PubMed] [Google Scholar]
- Tang AL, Hauff SJ, Owen JH, Graham MP, Czerwinski MJ, Park JJ, Walline H, Papagerakis S, Stoerker J, McHugh JB, et al. 2012. UM-SCC-104: A new human papillomavirus-16-positive cancer stem cell-containing head and neck squamous cell carcinoma cell line. Head Neck 34: 1480–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry F, Yaniv M 1987. The BPV1-E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. EMBO J 6: 3391–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP 2013. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh ST, Jin Y, Liu L, Wang J, Babrzadeh F, Gharizadeh B, Ronaghi M, Toh HC, Chow PK, Chung AY, et al. 2013. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis 34: 787–798 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 189: 12–19 [DOI] [PubMed] [Google Scholar]
- Wentzensen N, Vinokurova S, von Knebel Doeberitz M 2004. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res 64: 3878–3884 [DOI] [PubMed] [Google Scholar]
- Westra WH, Taube JM, Poeta ML, Begum S, Sidransky D, Koch WM 2008. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin Cancer Res 14: 366–369 [DOI] [PubMed] [Google Scholar]
- Wheelan SJ, Aizawa Y, Han JS, Boeke JD 2005. Gene-breaking: A new paradigm for human retrotransposon-mediated gene evolution. Genome Res 15: 1073–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JS, Weissfeld JL, Ragin CC, Rossie KM, Martin CL, Shuster M, Ishwad CS, Law JC, Myers EN, Johnson JT, et al. 2007. The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral Oncol 43: 701–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Nacu S 2010. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Chotewutmontri S, Wolf S, Klos U, Schmitz M, Durst M, Schwarz E 2013. Multiplex identification of human papillomavirus 16 DNA integration sites in cervical carcinomas. PLoS ONE 8: e66693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, McEwen GK, Margulies EH, Birney E 2009. Pebble and rock band: Heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PLoS ONE 4: e8407. [DOI] [PMC free article] [PubMed] [Google Scholar]