

Abstract
Intra-tumor heterogeneity is a hallmark of many cancers and may lead to therapy resistance or interfere with personalized treatment strategies. Here, we combined topographic mapping of somatic breakpoints and transcriptional profiling to probe intra-tumor heterogeneity of treatment-naïve stage IIIC/IV epithelial ovarian cancer. We observed that most substantial differences in genomic rearrangement landscapes occurred between metastases in the omentum and peritoneum versus tumor sites in the ovaries. Several cancer genes such as NF1, CDKN2A, and FANCD2 were affected by lesion-specific breakpoints. Furthermore, the intra-tumor variability involved different mutational hallmarks including lesion-specific kataegis (local mutation shower coinciding with genomic breakpoints), rearrangement classes, and coding mutations. In one extreme case, we identified two independent TP53 mutations in ovary tumors and omentum/peritoneum metastases, respectively. Examination of gene expression dynamics revealed up-regulation of key cancer pathways including WNT, integrin, chemokine, and Hedgehog signaling in only subsets of tumor samples from the same patient. Finally, we took advantage of the multilevel tumor analysis to understand the effects of genomic breakpoints on qualitative and quantitative gene expression changes. We show that intra-tumor gene expression differences are caused by site-specific genomic alterations, including formation of in-frame fusion genes. These data highlight the plasticity of ovarian cancer genomes, which may contribute to their strong capacity to adapt to changing environmental conditions and give rise to the high rate of recurrent disease following standard treatment regimes.
In recent years, tremendous progress has been made in the understanding of the complexity of the cancer genome (Stratton 2011). Studies including large numbers of patients per tumor type have identified recurrent mutations, copy number variants, epigenetic changes, and genomic rearrangements specific for certain cancer types (http://cancergenome.nih.gov/; The International Cancer Genome Consortium 2010; The Cancer Genome Atlas Research Network 2011).
Although more than 400 commonly mutated cancer genes have been identified (Futreal et al. 2004; Santarius et al. 2010), extensive genetic heterogeneity has been noticed across different cancer types and also within individual tumors (Stratton 2011; Yates and Campbell 2012). Intra-tumor heterogeneity is a result of the action of the evolutionary forces of mutation and selection (Stratton 2011; Yates and Campbell 2012). The traditional linear model of cancer evolution describes multiple, successive cycles of mutations and selection leading to malignant tumor cells, ultimately leading to metastases (Hanahan and Weinberg 2000; Klein 2009; Yates and Campbell 2012). In contrast, parallel evolution describes dissemination of tumor cells from the primary tumor as a continuous process occurring from very early on in tumor development. These disseminated cells may continue to evolve independent of the primary tumor, causing the formation of metastases genetically relatively distinct from the primary tumor and other metastases (Gray 2003; Klein 2009). Several studies have focused on spatial sampling of various cancer types to gain insight into the extent and complexity of tumor evolution (Campbell et al. 2010; Yachida et al. 2010; Gerlinger et al. 2012; Bashashati et al. 2013).
Here we studied intra-tumor heterogeneity in epithelial ovarian cancer. With an annual worldwide incidence of 220,000 and mortality of 140,000, epithelial ovarian cancer is the leading cause of death among women with gynecological malignancies and a disease in urgent need for improved treatment (Ferlay et al. 2010). Large-scale genomic analysis of ovarian cancer patients has uncovered only a few recurrently mutated genes, such as TP53 and mutations in BRCA1/BRCA2 (The Cancer Genome Atlas Research Network 2011). Ovarian cancers show a relatively high number of copy number variations and structural variations (SVs) (The Cancer Genome Atlas Research Network 2011; Malek et al. 2011; McBride et al. 2012). This may be explained by the high incidence of deregulation of genes in the homologous recombination pathway (BRCA1/BRCA2), which has provided opportunities for successful treatment with PARP inhibitors (Banerjee et al. 2010; McBride et al. 2012). Expression profiling has been instrumental to classify ovarian cancers and revealed molecular subtypes with prognostic relevance (Tothill et al. 2008; Verhaak et al. 2013). Despite these advances in understanding of ovarian cancer biology, the cure rate has not much improved (Ledermann and Kristeleit 2010; Vaughan et al. 2011).
We set out to understand the intra-tumor dynamics of treatment-naïve epithelial ovarian cancer by high-resolution analysis of genomic rearrangements. Because the effects of genomic rearrangements in tumor development are only poorly understood, we also examined the contribution of genomic rearrangements to intra-tumor differences in gene expression. We found that treatment-naïve epithelial ovarian cancers exhibit remarkably diverse patterns of genomic rearrangements, which in turn lead to intra-tumor changes in gene expression, including up-regulation of major cancer pathways in only subsets of samples from a single patient. These findings provide novel insight in potential mechanisms underlying treatment resistance.
Results
Topographic sampling of treatment-naïve epithelial ovarian cancer
Ovarian cancer is often discovered when the disease is already in an advanced stage, resulting in the presence of a unique metastasis pattern with cancer cells exfoliating throughout the abdominal cavity following the peritoneal fluid circulation route. The tumor mass is often large with metastases spread throughout the abdomen. Standard of care for such advanced ovarian cancer patients involves surgical cytoreduction before starting chemotherapy treatment. We obtained comprehensive tumor and whole blood samples from three treatment-naïve advanced epithelial ovarian cancer patients with high tumor loads (Table 1). Patients 1 and 3 were diagnosed with a serous adenocarcinoma, whereas patient 2 was diagnosed with a carcinosarcoma, which is a less frequently observed (<1%–4%) form of epithelial ovarian cancer (Rauh-Hain et al. 2013). Carcinosarcoma is characterized by the mixed histology of carcinomatous and sarcomatous components with a more aggressive behavior and a poorer prognosis when compared with serous adenocarcinomas (Supplemental Fig. 1; Harris et al. 2003).
Table 1.
Clinical data of epithelial ovarian cancer patients included in this study
For each patient, tumor biopsies were obtained during surgery from physically separated tumor sites in the abdomen with the final goal to obtain a representative set of samples (Supplemental Table 1; Fig. 1A). The tumor content of each sampling site was generally well above 50% based on histopathological measurements, although computational measurements by ASCAT indicated lower percentages (Supplemental Table 1; Supplemental Fig. 1; Van Loo et al. 2010). Particularly, the metastatic tumor biopsies from patient 1 (p1.IV-1, p1.IV-2, and p1.V-1) and the right ovary tumor sample from patient 3 (p3.III) have a relatively low tumor content. However, these samples were included in most of our analysis, because we could compensate for the lower tumor content by deep sequencing of identified genomic changes. A total of 34 samples (27 tumor, seven reference samples) were obtained and used for the analyses outlined below (Supplemental Table 2).
Figure 1.

Somatic genomic rearrangements detected in patient 1 (left), 2 (middle), and 3 (right). (A) Biopsy locations per patient. Ellipses indicate physically separated tumors; black dots represent biopsy locations. Ellipses are not indicative for tumor size. For patients 1 and 2, ellipses are colored according to the corresponding branch derived from the SV analysis (see panel C). Patient 1: ovaries (orange), om/per (blue). Patient 2: ovaries/pelvis (orange), om/per (blue). (Illustration © 2010 Terese Winslow, U.S. Govt. has certain rights.) (B) Bar chart representing the distribution of the frequency of breakpoints per patient. (C) Heat map and clustering analysis of the detected somatic breakpoints per patient. Rows represent breakpoints, red and yellow bars indicate the presence (red) or absence (yellow) of the breakpoint in a sample. Om/per, omentum/peritoneum. (D) Distribution of somatic rearrangement types per branch for patients 1 and 2 and for all patient 3 samples.
Heterogeneity of structural and copy number variation in treatment-naïve epithelial ovarian cancer
Ovarian cancer is notorious for its frequent genomic instability (The Cancer Genome Atlas Research Network 2011; McBride et al. 2012). Whole-genome mate-pair sequencing allows direct detection of genomic rearrangement breakpoints based on discordantly oriented and spaced mate-pairs (read pairs) (Medvedev et al. 2009). We performed whole-genome mate-pair sequencing using an insert size of ∼3 kb (Supplemental Fig. 2) for each of the biopsies in order to obtain a detailed and comprehensive representation of the genomic instability within tumor samples from three ovarian cancer patients. We used a breakpoint detection algorithm that simultaneously clusters discordant mate-pair sequencing reads from all tumor biopsies per patient (Kloosterman et al. 2011a), allowing us to genotype breakpoints that are present at low frequency with relatively high sensitivity, i.e., based on a single discordant read pair once a robust call is made in another sample of the same patient. For example, given the median physical genomic coverage of ∼50×, the data allow us to genotype a heterozygous breakpoint present in at least 5% and 14% of the tumor cells in samples with a tumor percentage of 90% and 30%, respectively (Supplemental Table 3). After stringent filtering and removal of SV calls present in matching normal control samples, we found between 120 and 369 somatic genomic rearrangements across the primary and metastatic tumor samples in three patients (Supplemental Table 4). We used PCR to validate a set of breakpoint calls and could confirm 95 out of 121 tested (>78% specificity) (Supplemental Fig. 3).
For each somatic breakpoint detected by mate-pair sequencing, we determined the number of tumor samples that carried the breakpoint. For patient 1, this revealed that only 2/369 somatic breakpoints are shared between all samples, whereas the majority was found to be shared between only four or five of the eight samples from this patient. In patient 2 the largest number of breakpoints is also shared between only four samples, but 34/120 breakpoints were shared between all nine tumor samples. In patient 3 the vast majority of breakpoints were shared between five and seven of the seven tumor samples (Fig. 1B). We then performed unsupervised hierarchical clustering using the breakpoint junctions detected across each of the biopsies per patient. For patients 1 and 2, this revealed two clusters of samples. Particularly patient 1 showed two extremely different branches. For both patients, one cluster contained all biopsies from the omentum and peritoneum, whereas the other cluster contained all biopsies from the ovaries, and for patient 2 also a biopsy from a tumor located in the pelvis. In contrast to the branching patterns detected in patient 1 and 2, a much more homogeneous pattern was detected in patient 3 (Fig. 1C). Several breakpoints overlap with cancer genes from the Cancer Gene Census, including NF1, FANCD2, and CDKN2A and these are all targeted by breakpoints present in only subsets of samples (Supplemental Table 4; Futreal et al. 2004). For FANCD2 and the cancer-related genes ERBB4 and ESR1, which are targeted by breakpoints in patient 2, we observed a sample-specific effect of the breakpoint on gene expression (Supplemental Fig. 4). Expression of ESR1 is a prognostic factor for survival in ovarian cancer (Zamagni et al. 2009).
Distinct patterns of genomic rearrangement classes are observed among different cancers (Stephens et al. 2009; Campbell et al. 2010; McBride et al. 2012). Analysis of breakpoint types per patient revealed that deletions comprised the largest subset (40%) in patient 1, which is consistent with recent findings indicating an excess of deletions in ovarian cancer with germline BRCA mutations (McBride et al. 2012), as was the case for this patient (see below). However, more detailed analysis of branch-specific breakpoints for patient 1 revealed a shift in rearrangement types between the two branches despite their shared BRCA status (Fig. 1D). The cluster of four omental/peritoneal metastatic tumors showed a higher percentage of somatic tandem duplication type rearrangements and inversions, and a lower percentage of deletions and interchromosomal rearrangements when compared with the cluster containing the tumors on the ovaries. In line with this, we observed an increase in head-to-tail breakpoint junctions at a cost of tail-to-head junctions for patient 1 in the omental/peritoneal samples when compared with the samples that originated from both ovaries (data not shown). A difference in rearrangement types was, however, not apparent for the branches in patient 2. For patient 3 we observed that the majority (>40%) of somatic SVs comprise tandem duplications, in line with previous studies (Fig. 1D; McBride et al. 2012). Inter-sample differences in rearrangement signatures were not detected for patient 3.
To get further support for the dynamic patterns of heterogeneity revealed by the somatic genomic breakpoints in the ovarian cancers studied here, we used SNP-array genotyping and copy number analysis. We performed unsupervised hierarchical clustering of allele frequencies derived from the SNP-array genotyping data. The analysis includes all SNPs with differences in allele frequencies across each of the biopsies per patient (∼10–30K SNPs) (Methods). Similar patterns of deviating allele frequencies for specific subsets of samples indicate shared ancestry, whereas diversity of these patterns rather suggests independent evolution. Heat map and clustering analysis of the allele frequencies of included SNPs confirmed the results from the mate-pair analysis for each patient (Supplemental Fig. 5).
Intra-tumor mutational profiles in ovarian cancer
Next, we screened coding sequences of a total of 2099 cancer genes across each of the biopsies (Supplemental Table 3). We validated all identified mutations on all tumor and matching normal tissue samples using PCR-based resequencing on the MiSeq at >1000× coverage. The MiSeq data were also used to derive or refine mutation frequencies (Supplemental Table 5). We detected 63 somatic single-nucleotide mutations in patient 1, and considerably fewer mutations in patients 2 and 3 (17 mutations per patient, Fig. 2A). In patient 1, we also identified a BRCA2 germline frameshift indel and concomitant LOH of chr 13 in the tumor biopsies.
Figure 2.

Somatic single-nucleotide mutation analysis results for patients 1, 2, and 3. (A) Regional distribution of mutations across tumor samples per patient. Blue gradient indicates the percentage of reads that carried the mutation. Gene colors indicate mutation impact: high, essential splice site or frame-shift (orange); medium, nonsynonymous (yellow); or silent, intronic, 5′ or 3′ UTR (white). (B) Distribution of the two TP53 missense mutations detected in patient 1 (P278L and I195N) across all tumor samples of this patient. (C) Kataegis as detected in patient 2 samples p2.VI-1 and p1.VI-2. The 12 single-nucleotide changes in FANCD2 coincide with a genomic breakpoint, which is solely detected in these samples. (D) Transitions versus transversions for patient 1. All ovarian samples (primary tumor [p1.I-1 and p1.I-2] and metastases located in the other ovary [p1.II-1 and p1.II-2]) versus the omentum/peritoneum metastases (p1.IV-1, p1.IV-2, p1.IV-3, and p1.V-1).
All patients carried TP53 mutations. In patient 2 and 3 a single TP53 mutation was detected in all tumor samples per patient. Interestingly, we identified two different driver TP53 missense mutations (P278L and I195N) in patient 1, occurring at distinct tumor locations. Both of these mutations have been described in the COSMIC database (Forbes et al. 2010). Only the samples derived from the right ovary (p1.I-1 and p1.I-2, the presumed primary tumor), contained both TP53 mutations, albeit I195N was detected at low frequency (1%–9% vs. 33%–77% for P278L) (Fig. 2B).
We observed 19 mutations that were unique to only a single ovary tumor sample in patient 1 (private mutations). However, none of the four metastases in the omentum and peritoneum (p1.IV-1 to p1.IV-3 and p1.V-1) carried private mutations. In fact, all mutations identified in the omentum and peritoneum cluster of samples were ubiquitous and, with the exception of a mutation in DLL1, all variants were also identified in the samples at the right ovary. For patient 2, 12 of the 17 mutations were in FANCD2 and all 12 occurred in samples p2.VI-1 and p2.VI-2 within a window of 1.2 kb and comprising characteristic TpCpX trinucleotides, likely resulting from kataegis (Nik-Zainal et al. 2012). Similar to kataegis described in breast cancer, these mutations coincided with SV uniquely present in these two tumor samples (Fig. 2C). Most of the other coding variants identified in patient 2 were shared between all samples. The majority of mutations in patient 3 were ubiquitous. Two mutations occurred in the tumor suppressor gene TSC1 (one missense, P141R, and one essential splice mutation). The TSC1 splice site mutation leads to truncation of the TSC1 transcript, suggesting deregulation of mTOR signaling as a possible contributor to tumor development in patient 3 (Supplemental Fig. 6; Dobbin and Landen 2013). An additional mutation was found in CSMD3 in this patient, which is frequently mutated in ovarian cancer and non-small cell lung cancer, although the functional role of this gene in tumor formation is not clear (The Cancer Genome Atlas Research Network 2011; Liu et al. 2012). Only two private cancer gene single-nucleotide mutations were found in patient 3 (sample p3.IV).
Overall, the single-nucleotide mutation data revealed a similar pattern of genetic heterogeneity as the structural and copy number variation data for patient 1; one cluster of mutations occurred at metastatic tumor sites in the omentum and peritoneum and another cluster of mutations was found in the tumors in the left ovary. Both clusters shared mutations with the presumed primary tumor samples (p1.I-1 and p1.I-2). Interestingly, we found a difference in the Transition/Transversion (Ti/Tv) ratio for the two branches in this patient (Fig. 2D), suggesting that distinct mutational forces acted in different branches of the ovarian tumor in patient 1 (Alexandrov et al. 2013). For patients 2 and 3 we found much fewer mutations and several of these were present in all samples. A mutation in BBS4 further supported the branching pattern observed in patient 2.
Gene expression differences across ovarian cancer biopsies reveal intra-tumor subtypes and branch-specific pathway activation
Gene expression profiling of ovarian cancer enabled classification in distinct subtypes associated with differences in survival and therapy resistance (Tothill et al. 2008; Verhaak et al. 2013). We performed RNA sequencing to measure gene expression across each of the tumor biopsies and we detected between 1000 and 1300 differentially expressed genes per sample compared with all other samples of the same patient (Supplemental Table 6). Hierarchical clustering of gene expression differences for each of the patients revealed two major branches for both patient 1 and patient 2 (Supplemental Fig. 5). For patient 3 the clustering of differentially expressed genes across tumor biopsies did not reveal any distinct subgroups. The clustering of samples based on RNA expression differences further substantiated intra-tumor diversity as observed based on genomic breakpoints.
We used the normalized coverage for 1500 genes that define six different epithelial ovarian cancer subtypes to classify each of the tumor biopsies from patients 1 to 3 (Tothill et al. 2008; The Cancer Genome Atlas Research Network 2011). The tumor biopsies from patient 1 fall apart into distinct subtypes following the branching we observed based on clustering of genomic and transcriptomic data: The samples from the omentum and peritoneum clearly display the C1 (high stromal) signature, overlapping with the C2 (high immune) signature as described before (Tothill et al. 2008), whereas samples from the ovaries rather fall into the C4 (low stromal response) category although some expression of genes in the high immune signature can also be observed (Fig. 3A). The samples from patient 3 display the C2 and C4 gene expression signatures. Patient 2 samples explicitly show the C5 (mesenchymal) signature, as expected from the histological examination, which indicated a carcinosarcoma (Supplemental Fig. 1). The distant metastases of patient 2 are different from the ovary and pelvis samples as they also show overlap with the C2 category.
Figure 3.

(A) Heat map of the percentage concordance of each sample with the subtypes of ovarian cancer presented by Tothill and colleagues (Futreal et al. 2004; Tothill et al. 2008; Santarius et al. 2010). C1, high stromal response; C2, high immune signature; C3, low malignant potential (LMP) signature; C4, low stromal response; C5, mesenchymal signature; C6, low grade endometrioid. (B) Allele frequencies of coding mutations in RNA and DNA for patient 1. (C) Branch-specific expression differences of genes involved in major signaling pathways for patient 1 (top) and patient 2 (bottom).
To determine whether single-nucleotide changes observed in the genome were also found in expressed transcripts we analyzed the frequency for each of the identified mutations among RNA sequencing reads for patient 1 (Fig. 3B). This analysis showed that some single-nucleotide changes present at the DNA level are not expressed. In addition, we also find that alleles with single-nucleotide variants detected at low frequencies at the DNA level are expressed at very high levels. For example, the shared TP53 I195N variant and the private BBS1 and MEN1 variants are expressed at a much higher frequency in the RNA, suggesting that they are relevant for tumor growth.
Based on the top 5% most significantly differentially expressed genes we used Cytoscape software to evaluate whether specific cellular pathways or processes have altered expression in any of the branches or samples (Shannon et al. 2003). All branches showed activation of pathways or processes related to cancer development compared with the total pool of non-tumor samples of all three patients, such as the different aspects of cell division (e.g., cell cycle checkpoints, DNA replication, chromosome segregation) and growth factor and p53 signaling (Supplemental Table 6). Specific pathway activation was observed for the two branches in patients 1 and 2 (Fig. 3C; Supplemental Table 6). The samples in the ovarian cluster from patient 1 expressed significantly higher levels of genes involved in ERBB signaling and post-translational protein modification compared with the samples in the omentum/peritoneum cluster. Up-regulation of Hedgehog/WNT/Cadherin genes was observed in the ovarian and pelvic samples of patient 2. Interestingly, samples from the omentum and the peritoneum in both patients 1 and 2 had many pathways commonly up-regulated compared with the other samples in these patients, including chemokine signaling and cytokine–cytokine receptor interactions, immune response, extracellular matrix organization, and integrin signaling. Finally, cell adhesion (CAM) was one of the processes enriched for in the ovary and pelvis samples in patients 1 and 2, although the exact genes up-regulated in the samples from these two patients differ.
Genomic heterogeneity causes intra-tumor differences in gene expression
The marked differences in gene expression observed across biopsies from the same patient prompted us to analyze the contribution of genomic rearrangements to these differences, an aspect of tumor biology which is poorly understood. Based on copy number profiling we identified 14 large copy number gains and losses (range: 0.97–28 Mb) that were only present in subsets of samples from patient 1 and patient 2 (Supplemental Table 7). For each of these copy number changes we determined the mean of the differences in log2 ratios from SNP-arrays and compared these with the mean of the log2 ratios derived from the RNA sequencing. We observed a strong correlation between copy number changes and gene expression changes based on pairwise comparisons (Fig. 4A), indicating that gene expression is strongly influenced by DNA copy number. However, on a genome-wide basis, only 1.5%–1.8 % of the differentially expressed genes are within the boundaries of the large copy number changes for patients 1 and 2, respectively. Thus other factors, such as mutations, translocations, epigenetic changes, or secondary effects are likely contributing to the intra-tumor gene expression differences.
Figure 4.
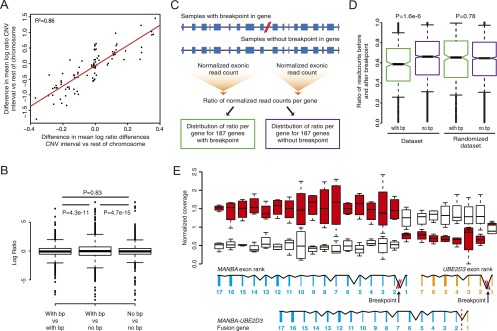
Intra-tumor differences in gene expression resulting from genomic rearrangements in patient 1. (A) Pairwise comparison of copy number changes and gene expression changes. (B) Boxplot showing log ratios derived from pairwise comparisons of patient 1 samples, categorized in three bins: (1) Both samples have a breakpoint, (2) one sample has the breakpoint and the other does not have the breakpoint, (3) both samples do not have a breakpoint. Statistical testing of differences in variance was performed using Levene’s test. (C) Schematic representation of a method used to detect expression differences of exons before and after a breakpoint in a gene. Per gene, the ratio of the normalized exonic read count before and after the breakpoint was determined for each of the samples from patient 1. Ratios were separated in two bins: one containing ratios derived from genes with a breakpoint and one containing ratios derived from genes without a breakpoint. (D) Boxplot of the distribution of ratios of the normalized exonic read count before and after a breakpoint for genes that contain a breakpoint (with bp) and genes that do not contain a breakpoint (no bp). The analysis was repeated by randomly assigning breakpoints to samples (randomized data set). Statistical testing was performed using a Mann-Whitney U-test. (E) Changes in gene expression for the exons of the MANBA and UBE2D3 gene exons in patient 1. In the presence of the deletion breakpoint a MANBA–UBE2D3 fusion gene is formed. (Red) Breakpoint present; (white) no breakpoint present.
To further study the effect of genomic breakpoints on gene expression, we utilized the precise breakpoint definition provided by mate-pair sequencing. We reasoned that an expression effect of a breakpoint should result in either a positive or negative change in gene expression in samples with the breakpoint, relative to samples without the breakpoint. To test this, we used the gene expression differences (log2 ratios) derived from pairwise comparisons based on each of the samples from patient 1 and categorized the comparisons in three bins: (1) Both samples have a breakpoint, (2) one sample has the breakpoint and the other does not have the breakpoint, and (3) both samples do not have a breakpoint. If genomic breakpoints have an effect on expression of the respective genes, we would anticipate an overall increase in fold changes in bin 2 versus bins 1 and 3. Indeed, we observed a significant increase of the variance of the distribution of fold changes in bin 2, indicating that on average, genomic rearrangement breakpoints affect gene expression both positively and negatively (Fig. 4B).
To get a more precise picture of the effects of SVs on gene expression, we measured the normalized read counts for exons located before and after an SV breakpoint in a gene (Fig. 4C). A shift in the ratio of the read count before and after the breakpoint would be expected if the expression of the exons before and/or after a breakpoint has changed as a result of the breakpoint. For example, part of a gene could be up-regulated due to fusion with another partner gene or a decrease in expression could be expected if one half of a gene is deleted. Furthermore, measuring this ratio allows us to solely detect the effect of a breakpoint in the gene and exclude influences of other factors on gene expression (e.g., neighboring breakpoints, promoter methylation). Figure 4D shows a boxplot of the distribution of ratios for the genes that do not contain a breakpoint and the genes that do contain a breakpoint, indicating a marked shift in distribution toward more extreme ratios for all genes containing a breakpoint. We repeated the same analysis by randomly assigning breakpoints to samples. In this case the distribution of ratios is the same for genes with and without breakpoints, indicating the specific effects of the breakpoints on gene expression (Fig. 4D). These results emphasize that expression measurements should not be determined only on a per gene basis, because subtle intra-gene expression differences due to rearrangements will be obscured for whole-gene measurements.
The changes in exon expression for samples with and without a deletion breakpoint in the MANBA and UBE2D3 genes (patient 1) illustrate the sensitivity of the ratio analysis (Fig. 4E). Exons at the 3′ end of MANBA are expressed higher in samples with the breakpoint relative to samples without the breakpoint and the reverse is true for exon 1. Similar effects were found for the UBE2D3 gene. An UBE2D3–MANBA in the frame fusion gene resulted from the somatic deletion in the ovary samples and the fusion transcript was expressed as verified by RT-PCR (Supplemental Fig. 7).
We systematically searched for genomic rearrangements that are predicted to result in gene fusions and found 28 putative fusions in patient 1 (Supplemental Table 4). For 12 of the predicted rearrangements, we designed PCR primers for RT-PCR across the breakpoint junctions and we could confirm expression of seven fusion transcripts of which four were in frame and differentially expressed in tumor biopsies (Supplemental Fig. 7). Among these is one fusion containing the MAST4 kinase. MAST kinases are involved in recurrent fusions in breast cancer and enhance cell proliferation (Robinson et al. 2011).
Discussion
We here show that treatment-naïve epithelial ovarian cancer, whether serous adenocarcinoma or carcinosarcoma, may display extensive intrinsic genomic and transcriptomic heterogeneity, leading to a broad variety and potentially functional lesion-specific deregulation of cellular pathways. The major genomic and transcriptomic differences were found between distant metastasis located at the omentum or peritoneum versus tumor samples at the ovaries and pelvis, substantiating previous evidence for intra-tumor heterogeneity in serous epithelial ovarian cancer (Khalique et al. 2007; Bashashati et al. 2013). The most striking heterogeneity was found for patient 1, where both the mutation and the SV data supported two subsets of tumor biopsies. This included two independent TP53 mutations, raising the question as to whether two separately initiated and evolving tumors had occurred in patient 1, with tissue from both tumors interwoven at the right ovary. So-called collision tumors have been described before and these are marked by histologically distinct tumors separated by stroma or basal lamina (Bige et al. 2009). Both ovaries are frequently affected in serous ovarian cancer (65%) (Cotran et al. 1999) and in rare cases this involves tumors with bi-focal origin (Abeln et al. 1995), which is a possibility we cannot fully exclude in our case. Both scenarios were not obvious from the histopathological examination. Interestingly, patient 1 harbored a germline BRCA2 mutation. BRCA2 functions in DNA repair, and disruption of BRCA2 leads to genomic instability (O’Donovan and Livingston 2010). Therefore, this mutation could possibly have promoted the very early separation of the tumor samples from this patient as opposed to the more coherent evolutionary patterns in patients 2 and 3.
Multi-site profiling of genomic changes allows estimation of the evolutionary course of cancer development, including timing of mutational events (Gerlinger et al. 2012; Yates and Campbell 2012). TP53 mutations were present in all samples from the three patients, indicating that these occurred early during tumor evolution. For patient 1, we observed two subsets of samples, which constitute two independent tumors or very early branched subclones (with independent TP53 mutations). This evolutionary pattern was supported by both mutation data and SV data. Within each of the branches we observed much more coherence than between the branches: We observed no unique mutations and just one unique rearrangement for the omentum and peritoneum samples. For the ovary samples of patient 1 we found several unique changes. This included 19 private mutations and 48 private genomic rearrangements all of which likely occurred late during tumor evolution, demonstrating continuous evolution at both the structural and mutational level in this branch. Furthermore, we observed indications of different mutational mechanisms operating in each of the two subsets in patient 1, both at the level of genomic rearrangements and point mutations. For patient 3, we observed only two private mutations, whereas all other mutations (15) were shared between samples. A large overlap between samples was also supported by the SV data from patient 3. However, we did observe 22 unique rearrangements, suggesting ongoing evolution at separate sites at the level of genomic rearrangements. For patient 2, the SV data showed the presence of two subsets of samples. However, a large fraction (34/120) of somatic SVs were found in all samples indicating a common evolutionary origin as opposed to the very early branching observed for patient 1. The common origin and branching in patient 2 was further supported by the presence of mutations shared by all samples and a unique coding mutation in the ovary/pelvis branch, respectively. In addition, we observed ongoing evolution in two pelvis samples based on the observation of a condensed cluster of mutations in FANCD2 coinciding with SVs, a mutational process which has been termed “kataegis” (Fig. 2C; Nik-Zainal et al. 2012). These data show that kataegis may act only regionally within the tumor of a single patient. Similarly, we have previously also observed that chromothripsis may exclusively occur in either primary, or metastatic tumor samples from the same patient (Kloosterman et al. 2011b), demonstrating that massive mutation mechanisms may occur late during tumor development and do not necessarily represent an initiating event. Whole-genome sequencing should reveal more single-nucleotide changes and provide further insight into possible differences in evolutionary timing relative to SVs in ovarian cancer.
There is a strong need for improved and targeted therapies for ovarian cancer to increase cure rates (Ledermann and Kristeleit 2010; Vaughan et al. 2011; Banerjee and Kaye 2013). Several targeted therapies are being tested, but careful selection of patients for targeted treatment is essential (Smolle et al. 2013). We show here that there is major intra-tumor heterogeneity concerning expression of cellular pathways, some of which are candidates for targeted treatment. For example, overexpression of the Hedgehog pathway was observed in a subset of metastases in the omentum and pelvic region of patient 2, compared with other pelvic lesions and tumor sites in the ovaries. High expression of the Hedgehog transcription factor gene GLI1 is associated with poor survival in advanced serous ovarian cancer (Ciucci et al. 2013) and Hedgehog components are deregulated in various sarcomas, presenting new treatment possibilities such as Hedgehog ligand antagonists and inhibition of Gli1 transcription activity (Kelleher et al. 2012). Also, we detected strong up-regulation of integrin pathway members in peritoneum and omentum metastases in patient 1, as well as elevated expression of inflammatory chemokines and cytokines primarily found in metastases in the omentum of both patients 1 and 2. Expression differences in these genes in metastatic lesions compared with primary tumors have previously been observed (Davidson 2007). These may provide an attractive target for treatment of ovarian cancer (Mantovani et al. 2008; Vaughan et al. 2011; Sawada et al. 2012), because the vast majority of patients die as a consequence of metastatic disease, while the primary tumor is often completely removed during debulking surgery.
The cancer genome harbors a wide variety of genomic alterations. Particularly the contribution of structural genomic rearrangements to tumor development is poorly understood. We here demonstrate that intra-tumor heterogeneity may involve cancer genes disrupted by genomic breakpoints present in only a subset of tumor masses. Furthermore, we associated the intra-tumor expression differences with genomic rearrangement breakpoints and found that effects may range from altered expression due to copy number changes of entire genes to very subtle effects involving breakpoints affecting only part of a gene, all occurring within a single patient. These data show that the effects of genomic rearrangements are profound, contribute to intra-tumor heterogeneity, and may be equally important as coding mutations for tumor development.
Because our study only covered multi-site analysis of three ovarian cancer patients, it remains to be seen how representative the identified genomic and transcriptomic characteristics will be in a larger sample set. Large-scale follow-up studies should be conducted to determine the rate of extreme intra-tumor heterogeneity in ovarian cancer. This aspect of tumor biology requires further attention to fully understand escape routes as a response to treatment and improve survival rates.
Methods
Patient sampling and consent
All patients included are epithelial ovarian cancer FIGO stage III/IV patients undergoing primary cytoreductive debulking according to the standard of care (Table 1). During primary debulking, tumor samples were obtained from thoroughly documented locations. Patients 1 and 2 underwent successful optimal debulking. In patient 3, optimal debulking appeared infeasible perioperatively and only partial cytoreduction was achieved. She later underwent successful cytoreduction after neo-adjuvant chemotherapy. We only included the tumor samples obtained from the first debulking for patient 3. In addition, DNA from blood and saliva was obtained from every patient as control samples (Oragene DNA kit, DNA Genotek Inc.). Tumor samples were immediately forwarded from the operating room to the pathology department. If feasible, multiple core and peripheral tumor samples of each individual metastasis or primary tumor were snap-frozen in liquid isopentane within 1 h. Tissue was processed into frozen sections and stained by Hematoxylin & Eosin (H&E) (Supplemental Fig. 1). A pathologist reviewed all slides and confirmed tumor type, estimated tumor cell percentage, and amount of necrosis (Supplemental Table 1). This study was approved by the ethics committee of the UMC Utrecht, The Netherlands. Patients could indicate in a specific section of the informed consent form that they wanted to be informed about incidental findings in their germline DNA that could affect their health, or the health of their relatives. All patients signed informed consent before debulking.
DNA, RNA isolation
Fresh frozen samples were homogenized and subsequently split for independent DNA and RNA isolation. DNA was isolated using the Qiagen Genomic DNA kit (Qiagen). Total RNA was isolated using TRIzol reagent (Life Technologies). After isolation, DNA samples were stored at −20°C, RNA samples at −80°C.
Mate-pair sequencing
Mate-paired libraries were generated from 5 to 10 μg of DNA isolated from tumor and control samples using the 5500 SOLiD Mate-Paired library kit (Life Technologies). Samples were sheared to 3-kb fragments by Hydroshear DNA shearing (Digilab). Per library, 2 × 50-bp mates were sequenced on a SOLiD 5500xl or SOLiD WildFire instrument. Forward and reverse tags were mapped independently (samse) to the reference genome (GRCh37) using BWA software and settings –c –l 25 –k 2 –n 10 (Li and Durbin 2009). Discordant reads were clustered using in-house software as described previously (Kloosterman et al. 2011a). The software is available from https://github.com/Vityay/1-2-3-SV. In a first step, we estimated the insert size distribution and location of discordant mate-pairs. This was done separately for each sample. Furthermore, PCR duplicates, reads with mapping quality 0, and nonuniquely mapped reads were removed from further analysis. As a second step clustering of discordant pairs was done for all samples from each patient together. Two pairs are considered to belong to the same cluster when the distance between coordinates of their 5′ tags together with the distance between 3′ tags does not exceed the median distance of the library with the largest insert size. The search is continued until no clusters with at least five clones in at least one of the samples can be found. As analysis of discordant read pairs does not give exact breakpoints of structural variants, the output lists genome segments containing each breakpoint along with information about the source of the discordant pairs (samples) and the properties of the cluster as outlined in Supplemental Table 4. The orientation of the different mate-pair tags in a cluster relative to each other is indicated by H (or h for the minus strand) when the tag has its “head” side (the side that points toward the start of the chromosome) opposed to the pairing tag and T (or t for the minus strand) when a tag has its “tail” side (the side that points toward the end of the chromosome) opposed to the pairing tag. The clustering results in calling of intrachromosomal rearrangements (deletion type, inverted, tandem duplication type) and interchromosomal rearrangements. To select for somatic variants, all genomic rearrangement breakpoints were filtered for normal tissue samples (blood, muscle, tuba) and an in-house database of mate-pair sequencing data from healthy individuals. To achieve high-quality calling of somatic structural variants, we required at least five independent discordant sequence reads derived from at least one tumor sample (Kloosterman et al. 2011a,b). For all breakpoint calls consistent with these criteria, presence of the breakpoint in other samples from the same patient was determined based on presence of at least one overlapping discordant read pair with the same orientation.
Primers for PCR confirmation of somatic breakpoints were designed based on mate-pair sequencing data. PCRs were performed under standard conditions as described before (Kloosterman et al. 2011a).
Cancer gene resequencing
SNVs and indels were detected by targeted sequencing of a total of 2099 cancer genes. First, samples were interrogated by a designed “Cancer mini-genome” consisting of 1977 cancer genes. Barcoded fragment libraries were generated from 2 μg of isolated DNA from tumor and control samples as previously described (Harakalova et al. 2011). Pools of libraries were enriched for 1977 cancer-related genes (Cancer mini-genome [Vermaat et al. 2012]) using SureSelect technology. Enriched libraries were sequenced on a SOLiD 5500xl or SOLiD WildFire instrument according to the manufacturers’ protocol. Furthermore, the exons within a subset of 409 oncogenes and tumor suppressor genes were interrogated by the Ion Ampliseq Comprehensive Cancer Panel (Life Technologies). Libraries were constructed from 40 ng of isolated DNA for each sample using standard AmpliSeq procedures. Barcoded libraries were pooled and sequenced on the Ion PGM Sequencer (Life Technologies).
Ion Torrent reads were aligned to the human reference genome version 19 (GRCh37) using Tmap. Variant calling on Ion Torrent data was performed using Strelka (Saunders et al. 2012). SOLiD reads were mapped on the same genome version, using BWA (–c –l 25 –k 2 –n 10) and variant calling was done using a custom pipeline identifying variants with at least 10× coverage, a 15% allele frequency, and multiple (>2) occurrences in the seed (the first 25-bp most accurately mapped part of the read) as well as support from independent reads (>3). All variant positions identified in either SOLiD or Ion Torrent data were subsequently genotyped in the raw data sets of both techniques for all samples using samtools mpileup, to ensure the presence or absence of possible low-frequency variants. Validation of single-nucleotide mutations and indels was performed by PCR amplification of mutation loci followed by Nextera XT library prep and sequencing on MiSeq (Illumina).
SNP-array analysis
For each sample, 200 ng DNA was used as input for copy number profiling using Cyto12 SNP arrays according to standard procedures (Illumina). Genomic events were identified by applying ASCAT processing (Allele Specific Copy Number Analysis of Tumors) with Nexus Copy Number 6.0 (BioDiscovery). Briefly, all signals in tumor samples similar to those in the provided reference sample are excluded from analysis, increasing specificity in detecting additional events in tumor samples.
Clustering of SNP array data was done by calculating a Euclidean distance matrix based on B-allele frequencies of all SNP positions showing a heterozygous genotype in the reference samples (allele frequency 20%–80%), and performing hierarchical clustering on these data using standard R functions.
RNA sequencing
Total RNA was processed with the Poly(A) Purist Kit (Life Technologies) to select for poly(A)+ RNA. Next, the mRNA-ONLY Eukaryotic mRNA Isolation kit (Illumina) was used to select for 5′ capped mRNA. Paired-end libraries were constructed from 8–30 μg total RNA per sample, using the SOLiD total RNA-seq kit (Life Technologies). Libraries were barcoded and sequenced on a SOLiD 5500xl instrument in paired-end mode (50 × 35 bp). Forward (F3) and reverse (F5) reads were mapped independently to the human reference genome (GRCh37) using BWA (–c –l 25 –k 2 –n 10) (Li and Durbin 2009). Coverage per gene was determined by adding up read counts of all coding regions as determined by BEDTools 2.16.2 multicov (Quinlan and Hall 2010).
Because of the lack of a true reference sample, i.e., healthy tissue from the ovaries of each patient, we compared each sample separately against the pool of all other samples of the same patient and determined gene expression differences within patients using the DEGseq R package (Wang et al. 2010). To identify the most significant expression differences, DEGseq output was first corrected for multiple testing by selecting genes with a P-value < 4.6399 × 10−8 (0.001 divided by 21552 [the number of tested genes]) and extracting the top 5% and bottom 5% of normalized log ratios. Normalized coverages of the genes emerging from this analysis were used to cluster samples based on Poisson Mixture Models generated with the HTSCluster R package (Rau et al. 2011). Pathway and GO biological process enrichment of up-regulated genes within the thus created clusters was determined by comparing the core samples in these clusters to a pool of all reference samples from our three patients and finally to each other by DEGseq and selecting the top 5% genes as described above. Resulting gene lists were analyzed through the Reactome FI Cytoscape Plugin (Shannon et al. 2003), and pathways or processes containing at least two up-regulated genes and a false discovery rate (FDR) <0.1 were reported as being affected. We performed molecular subtyping of samples by calculating the percentage of concordance of up- and down-regulated genes (compared with the common reference pool) with the profiles presented by Tothill and colleagues (Tothill et al. 2008; The Cancer Genome Atlas Research Network 2011).
Data access
The SNP-array data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE47633. The sequencing data have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena/) under accession number ERP003455.
Competing interest statement
Timothy T. Harkins and Clarence C. Lee are full-time employees of Life Technologies, a corporation that is commercializing SOLiD and Ion Torrent sequencing technology.
Acknowledgments
We thank Nicolle Besselink, Nico Lansu, and Ewart de Bruijn for generating sequencing data on SOLiD WildFire, Lars van der Veken for support with analysis of SNP-array data, and Maarten van Iterson for help with RNA-seq analysis. This work was financially supported by the Cancer Genomics Centre, which is part of the Netherlands Genomics Initiative, and the personalized cancer care priority program of the University Medical Centre Utrecht. We would like to acknowledge the Netherlands Center for Personalized Cancer Treatment for providing infrastructure access and expertise.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.161026.113.
References
- Abeln EC, Kuipers-Dijkshoorn NJ, Berns EM, Henzen-Logmans SC, Fleuren GJ, Cornelisse CJ 1995. Molecular genetic evidence for unifocal origin of advanced epithelial ovarian cancer and for minor clonal divergence. Br J Cancer 72: 1330–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR 2013. Deciphering signatures of mutational processes operative in human cancer. Cell Rep 3: 246–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Kaye SB 2013. New strategies in the treatment of ovarian cancer: Current clinical perspectives and future potential. Clin Cancer Res 19: 961–968 [DOI] [PubMed] [Google Scholar]
- Banerjee S, Kaye SB, Ashworth A 2010. Making the best of PARP inhibitors in ovarian cancer. Nat Rev Clin Oncol 7: 508–519 [DOI] [PubMed] [Google Scholar]
- Bashashati A, Ha G, Tone A, Ding J, Prentice LM, Roth A, Rosner J, Shumansky K, Kalloger S, Senz J, et al. 2013. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J Pathol 231: 21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bige O, Demir A, Koyuncuoglu M, Secil M, Ulukus C, Saygili U 2009. Collision tumor: Serous cystadenocarcinoma and dermoid cyst in the same ovary. Arch Gynecol Obstet 279: 767–770 [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin M-L, et al. 2010. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467: 1109–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network 2011. Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciucci A, De Stefano I, Vellone VG, Lisi L, Bottoni C, Scambia G, Zannoni GF, Gallo D 2013. Expression of the glioma-associated oncogene homolog 1 (gli1) in advanced serous ovarian cancer is associated with unfavorable overall survival. PLoS ONE 8: e60145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotran RS, Kumar V, Collins T, Robbins SL 1999. Robbins pathologic basis of disease. W.B. Saunders Company, Philadelphia [Google Scholar]
- Davidson B 2007. Anatomic site-related expression of cancer-associated molecules in ovarian carcinoma. Curr Cancer Drug Targets 7: 109–120 [DOI] [PubMed] [Google Scholar]
- Dobbin ZC, Landen CN 2013. The importance of the PI3K/AKT/MTOR pathway in the progression of ovarian cancer. Int J Mol Sci 14: 8213–8227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008 v2.0 2010. Cancer incidence and mortality worldwide: IARC CancerBase No. 10. International Agency for Research on Cancer, Lyon. http://globocan.iarc.fr [DOI] [PubMed]
- Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, et al. 2010. COSMIC (the Catalogue of Somatic Mutations in Cancer): A resource to investigate acquired mutations in human cancer. Nucleic Acids Res 38: D652–D657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR 2004. A census of human cancer genes. Nat Rev Cancer 4: 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JW 2003. Evidence emerges for early metastasis and parallel evolution of primary and metastatic tumors. Cancer Cell 4: 4–6 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA 2000. The hallmarks of cancer. Cell 100: 57–70 [DOI] [PubMed] [Google Scholar]
- Harakalova M, Mokry M, Hrdlickova B, Renkens I, Duran K, van Roekel H, Lansu N, van Roosmalen M, de Bruijn E, Nijman IJ, et al. 2011. Multiplexed array-based and in-solution genomic enrichment for flexible and cost-effective targeted next-generation sequencing. Nat Protoc 6: 1870–1886 [DOI] [PubMed] [Google Scholar]
- Harris MA, Delap LM, Sengupta PS, Wilkinson PM, Welch RS, Swindell R, Shanks JH, Wilson G, Slade RJ, Reynolds K, et al. 2003. Carcinosarcoma of the ovary. Br J Cancer 88: 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Cancer Genome Consortium 2010. International network of cancer genome projects. Nature 464: 993–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher FC, Cain JE, Healy JM, Watkins DN, Thomas DM 2012. Prevailing importance of the hedgehog signaling pathway and the potential for treatment advancement in sarcoma. Pharmacol Ther 136: 153–168 [DOI] [PubMed] [Google Scholar]
- Khalique L, Ayhan A, Weale ME, Jacobs IJ, Ramus SJ, Gayther SA 2007. Genetic intra-tumour heterogeneity in epithelial ovarian cancer and its implications for molecular diagnosis of tumours. J Pathol 211: 286–295 [DOI] [PubMed] [Google Scholar]
- Klein CA 2009. Parallel progression of primary tumours and metastases. Nat Rev Cancer 9: 302–312 [DOI] [PubMed] [Google Scholar]
- Kloosterman WP, Guryev V, van Roosmalen M, Duran KJ, de Bruijn E, Bakker SCM, Letteboer T, van Nesselrooij B, Hochstenbach R, Poot M, et al. 2011a. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum Mol Genet 20: 1916–1924 [DOI] [PubMed] [Google Scholar]
- Kloosterman WP, Hoogstraat M, Paling O, Tavakoli-Yaraki M, Renkens I, Vermaat JS, van Roosmalen MJ, van Lieshout S, Nijman IJ, Roessingh W, et al. 2011b. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol 12: R103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledermann JA, Kristeleit RS 2010. Optimal treatment for relapsing ovarian cancer. Ann Oncol (Suppl 7) 21: vii218–vii222 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Morrison C, Wang L, Xiong D, Vedell P, Cui P, Hua X, Ding F, Lu Y, James M, et al. 2012. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis 33: 1270–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek JA, Mery E, Mahmoud YA, Al-Azwani EK, Roger L, Huang R, Jouve E, Lis R, Thiery J-P, Querleu D, et al. 2011. Copy number variation analysis of matched ovarian primary tumors and peritoneal metastasis. PLoS ONE 6: e28561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F 2008. Cancer-related inflammation. Nature 454: 436–444 [DOI] [PubMed] [Google Scholar]
- McBride DJ, Etemadmoghadam D, Cooke SL, Alsop K, George J, Butler A, Cho J, Galappaththige D, Greenman C, Howarth KD, et al. 2012. Tandem duplication of chromosomal segments is common in ovarian and breast cancer genomes. J Pathol 227: 446–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, Stanciu M, Brudno M 2009. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods 6: S13–S20 [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. 2012. Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan PJ, Livingston DM 2010. BRCA1 and BRCA2: Breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 31: 961–967 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau A, Celeux M, Martin-Magniette ML, Maugis-Rabusseau C 2011. Clustering high-throughput sequencing data with Poisson mixture models. Inria Saclay, Ile-de-France. Technical report RR-7786 [DOI] [PubMed] [Google Scholar]
- Rauh-Hain JA, Diver EJ, Clemmer JT, Bradford LS, Clark RM, Growdon WB, Goodman AK, Boruta DM, Schorge JO, Del Carmen MG 2013. Carcinosarcoma of the ovary compared to papillary serous ovarian carcinoma: A SEER analysis. Gynecol Oncol 131: 46–51 [DOI] [PubMed] [Google Scholar]
- Robinson DR, Kalyana-Sundaram S, Wu Y-M, Shankar S, Cao X, Ateeq B, Asangani IA, Iyer M, Maher CA, Grasso CS, et al. 2011. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat Med 17: 1646–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS 2010. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer 10: 59–64 [DOI] [PubMed] [Google Scholar]
- Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK 2012. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28: 1811–1817 [DOI] [PubMed] [Google Scholar]
- Sawada K, Ohyagi-Hara C, Kimura T, Morishige K-I 2012. Integrin inhibitors as a therapeutic agent for ovarian cancer. J Oncol 2012: 915140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T 2003. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolle E, Taucher V, Pichler M, Petru E, Lax S, Haybaeck J 2013. Targeting signaling pathways in epithelial ovarian cancer. Int J Mol Sci 14: 9536–9555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, McBride DJ, Lin M-L, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, et al. 2009. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462: 1005–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton MR 2011. Exploring the genomes of cancer cells: Progress and promise. Science 331: 1553–1558 [DOI] [PubMed] [Google Scholar]
- Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B, et al. 2008. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res 14: 5198–5208 [DOI] [PubMed] [Google Scholar]
- Van Loo P, Nordgard SH, Lingjærde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, et al. 2010. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci 107: 16910–16915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan S, Coward JI, Bast RC, Berchuck A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R, Etemadmoghadam D, et al. 2011. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat Rev Cancer 11: 719–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RGW, Tamayo P, Yang J-Y, Hubbard D, Zhang H, Creighton CJ, Fereday S, Lawrence M, Carter SL, Mermel CH, et al. 2013. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest 123: 517–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermaat JS, Nijman IJ, Koudijs MJ, Gerritse FL, Scherer SJ, Mokry M, Roessingh WM, Lansu N, de Bruijn E, van Hillegersberg R, et al. 2012. Primary colorectal cancers and their subsequent hepatic metastases are genetically different: Implications for selection of patients for targeted treatment. Clin Cancer Res 18: 688–699 [DOI] [PubMed] [Google Scholar]
- Wang L, Feng Z, Wang X, Wang X, Zhang X 2010. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26: 136–138 [DOI] [PubMed] [Google Scholar]
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. 2010. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467: 1114–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LR, Campbell PJ 2012. Evolution of the cancer genome. Nat Rev Genet 13: 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamagni C, Wirtz RM, De Iaco P, Rosati M, Veltrup E, Rosati F, Capizzi E, Cacciari N, Alboni C, Bernardi A, et al. 2009. Oestrogen receptor 1 mRNA is a prognostic factor in ovarian cancer patients treated with neo-adjuvant chemotherapy: Determination by array and kinetic PCR in fresh tissue biopsies. Endocr Relat Cancer 16: 1241–1249 [DOI] [PubMed] [Google Scholar]