

Abstract
Primordial dwarfism (PD) is a disease in which severely impaired fetal growth persists throughout postnatal development and results in stunted adult size. The condition is highly heterogeneous clinically, but the use of certain phenotypic aspects such as head circumference and facial appearance has proven helpful in defining clinical subgroups. In this study, we present the results of clinical and genomic characterization of 16 new patients in whom a broad definition of PD was used (e.g., 3M syndrome was included). We report a novel PD syndrome with distinct facies in two unrelated patients, each with a different homozygous truncating mutation in CRIPT. Our analysis also reveals, in addition to mutations in known PD disease genes, the first instance of biallelic truncating BRCA2 mutation causing PD with normal bone marrow analysis. In addition, we have identified a novel locus for Seckel syndrome based on a consanguineous multiplex family and identified a homozygous truncating mutation in DNA2 as the likely cause. An additional novel PD disease candidate gene XRCC4 was identified by autozygome/exome analysis, and the knockout mouse phenotype is highly compatible with PD. Thus, we add a number of novel genes to the growing list of PD-linked genes, including one which we show to be linked to a novel PD syndrome with a distinct facial appearance. PD is extremely heterogeneous genetically and clinically, and genomic tools are often required to reach a molecular diagnosis.
Growth is a fundamental trait of living organisms that is achieved primarily through a positive balance between cellular proliferation and apoptosis. In humans, disorders of growth are common reasons for referral to pediatric endocrinology because the hormonal axis plays a critical role in controlling growth (Daniel et al. 2008). However, many syndromes are known to cause severely stunted growth despite a normal hormonal axis, the most notable of which are skeletal dysplasias, although these represent a selective defect in bony growth. On the other hand, generalized growth deficiency states that are nonhormonal in etiology and are not related to a negative caloric balance offer a unique opportunity to explore factors that control organismal growth at a more fundamental level and are likely to contribute to the ∼90% of failure-to-thrive children in whom no specific etiology is identified (Jaffe 2011). Severe failure to thrive is commonly associated with a wide array of chromosomal aberrations, which attests to a role played by the genes in growth control, but assigning such a role to individual genes is not usually possible due to the nature of these chromosomal aberrations. On the other hand, single gene mutations that are associated with growth deficiency lend themselves readily to gene mapping strategies that have gained more speed and efficiency recently with the advent of next-generation sequencing.
Primordial dwarfism (PD) refers to severely stunted growth that has its onset prenatally. The condition is highly heterogeneous clinically, but the use of certain phenotypic aspects can help define clinical subgroups. For example, a distinct facial profile and associated microcephaly defines Seckel syndrome (Seckel 1960; Majewski and Goecke 1982). Other PD syndromes can also be readily recognized by their characteristic facial appearance, such as 3M syndrome and POC1A-related PD, both of which lack microcephaly as a diagnostic feature (Al-Dosari et al. 2012; Shaheen et al. 2012). This clinical classification allowed more homogeneous patient populations to be grouped together, which facilitated disease gene identification, especially in the last few years. The identification of these disease genes has unraveled novel pathways that control growth (Klingseisen and Jackson 2011). These pathways include (1) abnormal mitosis (CENPJ- and PCNT-related Seckel syndrome and microcephalic osteodysplastic primordial dwarfism) (Griffith et al. 2007; Al-Dosari et al. 2010), (2) abnormal IGF2 expression (Russel-Silver syndrome) (Netchine et al. 2007), (3) perturbed DNA damage response (ATR-, ATRIP- and RBBP8-related Seckel syndrome) (O'Driscoll et al. 2003; Qvist et al. 2011; Ogi et al. 2012), (4) defective spliceosomal machinery (RNU4ATAC [formerly referred to as U4atac]-related microcephalic osteodysplastic primordial dwarfism) (He et al. 2011), and (5) abnormal replication licensing (in Meier-Gorlin syndrome) (Bicknell et al. 2011; Kuo et al. 2012).
Little is known about the contribution of each of the above genes to the mutation burden in PD, although it is widely believed that its genetic heterogeneity is far from being fully captured (Klingseisen and Jackson 2011). Since almost all known PD disease genes are autosomal recessive in nature and since the highly consanguineous nature of the Saudi population is likely to facilitate both the occurrence of these recessive mutations as well as their identification through autozygome analysis (as we have shown for CENPJ-related Seckel syndrome, POC1A-related PD, and 3M syndrome), we conducted a study on PD patients who were referred to the clinical genetics service in order to examine the contribution of known genes and to identify novel genes. This is the largest comprehensive genomic study on PD that we are aware of and it significantly expands the genetic heterogeneity of this disorder, as we show below.
Results
Clinical report
We have a used a broad definition of PD (height and weight > 3 SD below the mean at birth and persistent growth deficiency postnatally with or without associated microcephaly) due to a lack of agreed-upon criteria that define this phenotype. We specifically excluded those with karyotypic abnormality or frank skeletal dysplasia. For example, patients with 3M syndrome (originally described as an autosomal recessive primordial dwarfism phenotype) (Miller et al. 1975; Clayton et al. 2012) were included since they lack frank skeletal dysplasia. In total, 16 patients (14 families) met the operational definition of PD and had no evidence of karyotypic abnormality or frank skeletal dysplasia (Table 1; Supplemental Table S1). Typical Seckel facies were observed in two, whereas most (n = 14) had associated microcephaly but without Seckel facies. In addition, distinct facies were observed in PD_F4_IV:2 and PD_F5_IV:4 but did not fit any known syndrome (Fig. 1). Eleven of the families were multiplex, although only two families had a viable second affected relative. Autozygome-guided mutation analysis revealed the underlying mutation in three of the cases, whereas exome sequencing revealed a likely causal mutation in an additional five, as described below in detail.
Table 1.
Summary of the mutations identified in the study patients
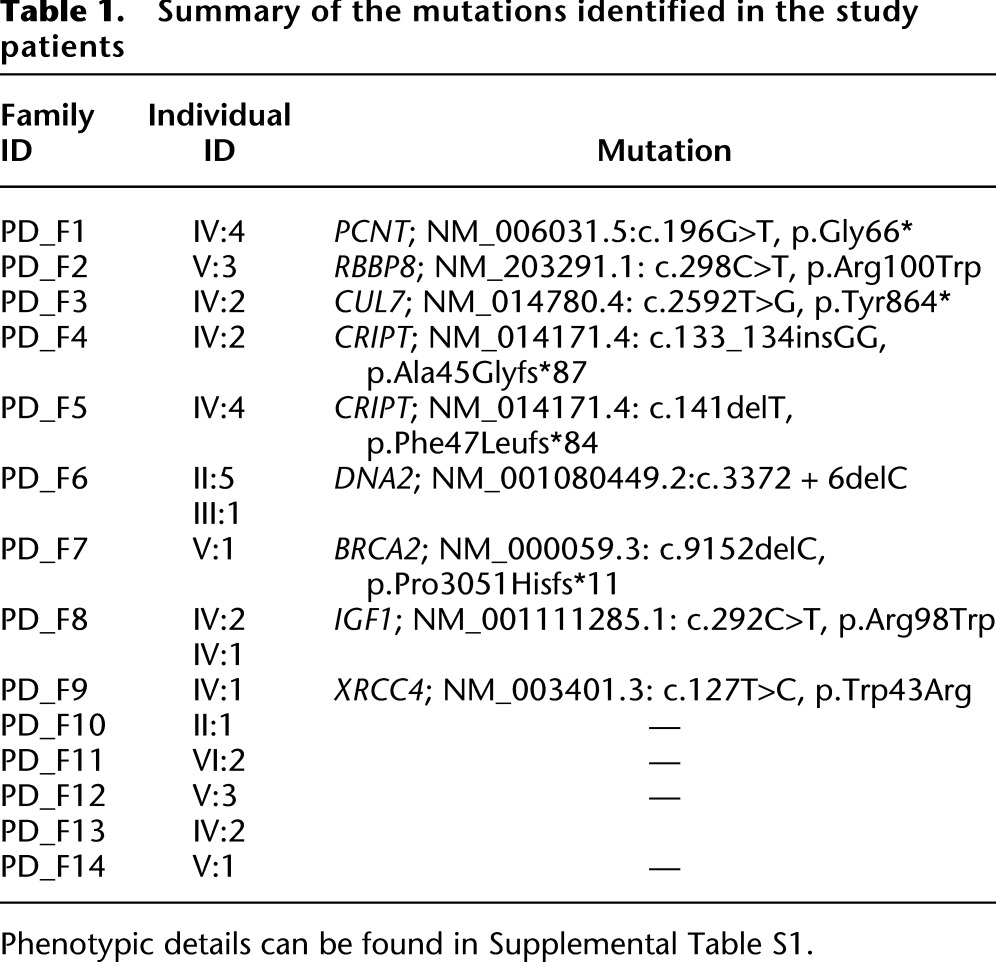
Figure 1.

Identification of a novel PD syndrome linked to CRIPT. (A,B) Pedigrees and clinical photographs of PD_F4_IV:2 and PD_F5_IV:4, respectively. The index is indicated by a red arrow, and asterisks denote individuals whose DNA was available for analysis. Note the strikingly similar clinical appearance (high forehead, mild proptosis, anteverted nares, flat nasal bridge, hypoplastic digits and talipes). Please also note the mottled hypopigmentation in PD_F4_IV:2. (C) Diagram of CRIPT (numbers indicate the number of exons, red color represents the coding exons while blue is the 5′ and 3′ UTR, and triangles indicate the sites of the mutations). (Upper panel) Sequence chromatogram showing the homozygous frameshift insertion in PD_F4_IV:2 and the heterozygous frameshift deletion in the parent of PD_F5_IV:4 (mutations are denoted by red lines).
Identification of novel PCNT, RBBP8, and CUL7 mutations
Only three patients had a positive result when autozygome-guided mutation analysis was used to identify the causative PD disease gene. PD_F1_IV:4 is a male offspring to first cousin parents with a previous daughter who died of cerebral hemorrhage due to a cerebral aneurysm and was diagnosed with Seckel syndrome and neurofibromatosis. The index was born preterm IUGR at 29 wk of gestation. At 12 yr, his weight was 19 kg and his height was 103 cm (Supplemental Table S1). PCNT and RBBP8 resided within the autozygome of this patient, so they were sequenced and a novel mutation (NM_006031.5:c.196G>T; p.Gly66*) was identified in PCNT (Table 1). PD_F2_V:3 is a 9-yr-old female born to second cousin parents with no family history (Supplemental Table S1). RBBP8 resided within the autozygome of this patient, so it was sequenced and a novel mutation (NM_203291.1:c.298C>T: p. Arg100Trp) (Table 1) was identified. It is interesting to note that our patient has brain calcification which was not reported previously in patients with the RBBP8 mutation (Supplemental Tables S1, S2). PD_F3_IV:2 had typical facial features of 3M syndrome, so we proceeded with sequencing the only 3M disease gene that resided within his autozygome and identified a novel mutation in CUL7 (NM_014780.4: c.2592T>G, p.Tyr864*). The similarities and differences between patients with PCNT, RBBP8, CUL7, and previously reported cases are highlighted in Supplemental Table S2.
Exome sequencing reveals novel candidate genes in PD
Identification of a novel PD syndrome caused by mutations in CRIPT
Exome sequencing on PD_F4_IV:2 revealed a variant in CRIPT (NM_014171.4: c.133_134insGG, p.Ala45Glyfs*87) as the only truncating variant that remained after filtering (see Methods; Supplemental Table S3). PD_F4_ IV:2 has a highly characteristic facial appearance that was shared by PD_F5_IV:4 (Fig. 1). Since PD_F5_IV:4 is deceased with no banked DNA, we directly sequenced CRIPT in the parents and found that they both carry a novel truncating mutation (NM_014171.4: c.141delT, p.Phe47Leufs*84) in the heterozygous state. The identification of two truncating homozygous mutations in two unrelated families with the same phenotype confirms that this is a distinct PD syndrome caused by mutations in CRIPT. Unfortunately, failure to propagate lymphoblasts and fibroblasts on the index patient did not permit us to study the effect of CRIPT deficiency on patient cells, although it hints at a proliferation defect.
Identification of a novel Seckel syndrome locus on 10q21.2-10q22.1 likely caused by DNA2 mutation
PD_F6_II:5 and PD_F6_III:1 are second-degree relatives (uncle and niece) with strikingly similar facial features highly consistent with the clinical phenotype of Seckel syndrome (Fig. 2). Autozygome analysis showed a single ROH shared between the two on chr10:63,850,661-71,470,390 (GRCh37/hg19) (Fig. 2B). Full sequencing of all the genes within this interval by exome sequencing (gaps in sequencing were filled by Sanger sequencing to ensure 100% coverage) revealed a novel homozygous mutation in DNA2 (NM_001080449.2;c.3372 + 6delC). This mutation predicts truncation of one transcript (ENST00000440722) and abnormal splicing of another two transcripts (ENST00000399180 and ENST00000358410), all of which are described as protein coding (Fig. 3A). Indeed, RT-PCR confirmed the predicted abnormal splicing (Fig. 3B). Cloning experiments on the RT-PCR products obtained from the patient RNA revealed the presence of five aberrant transcripts, all sharing the same abnormal donor site, while each had a different acceptor site. All five aberrant transcripts predict frameshift and/or truncation (Fig. 3B). Western blot analysis also showed marked reduction (∼50%) in the protein level (Fig. 3C).
Figure 2.

Identification of a novel Seckel syndrome locus linked to Chr10. (A) Pedigree and clinical photographs of the PD_F6 family. The index is indicated by a red arrow, and asterisks denote individuals whose DNA was available for analysis. Please note that the degree of consanguinity between II:1 and II:2 was not determined by parents (blue triangle) but was inferred by the size of the autozygome to be second- or third-degree consanguinity. The clinical photographs of the two affected (III:1 and II:5) show the typical Seckel “bird-like” face. (B) HomozygosityMapper reveals only one ROH on chromosome 10 shared between the two affected (red arrow), and AutoSNPa highlights a corresponding ROH of 7.6 Mb (boxed in red) exclusively shared between PD_F6_II:5 and PD_F6_III:1.
Figure 3.
Identification of a novel DNA2 mutation that causes increased senescence and DNA damage in a family with Seckel syndrome. (A) Upper panel shows DNA sequence chromatogram with the novel 1-bp deletion variant identified in the two affected members of PD_F6, and a schematic of the three transcripts of DNA2 affected by the mutation. The site of mutation on the sequence chromatogram is denoted by a red bar, and blue triangles denote the position of the variant on the three DNA2 transcripts. (B) Upper panel shows the gel image of the RT-PCR that reveals the presence of an aberrant band in the two patients as well as the reduction of the normal band in Pt1 and Pt2 (23% and 53%, respectively). Note the absence of the aberrant band in the two controls. Lower panel shows a cartoon of DNA2 mRNA (only exons 19–21 are shown) and the result of cloning experiments on the RT-PCR product obtained from the patient RNA which revealed the presence of five aberrant transcripts, all sharing the same abnormal donor site, while each had a different acceptor site. (C) Immunoblotting using antibody against DNA2. The protein level of DNA2 from cells derived from index individual (Pt) were 50% reduced in comparison to the normal type control (Ct), respectively (the average of two independent experiments was 60%). (D) Images of beta-gal-stained control cells (A) and patient (B) showing the presence of blue staining and flattening of patient cells (red triangle). Please note that patient and control fibroblasts had a comparable number of passages (6–7). (E) Representative images and the quantification graph in the assessment of “comet tail” lengths from patient cells transfected with shuttle (A) and DNA2 (B). There was a significant reduction in the number of cells with comet tails as well as the size of comet tails (P = 4.4 × 10−6) obtained from the patient cells after exogenous expression of DNA2. Scale bar = 50 μm.
We noticed that patient fibroblast cells were remarkably larger in size compared to control fibroblasts, and assume a more flat morphology, two key features of senescence. Therefore, we performed a senescence assay to test if the senescence pathway is activated in our patients as a consequence of DNA2 deficiency. Indeed, patient cells showed a blue beta-galactosidase staining in addition to the previously described abnormal morphology (“fried egg” appearance) (Fig. 3D).
Because DNA2 is known to play a role in DNA damage repair (Zheng et al. 2008), we set out to test patient cells' sensitivity to DNA damage. At baseline, comet tails of remarkable length were observed in patient cells compared to control cells. These results are highly suggestive that Seckel disease in this family is caused by marked DNA damage sensitivity due to DNA2 mutation. In order to establish a causal link, we attempted to rescue this phenotype by transfecting patient cells with a wild-type DNA2 vector. A significant and remarkable reduction in the tail length was observed in the patient cell transfected with DNA2 compared to the empty vector control (Fig. 3E).
Identification of XRCC4 and as a novel PD candidate disease gene
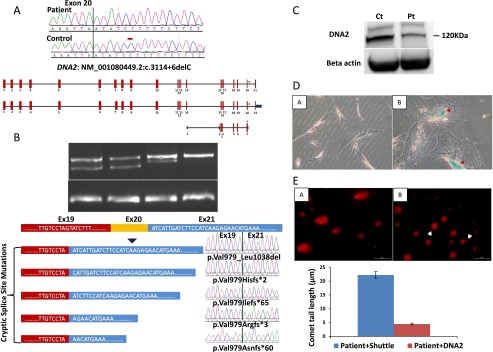
Exome sequencing in PD_F9_IV:1 with PD revealed a missense variant in XRCC4 (NM_003401.3:c.127T>C; p.Trp43Arg) as the most likely candidate from among the three variants that remained after filtering (Fig. 4B; Supplemental Table S3). The amino acid at position 43 is conserved along species down to zebrafish (Danio rerio) (Fig. 4C). The established role of XRCC4 in DNA damage repair and the embryonic lethal phenotype in the knockout mouse also support the candidacy of this variant (Gao et al. 1998; Ma et al. 2004). Indeed, we were able to show that XRCC4 knockdown in control cells resulted in a significant impairment in DNA damage following ionizing radiation (Fig. 4D) but not UV exposure, consistent with previously published work (Delacôte et al. 2002). On the other hand, the candidacy of the two other variants that remained after filtering (in VEGFC and AKAP11; see Supplemental Table S3) was not supported by available mouse data.
Figure 4.
(A–C) Pedigree and stacked Venn diagrams illustrating the exome filtration scheme, sequence chromatogram, and the multisequence alignment of orthologs of the mutation identified in XRCC4. (D) Representative images and the quantification graph in the assessment of “comet tail” lengths from fibroblast cells treated with either XRCC4 or scrambled siRNA 4 h post-IR (5 Gy), showing that XRCC4 deficiency results in impaired DNA damage repair (P = 1.18 × 10−10).
Identification of BRCA2 as a novel PD disease gene
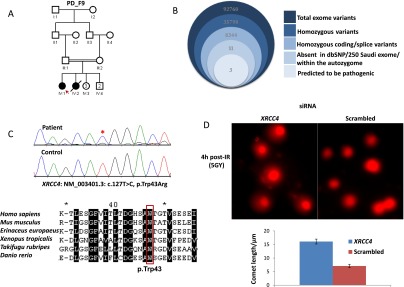
PD_F7_V:1 first presented at the age of 18 mo with severe failure to thrive and microcephaly (weight, length, and hc > 5 SD below the mean). She had markedly increased chromosomal breakage in response to DEB, but a bone marrow biopsy at the age of 3 yr was normal. She developed unilateral retinoblastoma, followed shortly by aggressive brain glioma, and died at the age of 3.5 yr. Exome sequencing revealed a homozygous truncating BRCA2 mutation (NM_000059.3:c.9152delC) (Fig. 5). Although short stature and microcephaly have been reported in association with biallelic BRCA2 mutations (Alter et al. 2007), this is the first instance in which frank PD is linked to BRCA2 (Supplemental Table S2).
Figure 5.
Pedigree, clinical photographs, and stacked Venn diagrams illustrating the exome filtration scheme and sequence chromatogram of the mutation identified in BRCA2 PD_F7. The index is indicated as a red arrow, and asterisks denote individuals whose DNA was available for analysis.
Identification of a novel IGF1 mutation in PD
Although patients with biallelic IGF1 mutations and PD have been reported (Woods et al. 1996; Bonapace et al. 2003), the extreme rarity of mutations in this gene precluded us from recognizing IGF1 as a candidate PD gene in the step of autozygome-guided mutation analysis, and it was only after exome sequencing that a novel mutation (NM_001111285.1.:c.292C>T: p.Arg98Trp) was identified. This is only the fifth biallelic mutation in IGF1 reported to date.
Exome sequencing on five additional PD patients (in whom autozygome-guided mutation analysis did not reveal any causal mutation) failed to identify any obvious candidate. A summary of the variants that remained after filtering in these five patients is shown in Supplemental Table S3, and the coordinates of the autozygome for each patient is given in Supplemental Table S4.
Discussion
The increasing number of molecular mechanisms that converge on PD as a final common phenotype underscores the complex network that controls growth as a fundamental aspect of life, particularly in complex organisms (Klingseisen and Jackson 2011). While some mechanisms can be readily linked to the phenotype, e.g., impaired mitosis in PCNT- and CENPJ-related PD, others are more difficult to interpret in the context of PD, such as impaired spliceosomal machinery. Clearly, the pace at which novel PD disease genes are identified and, consequently, novel molecular perturbations are revealed, is surpassing our capacity to establish mechanistic links. This study, the largest genomic study to date on PD patients, provides additional novel insights into the pathogenesis of PD by similarly highlighting novel disease genes, some of which cannot be neatly grouped under any of the molecular pathways known to be perturbed in PD. Another challenge in PD research is the case definition which is largely based on expert opinion rather than empirical data. By applying a relatively broad definition of PD uniformly across a relatively large cohort, we were able to provide much needed data on the clinical utility of this definition in the context of molecular characterization of patients with this condition. This definition clearly enabled us to capture patients with mutations in known PD genes. It also allowed us to identify patients with mutations in compelling candidate genes that can reasonably be placed within a growing “PD gene network.” One may argue that the one-third of families that we failed to solve in this study could be decreased if a more stringent definition were to be used. We propose that this is best addressed by comparing our results to future series that similarly apply a particular definition of PD uniformly across a large cohort.
CRIPT is a microtubule-binding protein that is mainly studied in the context of its bridging function between the cytoskeleton and other proteins, particularly synaptic proteins such as PSD-95 (Passafaro et al. 1999). However, the finding that Cript−/− mice die at embryonic day 8 strongly suggests that it plays a more general developmental role and is consistent with the phenotypic consequence of knocking out other known PD disease genes, e.g., Cenpj and Pcnt (Wu and Tang 2012). Both patients with CRIPT homozygous truncation have a highly consistent and clinically recognizable phenotype of PD that is characterized by frontal bossing, high forehead, sparse hair and eyebrows, telecanthus, mild proptosis (staring look), upturned nostrils, and hypoplastic terminal phalanges with brachydactyly. Although the genetic evidence is compelling (two independent homozygous truncating mutations in two patients with the same phenotype), it remains unclear, mechanistically, how deficiency of this microtubule-binding protein can have such a profound effect on body size. On the other hand, DNA2 is a compelling candidate PD disease gene both biologically and genetically. We show that a single autozygous interval is shared between the affected members of a multiplex consanguineous family, which in itself is strong evidence for a locus for Seckel syndrome that has not been described before. Full sequencing of all coding regions within this interval revealed a truncating mutation in DNA2 as the only novel variant therein. DNA2 is a highly conserved 1146-aa protein that possesses nuclease and ATPase domains in its N-terminal half and a helicase domain in its C-terminal half. Its yeast ortholog unwinds double-stranded DNA to generate single-stranded DNA in the process of providing a template for base-excision repair but also during DNA replication, and it has been shown to play a role in replication checkpoint initiation (Ayyagari et al. 2003; Zhu et al. 2008; Mimitou and Symington 2009; Kumar and Burgers 2013). In fact, DNA2 deletion mutants in yeast are nonviable (Budd et al. 2011). It is interesting to note that both parental pairs in this extended family had numerous miscarriages, suggesting that severe reduction in DNA2 is probably incompatible with life and that the survival of the two patients was the consequence of favorable transcriptional aberration that allowed a sufficient amount of DNA2 to be made beyond a particular threshold. Consistent with this essential role, our data clearly show drastically increased DNA damage in patient cells, that is rescued by overexpressing wild-type DNA2, and enhanced senescence. These molecular pathways underlie the pathogenesis of a number of PD syndromes as described above. Very recently, a group described three heterozygous missense variants in the setting of a mitochondrial myopathy and it was proposed that these variants impair DNA2's capacity to repair damage in mitochondrial DNA (Ronchi et al. 2013). Several explanations can be advanced to reconcile this apparent discrepancy. First, different phenotypes resulting from dominant and recessive mutations in the same gene are known to exist. Second, we propose that the effect of our mutation is a direct deficiency of DNA2, whereas the three alleles described by Ronchi et al. (2013) may exert an allele-specific effect on protein function. Lastly, it is possible that the variants described by Ronchi et al. (2013), or the one described by us, are not causal to the observed phenotypes. However, the latter is unlikely because our mutation was not identified in 250 local Saudi exomes, 131 Saudi controls by direct sequencing (including 31 tribe-specific controls), or in EVS. Clearly, reports of additional mutations in DNA2 will clarify the involvement of DNA2 in PD pathogenesis.
The evidence supporting candidacy of XRCC4 as a PD disease gene is strongly supported by the literature. XRCC4 plays a critical role in nonhomologous end joining (NHEJ) the primary mechanism for repairing DNA double-strand breaks (DSBs). Specifically, XRCC4 complexes with NHEJ1 to act as a bridging and a ligase complex to initiate the repair of double-stranded DNA breaks (Ahnesorg et al. 2006). Indeed, we show that knockdown of XRCC4 in control fibroblasts results in significant impairment of DNA repair following IR-induced damage. In accordance with the recognized role of this critical pathway to growth, knockout mice for Xrcc4 die in late gestation, with marked reduction in body size (Gao et al. 1998). Mouse embryonic fibroblasts (MEFs) derived from these mice exhibit gross growth defects and premature senescence (Gao et al. 1998). Finally, XRCC4 is known to complex with DNA ligase 4 (LIG4) to be recruited together with double-strand DNA breaks to initiate repair, and mutations in LIG4 are known to cause microcephaly and severe growth deficiency in humans (LIG4 syndrome) (Girard et al. 2004). In fact, mutations that remove the XRCC4 binding domain in LIG4 are known to be severe null mutations (Buck et al. 2006). Thus, it appears likely that the variant we observed in XRCC4 is involved in the pathogenesis of PD in our patient.
BRCA2 is a well-known tumor suppressor, and heterozygous mutations are an established, albeit rare, risk factor of breast and ovarian cancer (Roy et al. 2012). Homozygous BRCA2 mutations, on the other hand, represent a distinct complementation group of Fanconi anemia, a bone marrow failure syndrome that is distinguished by a characteristic set of associated congenital malformations (Howlett et al. 2002). Although short stature is a known manifestation of Fanconi anemia, it is usually in the range of 2.35 SD below the mean, according to a very large study on the anthropometric evaluation of 54 patients with Fanconi anemia (Wajnrajch et al. 2001). The patient we describe here has frank PD with growth parameters, including head circumference, more than 5 SD below the mean. The finding of a normal bone marrow is unusual because even though hematological evidence of bone marrow failure has an age-dependent penetrance, bone marrow pathology is usually evident early on. This patient's severe PD is in fact consistent with the murine phenotype of biallelic loss of function of Brca2 (Ludwig et al. 1997). The paradox between uncontrolled proliferation with monoallelic loss of function and markedly reduced proliferation with biallelic loss of function is poorly understood, but we note that we have recently described a similar phenomenon for another tumor suppressor gene known as LARP7 (Alazami et al. 2012).
The remaining PD patients harbored a number of variants that we could not prioritize based on available evidence, so it is possible that they may or may not be relevant. For example, an Xirp2 knockout mouse is much smaller that its wild-type and heterozygous littermates (Wang et al. 2010). Clearly, our results demonstrate the need for additional studies on larger patient cohorts in order to unravel more disease genes in this markedly genetically heterogeneous disorder.
In summary, our data show that previously identified PD disease genes appear to account for only a subset of PD patients and that many more novel disease genes await discovery. We provide evidence to support the candidacy of a number of such novel disease genes, and future studies are likely to uncover more. PD patients require careful clinical phenotyping, and genomic analysis is often required to reach a specific molecular diagnosis, which is not only helpful in elucidating the molecular pathogenesis further but also empowers the affected families to pursue various reproductive options not previously available to them.
Methods
Human subjects
PD was defined as height and weight more than 3 SD below the mean for age at the time of birth that persist or worsen post-natally, with or without microcephaly. Patients with known chromosomal aberrations were excluded as well as those with frank skeletal dysplasia. All patients underwent a thorough clinical genetics evaluation and basic workup that consisted of hormonal studies, metabolic profile, and skeletal survey. Additional tests were ordered as necessitated by the clinical presentation. For example, chromosomal breakage studies were ordered if there was significant microcephaly. Patients were enrolled in an IRB-approved protocol with written informed consent. Additional consent to publish facial photos was also obtained in selected cases. Venous blood was collected in EDTA and, whenever possible, in sodium heparin tubes for DNA extraction and establishment of EBV-transformed lymphoblastoid cell lines, respectively. Skin biopsies were obtained in selected cases with proper consent.
Autozygome-guided mutation analysis
Determination of the entire set of autozygous intervals per individual genome (autozygome) was made possible by genome-wide genotyping on the Axiom SNP Chip (Affymetrix) platform, followed by calculating runs of homozygosity (ROH) that are >2 Mb in size and span >107 SNPs using autoSNPa as described before (Carr et al. 2006; Alkuraya 2012). The entire autozygome (or shared autozygome in the case of multiple available relatives) was examined for known PD disease genes, which were then Sanger- sequenced. If no mutations were identified or if the autozygome did not overlap with known PD disease genes, exome sequencing was pursued.
Exome sequencing and variant analysis
Exome capture was performed using the TruSeq Enrichment kit (Illumina) following the manufacturer's protocol. Samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using an Illumina HiSeq 2000 sequencer. The reads were mapped against UCSC hg19 (Kent et al. 2002) (http://genome.ucsc.edu/) by BWA (Li and Durbin 2009) (http://bio-bwa.sourceforge.net/). The SNPs and indels were detected by SAMtools (Li et al. 2009) (http://samtools.sourceforge.net/). Resulting variants were filtered using the autozygome coordinates and other filters as described before (Alkuraya 2012). Briefly, we only considered coding/splicing homozygous variants that are absent in dbSNP132 and 250 in-house Saudi exomes, and that reside within the autozygome of each patient in simplex cases or the shared autozygome of affected members of a given family in multiplex cases.
Immunoblot analysis
Total protein was extracted from patient and control fibroblasts. Anti-DNA2 antibody was purchased from Abcam (ab96488). The membrane was blocked with 5% milk powder in PBST for 1 h at room temperature (RT) and incubated with the primary antibody (dilution 1:1000) overnight at 4°C, followed by stringency washes and treatment with secondary antibody for signal detection. The intensity of the specific protein bands was analyzed with the National Institutes of Health (NIH) ImageJ software.
Comet assay and expression of DNA2
Patient fibroblast cells were seeded on a six-well dish and transfected with Myc-DDK-tagged ORF clone of Homo sapiens DNA2 (Origene, RC217395). Myc-tagged empty plasmid was used as a control. Thirty six hours later, the comet assay was performed to detect the DNA damage as follows. Roughened microscope slides were dipped briefly into 1.5% hot normal-melting agarose (NMA). The slides were dried and coated with 300 mL of 1.0% NMA in PBS, and the agarose was allowed to solidify. Subsequently, fibroblasts were mixed with 95 mL of 0.75% low-melting-point agarose. The mixture was spread on the slide using a cover slip and then allowed to solidify. After removal of the cover slip, the slides were immersed in freshly prepared cold lysing solution for at least 2 h. The alkaline unwinding, electrophoresis, and neutralization steps were performed by removing the slides from the lysis solution and placing them in the electrophoresis chamber, which was then filled with freshly made alkaline buffer. The cells were exposed to alkali for 30 min to allow for DNA unwinding and the expression of alkali-labile sites. Subsequently, the DNA was electrophoresed for 30 min at 300 mA and 25 V. After electrophoresis, the slides were placed in a horizontal position and washed to neutralize the excess alkali, followed by the addition of ethidium bromide and analysis using a fluorescence microscope. Tail lengths were measured and cells were scored.
Senescence assay
The senescence assay was performed using a senescence beta-galactosidase staining kit (#9860; Cell Signaling) following the manufacturer's protocol.
siRNA gene silencing and IR
Control fibroblast cells isolated from two normal individuals were transfected with two different siRNAs (s14951 and s14949, Ambion) against XRCC4 using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Scrambled control siRNA was used in parallel. Briefly, 0.1 nmole of siRNA and Lipofectamine were diluted in Opti-MEM media, and the mixture was added to the cells and incubated for 48 h. Cells were then irradiated with 5 Gy ionizing radiation (IR) by XRAD 320 (Precision X-Ray Inc.), followed by incubation for 4 h at 37°C (5% CO2), and then a comet assay was performed as described above. The efficiency of XRCC4 knockdown was evaluated by real time PCR.
Data access
Exome variants and array data sets included in the paper are available through the link http://sgp.kfshrc.edu.sa/bioinf/db/variants/dg/downloads. Novel variants were submitted to NCBI's ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/; accession numbers: SCV000108550, SCV000108551, SCV000108552, SCV000108553, SCV000108405, SCV000108406, SCV000108407, SCV000108408, SCV000108409) and LOVD (http://www.lovd.nl/3.0/home; variant IDs: 0000022851, 0000022850, 0000022849, 0000022848, 0000022846, 0000022844, 0000022845, 0000022852).
Acknowledgments
We thank the patients and their families for their enthusiastic participation. We are grateful to Niema Ibrahim, Mais Hashem, and Firdous Abdulwahab for their help as clinical research coordinators. We also thank the staff at the Genotyping and Sequencing Core Facilities at KFSHRC for their technical help. This work was supported by a DHFRM Collaborative Research Grant (FSA).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.160572.113.
References
- Ahnesorg P, Smith P, Jackson SP 2006. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124: 301. [DOI] [PubMed] [Google Scholar]
- Al-Dosari MS, Shaheen R, Colak D, Alkuraya FS 2010. Novel CENPJ mutation causes Seckel syndrome. J Med Genet 47: 411–414 [DOI] [PubMed] [Google Scholar]
- Al-Dosari MS, Al-Shammari M, Shaheen R, Faqeih E, Alghofely MA, Boukai A, Alkuraya FS 2012. 3M syndrome: An easily recognizable yet underdiagnosed cause of proportionate short stature. J Pediatr 161: 139–145.e1 [DOI] [PubMed] [Google Scholar]
- Alazami AM, Al-Owain M, Alzahrani F, Shuaib T, Al-Shamrani H, Al-Falki YH, Al-Qahtani SM, Alsheddi T, Colak D, Alkuraya FS 2012. Loss of function mutation in LARP7, chaperone of 7SK ncRNA, causes a syndrome of facial dysmorphism, intellectual disability, and primordial dwarfism. Hum Mutat 33: 1429–1434 [DOI] [PubMed] [Google Scholar]
- Alkuraya FS. 2012. Discovery of rare homozygous mutations from studies of consanguineous pedigrees, Chapter 6: Unit 6.12. In Current protocols in human genetics (ed. Haines JL, et al). Wiley, Hoboken, NJ. [Google Scholar]
- Alter BP, Rosenberg PS, Brody LC 2007. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet 44: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyagari R, Gomes XV, Gordenin DA, Burgers PM 2003. Okazaki fragment maturation in yeast I. Distribution of functions between FEN1 and DNA2. J Biol Chem 278: 1618–1625 [DOI] [PubMed] [Google Scholar]
- Bicknell LS, Bongers EM, Leitch A, Brown S, Schoots J, Harley ME, Aftimos S, Al-Aama JY, Bober M, Brown PA 2011. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet 43: 356–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonapace G, Concolino D, Formicola S, Strisciuglio P 2003. A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J Med Genet 40: 913–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck D, Moshous D, de Chasseval R, Ma Y, le Deist F, Cavazzana-Calvo M, Fischer A, Casanova JL, Lieber MR, de Villartay JP 2006. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur J Immunol 36: 224–235 [DOI] [PubMed] [Google Scholar]
- Budd ME, Antoshechkin IA, Reis C, Wold BJ, Campbell JL 2011. Inviability of a DNA2 deletion mutant is due to the DNA damage checkpoint. Cell Cycle 10: 1690–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr IM, Flintoff KJ, Taylor GR, Markham AF, Bonthron DT 2006. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum Mutat 27: 1041–1046 [DOI] [PubMed] [Google Scholar]
- Clayton PE, Hanson D, Magee L, Murray PG, Saunders E, Abu-Amero SN, Moore GE, Black GCM 2012. Exploring the spectrum of 3-M syndrome, a primordial short stature disorder of disrupted ubiquitination. Clin Endocrinol 77: 335–342 [DOI] [PubMed] [Google Scholar]
- Daniel M, Kleis L, Cemeroglu AP 2008. Etiology of failure to thrive in infants and toddlers referred to a pediatric endocrinology outpatient clinic. Clin Pediatr (Phila) 47: 762–765 [DOI] [PubMed] [Google Scholar]
- Delacôte F, Han M, Stamato TD, Jasin M, Lopez BS 2002. An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res 30: 3454–3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J 1998. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95: 891–902 [DOI] [PubMed] [Google Scholar]
- Girard PM, Kysela B, Harer CJ, Doherty AJ, Jeggo PA 2004. Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: The impact of two linked polymorphisms. Hum Mol Genet 13: 2369–2376 [DOI] [PubMed] [Google Scholar]
- Griffith E, Walker S, Martin C-A, Vagnarelli P, Stiff T, Vernay B, Al Sanna N, Saggar A, Hamel B, Earnshaw WC 2007. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat Genet 40: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Liyanarachchi S, Akagi K, Nagy R, Li J, Dietrich RC, Li W, Sebastian N, Wen B, Xin B 2011. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science 332: 238–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, et al. 2002. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297: 606–609 [DOI] [PubMed] [Google Scholar]
- Jaffe AC 2011. Failure to thrive: Current clinical concepts. Pediatr Rev 32: 100–108 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingseisen A, Jackson AP 2011. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev 25: 2011–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Burgers PM 2013. Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev 27: 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AJ, Song J, Cheung P, Ishibe-Murakami S, Yamazoe S, Chen JK, Patel DJ, Gozani O 2012. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 484: 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup 2009. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A 1997. Targeted mutations of breast cancer susceptibility gene homologs in mice: Lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev 11: 1226–1241 [DOI] [PubMed] [Google Scholar]
- Ma Y, Lu H, Tippin B, Goodman MF, Shimazaki N, Koiwai O, Hsieh CL, Schwarz K, Lieber MR 2004. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol Cell 16: 701–713 [DOI] [PubMed] [Google Scholar]
- Majewski F, Goecke T 1982. Studies of microcephalic primordial dwarfism I: Approach to a delineation of the Seckel syndrome. Am J Med Genet 12: 7–21 [DOI] [PubMed] [Google Scholar]
- Miller JD, McKusick VA, Malvaux P, Temtamy S, Salinas C 1975. The 3-M syndrome: A heritable low birthweight dwarfism. Birth Defects Orig Artic Ser 11: 39–47 [PubMed] [Google Scholar]
- Mimitou EP, Symington LS 2009. DNA end resection: Many nucleases make light work. DNA Repair (Amst) 8: 983–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netchine I, Rossignol S, Dufourg M-N, Azzi S, Rousseau A, Perin L, Houang M, Steunou V, Esteva B, Thibaud N 2007. 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell-Silver syndrome: Clinical scoring system and epigenetic-phenotypic correlations. J Clin Endocrinol Metab 92: 3148–3154 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA 2003. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Ogi T, Walker S, Stiff T, Hobson E, Limsirichaikul S, Carpenter G, Prescott K, Suri M, Byrd PJ, Matsuse M 2012. Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel Syndrome. PLoS Genet 8: e1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passafaro M, Sala C, Niethammer M, Sheng M 1999. Microtubule binding by CRIPT and its potential role in the synaptic clustering of PSD-95. Nat Neurosci 2: 1063–1069 [DOI] [PubMed] [Google Scholar]
- Qvist P, Huertas P, Jimeno S, Nyegaard M, Hassan MJ, Jackson SP, Børglum AD 2011. CtIP mutations cause Seckel and Jawad syndromes. PLoS Genet 7: e1002310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi D, Di Fonzo A, Lin W, Bordoni A, Liu C, Fassone E, Pagliarani S, Rizzuti M, Zheng L, Filosto M 2013. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am J Hum Genet 92: 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Chun J, Powell SN 2012. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat Rev Cancer 12: 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seckel HPG. 1960. Bird-headed dwarfs: Studies in developmental anthropology including human proportions. CC Thomas, Springfield, IL. [Google Scholar]
- Shaheen R, Faqeih E, Shamseldin HE, Noche RR, Sunker A, Alshammari MJ, Al-Sheddi T, Adly N, Al-Dosari MS, Megason SG, et al. 2012. POC1A truncation mutation causes a ciliopathy in humans characterized by primordial dwarfism. Am J Hum Genet 91: 330–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajnrajch MP, Gertner JM, Huma Z, Popovic J, Lin K, Verlander PC, Batish SD, Giampietro PF, Davis JG, New MI 2001. Evaluation of growth and hormonal status in patients referred to the International Fanconi Anemia Registry. Pediatrics 107: 744–754 [DOI] [PubMed] [Google Scholar]
- Wang Q, Lin JL-C, Reinking BE, Feng H-Z, Chan F-C, Lin C-I, Jin J-P, Gustafson-Wagner EA, Scholz TD, Yang B 2010. Essential roles of an intercalated disc protein, mXinß, in postnatal heart growth and survival. Circ Res 106: 1468–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods KA, Camacho-Hubner C, Savage MO, Clark AJ 1996. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med 335: 1363–1367 [DOI] [PubMed] [Google Scholar]
- Wu KS, Tang TK 2012. CPAP is required for cilia formation in neuronal cells. Biol Open 1: 559–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Zhou M, Guo Z, Lu H, Qian L, Dai H, Qiu J, Yakubovskaya E, Bogenhagen DF, Demple B, et al. 2008. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol Cell 32: 325–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung W-H, Shim EY, Lee SE, Ira G 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double strand break ends. Cell 134: 981. [DOI] [PMC free article] [PubMed] [Google Scholar]