

Abstract
There is increasing interest in clinical genetics pertaining to the utilization of high-throughput sequencing data for accurate diagnoses of monogenic diseases. Moreover, massive whole-exome sequencing of tumors has provided significant advances in the understanding of cancer development through the recognition of somatic driver variants. To improve the identification of the variants from HTS, we developed VariantMaster, an original program that accurately and efficiently extracts causative variants in familial and sporadic genetic diseases. The algorithm takes into account predicted variants (SNPs and indels) in affected individuals or tumor samples and utilizes the row (BAM) data to robustly estimate the conditional probability of segregation in a family, as well as the probability of it being de novo or somatic. In familial cases, various modes of inheritance are considered: X-linked, autosomal dominant, and recessive (homozygosity or compound heterozygosity). Moreover, VariantMaster integrates phenotypes and genotypes, and employs Annovar to produce additional information such as allelic frequencies in the general population and damaging scores to further reduce the number of putative variants. As a proof of concept, we successfully applied VariantMaster to identify (1) de novo mutations in a previously described data set, (2) causative variants in a rare Mendelian genetic disease, and (3) known and new “driver” mutations in previously reported cancer data sets. Our results demonstrate that VariantMaster is considerably more accurate in terms of precision and sensitivity compared with previously published algorithms.
The recent advances of high-throughput sequencing (HTS) technologies have provided new diagnostic opportunities in the field of clinical genetics (Makrythanasis and Antonarakis 2012). An unprecedented amount of sequence data has been collected on multiple genetic disorders, including cancers, aiming to identify the causative variants. However, extensive natural germline variation of each individual makes the detection of disease-causing variants a complicated task (Cooper and Shendure 2011).
For genetic transmissible or de novo diseases, significant help comes from sequencing the genomes (or the exomes) of the parents and close relatives of affected individuals. Indeed, the integration of familial information substantially improves the identification of causative variants. In cancer genetics, analogous strategies have already been applied to extract somatic driver mutations in tumors through the comparison of tumor and normal samples from the same individual (Nikolaev et al. 2012).
So far, few computational approaches to exploit this additional information have been proposed and most of them are tailored to specific tasks. For instance, FamSeq (Peng et al. 2013) and PolyMutt (Li et al. 2012a) use relatedness to call variants segregated in familial pedigrees, but they are not designed for case-control studies and do not integrate phenotypes in the analysis or provide any filtering option to prioritize the variants. On the other hand, algorithms like VarScan (Koboldt et al. 2012) have been specifically conceived to extract somatic mutations from tumor samples. To our knowledge, the only tool that filters and processes familial and matched tumor-germline data sets is KGGSeq (Li et al. 2012b). However, KGGSeq, as well as the previously mentioned algorithms, relies solely on processed data (i.e., VCF files produced by algorithms such as SAMtools [Li et al. 2009] or GATK [McKenna et al. 2010]). In fact, the limitations of the current sequencing technologies and variant calling algorithms may result in a non-negligible amount of erroneous (false-positive and false-negative) variant calls (Knocking on the clinic door. [Editorial] 2012). Here we propose VariantMaster, a novel utility that implements an original methodology to evaluate the status (presence or absence) of a variant in familial or case-control contexts. This feature, along with robust control of modes of inheritance and a highly flexible and customizable system of variant filtration, makes it a powerful and accurate tool to identify causative variants in familial, sporadic germline, and somatic genetic disorders, including cancers. VariantMaster also allows for the search of causative variants in one or more recurrently mutated genes in a pool of unrelated individuals sharing the same phenotype (Supplemental Fig. 1).
Results
Current procedures to identify causative genomic or somatic variants are composed of two independent steps: (1) variant calling (single or multisample and/or pedigree aware) and (2) prioritization of candidate variants.
VariantMaster implements a fundamentally different approach. It takes as input the variants called in each affected individual with standard tools (e.g., SAMtools, GATK) and, given this information, statistically estimates the conditional probability that these variants are present or absent in the other family members (conditional variant calling). The central goal is to isolate variants or combination of variants that are specific to affected individuals (or somatic, i.e., specific to tumors).
For this, VariantMaster uses raw sequence data information available in BAM files (binary sequence alignment/map format) in addition to the variants reported in the VCF files. BAM and VCF files can be generated using standard tools such as BWA, SAMtools, or GATK with default parameters. More specifically, for each variant in each affected individual, the algorithm estimates the strand bias and the probability that each family member is a carrier accounting for the respective fraction of supporting reads and the corresponding base call error rate (for a detailed description, see Methods).
This procedure requires non-negligible computational time per variant. However, depending on the type of the analysis, several variants can be excluded a priori based on annotation data. Therefore, the algorithm initially annotates, through Annovar, all the given variants and allows the user to define the prioritization filters appropriate to the specific analysis. In general, this approach considerably reduces the burden of variants to process, providing a twofold advantage: reduced processing time and clearer readouts for the user. For example, the identification of a candidate coding variant of a rare, recessive Mendelian disease in a family trio by filtering for a minimum allelic frequency (MAF) < 2%, requires a computational time of ∼10 min compared with >1 h using an unfiltered variant list. VariantMaster allows the user to customize the processing of all available genetic annotations. An example of an accurate prioritization of causative variants for Mendelian disorders may require the following filters: (1) quality score (some conservative thresholds might be set to QS > 50 for SAMtools calls, QS > 300 for GATK calls, and QS > 600 for PINDEL calls); (2) allelic frequency (MAF) <2%, if available, in databases such as 1000 Genomes (www.1000genomes.org), Exome Variant Server (evs.gs.washington.edu/EVS), and dbSNP (www.ncbi.nlm.nih.gov/SNP); (3) removal of synonymous nonsplicing variants; and (4) removal of variants in regions of segmental duplication. Additional filtering could include the selection of pathogenic variants according to scores provided by SIFT (Kumar et al. 2009), PolyPhen-2 (Adzhubei et al. 2010), MutationTaster (Schwarz et al. 2010), LRT (Chun and Fay 2009), and others. Some examples of filter configurations are provided in the Supplemental Material.
VariantMaster has been conceived to be flexible with respect to the availability of input data. In some cases, whole-genome or whole-exome sequences of some individuals are not available due to budget limitations or because it is impossible to collect DNA. For this reason, VariantMaster has the additional option to analyze families where data for one or more individuals (i.e., parents) are missing. This specific feature was successfully applied to the identification of a recessive causative variant to an incomplete pedigree where two affected siblings and one unaffected sister were exome sequenced (Moore et al. 2013). More importantly, VariantMaster can deal with exomes sequenced at different coverages, allowing cost-effective experimental designs. Naturally, a trade-off with the increase of false positives should be taken into account (see application examples 2 and 3).
In order to comprehensively evaluate the effectiveness of VariantMaster, we designed different test cases. We performed conditional variant calling and de novo discovery on the HapMap CEU trio, as well as causative recessive variant identification on a familial case using GATK, VariantMaster, KGGSeq, FamSeq, and PolyMutt. Finally, we challenged VariantMaster with VarScan on molecular cancer profiling.
Proof of concept: Conditional variant calling
VariantMaster implements a statistical procedure to determine the presence or absence of a specific variant in a family member conditional to its presence in one (or more) affected relatives (for a detailed description, see Methods). In order to prove the reliability of this engine, we downloaded the HapMap WGS data sets (BAM) of the CEU trio (NA12891, father; NA12892, mother; NA12878, child) from the 1000 Genomes Project and the corresponding HapMap genotyped SNPs from ftp://ftp.ncbi.nlm.nih.gov/hapmap/ as reference. We performed the multisample variant calling using the standard GATK Unified Genotyper with default parameters. Resulting variants presented with an average coverage of 90×. We then used PolyMutt and FamSeq independently to refine the calls based on relatedness and, in parallel, used VariantMaster to call the variant in the parents given those called in the child. Only variants genotyped in the HapMap reference were considered for the analysis. Each algorithm provided the accuracy from the number of correct calls (i.e., concordant with the genotyped SNPs). We repeated the procedure by randomly decreasing the read coverage of the parents (50%, 30%, 25%, 10%, 7.5%, 5%). The resulting accuracies are reported in Figure 1. Notably, differences among the algorithms were appreciable when the coverage fell below 10% of its original value (corresponding to ∼10× on average). For lower coverages, PolyMutt performed better than GATK; however, FamSeq did not show any significant advantage. Remarkably, VariantMaster yielded the best overall performance, scoring >94% of correct calls even at an average parental coverage of ∼5× (5%).
Figure 1.

Conditional variant calling. VariantMaster, PolyMutt, and FamSeq correct variant calls (accuracy) of parents' WGS data—given the variants called by the GATK Unified Genotyper in the child—have been compared at decreasing read coverages in parents' data.
Application example 1: Identification of de novo variants
We challenged VariantMaster with the extraction of de novo variants in the whole-exome sequencing data of the child of the healthy CEU trio, and we compared the results with those obtained using a combination of KGGSeq alone or in conjunction with PolyMutt (with the de novo option) or FamSeq on the same data set. A previous study on this data set reported six validated de novo variants, of which one is germline and five are somatic variants generated “by the age of the cell lines (number of passages), the mutagenicity of the cell culture conditions, and/or the clonality of the cell lines” (Conrad et al. 2011). Raw BAM files were processed as reported in the Methods and Supplemental Material. Briefly, raw reads were mapped to the hg19 reference, and subsequent BAM and VCF file generations were performed using a standard pipeline based on BWA (Li and Durbin 2009), followed by the GATK Unified Genotyper. By using the six validated de novo variants as given, we estimated the impact of false positives by calculating precision = true_positives/(true_positives + false_positives) and sensitivity with respect to an increasing threshold over the quality score (QS), as provided by GATK. At low QS (50–300), VariantMaster was the only tool that identified all six variants (100% [6/6] sensitivity, precision from 19% [6/31] to 36% [6/17]). One true variant, called with a score of 329 by GATK, was filtered out at QS > 300 (83% [5/6] sensitivity, 31% [5/16] precision). By increasing the QS threshold, VariantMaster rapidly reduced the number of false positives, thereby increasing its precision up to 83% (5/6) and obtaining a sensitivity of 83% (5/6). KGGSeq started with 83% (5/6) sensitivity and 13% (5/39) precision. It partially benefited from PolyMutt prefiltering at low QS (83% [5/6] sensitivity, 17% [5/30] precision), since it lost one true positive variant at QS > 1000 due to PolyMutt QS recalibration (66% [4/6] sensitivity, 29% [4/14] precision). However, no changes to KGGSeq performance were observed when FamSeq prefiltering was applied. It is worth noting that replacing the QS with the de novo quality (DQ) index provided by PolyMutt did not affect the outcome. At the highest QS, KGGSeq alone reported 83% (5/6) sensitivity and 42% (5/12) precision. PolyMutt + KGGSeq attained 66% (4/6) sensitivity and 31% (4/13) precision. In general, for any QS threshold, VariantMaster outperformed all the other methodologies in terms of precision. We repeated the same analysis with VariantMaster after reducing the parental read coverage to 25% (corresponding to 16× on average). As expected, for all QS threshold we observed an increase in the amount of false positives. However, for high-quality calls, VariantMaster yielded only three false positives in this low-coverage condition (precisions range from 15% [6/39] to 63% [5/8]). All results have been reported in Figure 2. Taken together, we conclude that VariantMaster is more precise and more accurate than KGGSeq alone or in combination with PolyMutt or FamSeq for the identification of de novo variants.
Figure 2.

Extraction of de novo mutations from the CEU trio. Precisions obtained by VariantMaster, KGGSeq, and PolyMutt + KGGSeq, as functions of increasing quality score thresholds, have been plotted. Losses of true de novo variants have been emphasized with color-matching arrows. Precisions obtained by VariantMaster with a reduction to 25% coverage (∼10×) in the unaffected individuals are shown in green.
Application example 2: Identification of causative variants in a familial case
In order to test the algorithm in a clinical context, we attempted to identify putative causative variants in a nuclear family where two affected children (males) are affected by severe mental retardation and ataxia. The parents and the sister are not affected. The pedigree structure suggests either an X-linked or a recessive model of segregation (Supplemental Fig. 2). DNA of all family members was extracted, processed, and exome-sequenced as described above. For the identification of candidate variants respecting the X-linked and recessive inheritance, we ran VariantMaster and KGGSeq, alone and in combination with PolyMutt and FamSeq. All algorithms were run with analogous filter settings and for the X-linked and recessive analysis (including compound heterozygosity).
As done previously, we performed an incremental thresholding on variant quality score, as reported by GATK, to count the number of candidate variants provided by each method (Fig. 3).
Figure 3.

Causative recessive and X-linked variants' identification in a family presenting with a Mendelian disorder characterized by mental retardation, ataxia, and epilepsy. The number of candidate variants as a function of increasing quality score thresholds obtained by VariantMaster, KGGSeq, PolyMutt + KGGSeq, and FamSeq + KGGSeq is shown. Results obtained by VariantMaster after reduction to 25% coverage (∼10×) in the unaffected individuals are shown in green. All methods except PolyMutt + KGGSeq, which lost the SLC9A6 variant at QS > 700 (black cross) and the ADSL variant at QS > 1000 (red cross), identified the two causative variants at each threshold (for details, see text).
At higher thresholds, VariantMaster identified three hemizygous variants for the X-linked model in COL4A5, IRS4, and SLC9A6 present in both affected brothers, in heterozygosity in the unaffected mother, and absent in the father. Among them, only one variant, in SLC9A6, is considered potentially damaging by all SIFT, PolyPhen-2, LRT, and MutationTaster (Supplemental Table 1). In addition, this variant is highly conserved (GERP++ score >5), and the unaffected female is not a carrier of this mutation. Interestingly, the mutation in SLC9A6 (p.R536Q) has already been observed in a study on mental retardation (Tarpey et al. 2009), and other mutations on this gene have been associated with mental retardation, ataxia, and epilepsy (Online Mendelian Inheritance in Man [OMIM]: 300231, Mental Retardation, X-linked, syndromic, Christianson-type; MRXSCH) (Gilfillan et al. 2008). This mutation was subsequently found in a healthy uncle, making its clinical significance unclear. When the recessive model was imposed, VariantMaster remarkably yielded only one variant (Supplemental Table 2). The rare variant rs119450941 (p.R486H) in ADSL (MAF < 0.0001)—reported as damaging by SIFT, MutationTaster, and LRT and probably damaging by PolyPhen-2—has already been identified in several patients with mental retardation (Marie et al. 2000; Race et al. 2000; Edery et al. 2003; Jurecka et al. 2008) and considered as causative for the adenylosuccinate lyase deficiency (OMIM: 103050). Clinical phenotypes of this disease are ataxia, psychomotor retardation, autistic features, epilepsy, and mental retardation. After conventional validation, ADSL p.R486H was then considered the causative mutation, although with our current knowledge, we cannot exclude some involvement of the SLC9A6 variant as a modifier of the phenotype.
Considering ADSL(p.R486H) and SLC9A6(p.R536Q) as causative variants, VariantMaster largely yielded the best precision when compared with KGGSeq alone or in combination with PolyMutt or FamSeq (Fig. 3). Although FamSeq did not significantly affect KGGSeq performances, PolyMutt considerably improved KGGSeq false-positive rejection but led to a loss of the SLC9A6 variant at QS = 700 and the loss of ADSL variant at QS = 1000.
We repeated the analysis with VariantMaster after reducing the healthy relatives' read coverage to 25% of the original value (corresponding to 13× on average). As previously observed, the increase of false positives was more effective at low QS. At QS > 700, the number of candidate variants reduced to that obtained with full read coverage data.
Taken together, we conclude that VariantMaster is both more efficient and accurate than KGGSeq, PolyMutt + KGGSeq, and FamSeq + KGGSeq for the identification of causative variants in familial cases.
Application example 3: Identification of somatic variants in paired tumor-germline samples
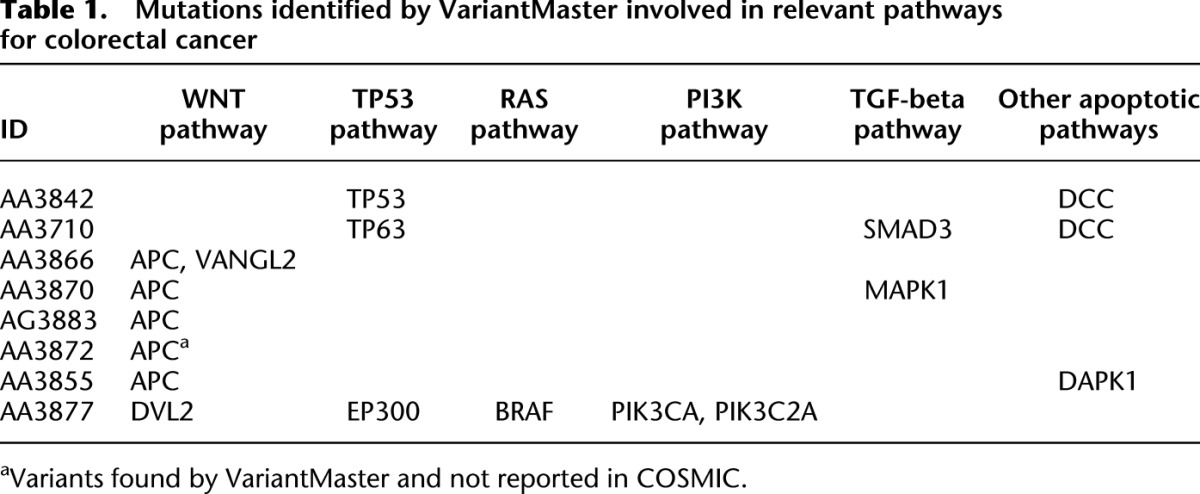
We tested VariantMaster on the identification of somatic variants from 16 random samples (BAM format) corresponding to eight sequenced exomes of colorectal cancer and the eight matched exomes of germline controls downloaded from the TGCA consortium (Table 1; The Cancer Genome Atlas Network 2012). Raw reads were mapped with BWA, and SAMtools was used for variant extraction. VariantMaster was set to consider likely functional variants (nonsense, missense, frameshift, and splicing) with a minimal coverage of 3× in the tumors (for details, see Supplemental Material). After annotation and filtering, the algorithm identified 2927 putative somatic variants, 1522 (52%) of these being already validated as somatic by the TCGA consortium and registered in the COSMIC database (http://www.sanger.ac.uk/genetics/CGP/cosmic). In agreement with previously published data (The Cancer Genome Atlas Network 2012), VariantMaster reported the recurrent mutated APC gene in four out of eight samples (AA3855, AA3866, AA3870, and AG3883). Remarkably, the algorithm identified two novel putative driver mutations: a new somatic APC frameshift deletion (c.4260delA) in AA3872 and a new AMER1 frameshift insertion (c.1099_1100insGGAG) in AA3870. Other putative driver variants reported by VariantMaster and already validated in COSMIC were in TP53 (AA3842), DCC (AA3842, AA3710), PIK3CA (AA3877), SMAD3 (AA3710), AMER1, BRAF-V600E, and EP300. Table 1 summarizes these results.
Table 1.
Mutations identified by VariantMaster involved in relevant pathways for colorectal cancer
As a comparison, we used VarScan (Koboldt et al. 2012), an algorithm specifically designed to identify somatic mutations in tumor samples. We ran VariantMaster and VarScan with default settings (for details, see Supplemental Material) over the eight TCGA samples. As reported in Table 2, VariantMaster outperformed VarScan in terms of sensitivity, with an average of 72% of correctly detected somatic variants (compared with 55% for VarScan), and in terms of false positive detection with an average precision of 40% compared with 18% (Fig. 4).
Table 2.
Performance comparison on somatic variants' identification in TCGA colorectal tumor samples
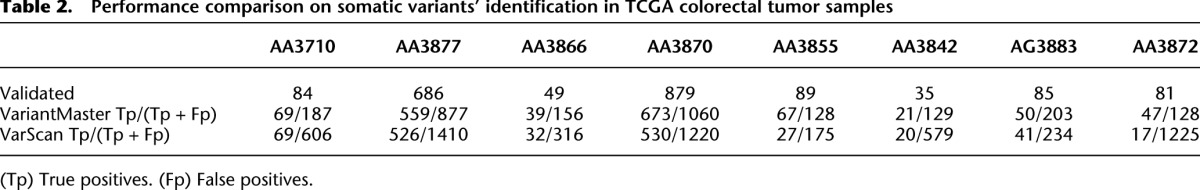
Figure 4.

Performance of VariantMaster on the extraction of somatic variants from eight TCGA colorectal cancer matched tumor-germline samples in comparison with VarScan. VariantMaster yielded better results in all analyzed samples. In particular, VariantMaster achieved an average sensitivity of 72%, significantly higher than VarScan (55%). Moreover, VariantMaster outperformed VarScan in terms of precision, obtaining an average of 40% compared with 18% obtained by VarScan.
Discussion
In this article, we present VariantMaster, a tool capable of identifying relevant variants in a broad range of applications: causative variants in familial cases, de novo mutations, and somatic variants in tumor samples. In order to sensibly reduce the impact of false positives, the algorithm implements a conditional variant calling procedure to evaluate the presence or the absence of each variant in all informative samples. With this approach, the algorithm accurately handles candidate variants even when respective genomic regions are not well covered. Indeed, as shown by our comparisons with previously published algorithms in different application cases, VariantMaster significantly increased the rejection of false positives while improving the discovery of true causative variants in familial cases and in matched tumor-germline experiments. Specifically, in the application example 3, VariantMaster was able to identify two novel putative variants in previously extensively analyzed and annotated data sets.
VariantMaster is designed to work with data produced by standard HTS pipeline based on widely adopted technologies and commonly used software such as BWA, GATK, and SAMtools with default parameters. In an ideal scenario, an average coverage of 30× to 50× per exome would guarantee high-quality variant calling and a consistent reduction of false-positive calls (Bamshad et al. 2011). However, we have shown that the VariantMaster approach can be adopted for cost-effective experiments with an acceptable loss in terms of sensitivity and precision, where the proband or the affected individuals (at least one) are sequenced at full coverage with the other family members sequenced at 10× to 15× coverage. The current release has been extensively tested on sequencing data produced on Illumina systems. It cannot be used with colorspace (i.e., SOLiD) data.
VariantMaster works as a stand-alone program or as part of a full pipeline. It requires at least 3 GB of RAM and fully processes all modes of inheritance (de novo, dominant, recessive, X-linked) for an annotated exome-sequenced trio (father, mother, proband) in <20 min on a standard computer. In general, VariantMaster takes 3–5 min of computation time per annotated sample.
VariantMaster has the potential to become an indispensable tool in the investigation of genetic diseases and molecular cancer profiles. The demonstrated effectiveness and flexibility in identifying a recessive causative mutation in a rare disease in a nuclear family and deleterious variants in a subset of TCGA colorectal tumor samples show its high potential for integration as part of a standard pipeline in either a research or diagnostic setting.
Methods
Input data
VariantMaster accepts BAM formatted files and processes lists of annotated or nonannotated variants (Variant Call Format, VCF). Nonannotated variants are processed with Annovar equipped with databases for genomic annotations; allelic frequencies in 1000 Genomes, dbSNP, and Exome Variant Server; as well as conservation tracks and pathogenicity scores.
Relationships (family pedigrees, tumor-normal or unrelated individuals) are provided to VariantMaster through a TFAM file (http://pngu.mgh.harvard.edu/∼purcell/plink/). VariantMaster can process one or more pedigrees of arbitrary complexity in one run. Filtering operators are defined for each analysis mode in a separate configuration file (for details, see Supplemental Material).
Carrier probability estimation and conditional variant calling
Given that a single nucleotide variant A with coordinates 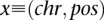 has been identified in the family member
has been identified in the family member 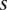 [described by the variable
[described by the variable 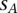 and
and 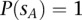 ] and
] and 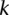 (>0) out of
(>0) out of 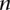 reads presenting with the allele A has been observed in the same site of the genome of another family member
reads presenting with the allele A has been observed in the same site of the genome of another family member 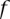 [expressed with
[expressed with 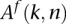 ], we would like to estimate the probability that
], we would like to estimate the probability that 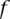 is effectively a carrier of the variant A (this event being described by the dichotomous variable
is effectively a carrier of the variant A (this event being described by the dichotomous variable 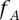 ). In other words, we need to calculate the conditional probability
). In other words, we need to calculate the conditional probability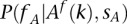 .
.
Indeed, by means of the chain rule,
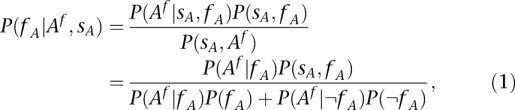 |
where we observed that 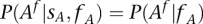 .
.
Moreover, since 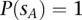 ,
,
where 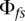 is the coefficient of relationship [=
is the coefficient of relationship [= 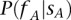 ] that can be calculated from the pedigree structure, and
] that can be calculated from the pedigree structure, and 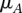 is the allelic frequency in the general population (= 0 if A has never been observed before).
is the allelic frequency in the general population (= 0 if A has never been observed before).
We estimated 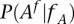 by considering the probability of observing one read with a specific variant in a locus
by considering the probability of observing one read with a specific variant in a locus 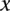 as being the same among all the individuals. Accordingly, we have
as being the same among all the individuals. Accordingly, we have
where 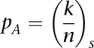 is the allelic ratio of the variant A as observed in
is the allelic ratio of the variant A as observed in 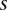 .
.
The term 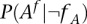 is the probability of observing k reads presenting with the allele A whether
is the probability of observing k reads presenting with the allele A whether 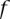 is not a carrier. The probability of erroneously observing A may be written in terms of the base calling error rate as given in Phred format by the sequencer, conditioned on the coverage
is not a carrier. The probability of erroneously observing A may be written in terms of the base calling error rate as given in Phred format by the sequencer, conditioned on the coverage 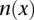 . Therefore, if A has been seen k times and
. Therefore, if A has been seen k times and 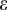 is the vector containing k equal error rates, we write the following:
is the vector containing k equal error rates, we write the following:
Replacing Equations 2a, 2b, and 2c in Equation 1 and simplifying, we eventually get
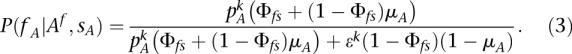 |
For practical application, we consider the individual 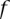 as a carrier of A if
as a carrier of A if 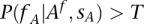 with
with 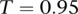 . However, this parameter can be changed by the user in the configuration settings. We arbitrarily set
. However, this parameter can be changed by the user in the configuration settings. We arbitrarily set 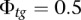 in tumor-germline matched analyses.
in tumor-germline matched analyses.
Exome sequencing, mapping, and variant calling
Exome capture of all members of the familial case was conducted using the SureSelect Human All Exon V3 or V4 kit (Agilent Technologies) according to manufacturer's recommendations. Three different genomic libraries were pooled and sequenced in one lane of an Illumina HiSeq 2000 sequencer using a 2 × 95-bp paired end indexing protocol. Specifically, the mapping of fastq reads was performed with BWA, with duplicate removal by SAMtools, against the hg19 reference. SNPs have been called by the GATK Unified Genotyper. A comprehensive schematic of the pipeline is represented in Supplemental Figure 1.
Whole-genome and whole-exome sequencing data (BAM) of the CEU trio (NA12878, daughter; NA12891, father; and NA12892, mother) were downloaded from ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/working/20120117_ceu_trio_b37_decoy/ and processed as previously described. HapMap genotyped SNPs were downloaded from ftp://ftp.ncbi.nlm.nih.gov/hapmap/genotypes/2010-08_phaseII + II.
Aligned reads (BAM) of eight paired tumor samples + germline were downloaded from https://tcga-data.nci.nih.gov/, and the respective annotations were obtained from COSMIC (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/). Variants were extracted as previously described. Functional variants from each tumor sample (nonsense, missense, frameshift, and splicing) with coverage > 3, QS > 15, and no constraints on frequency were filtered in by VariantMaster and considered as putative somatic mutations if not found in the related germline sample.
Further details regarding data processing and settings of KGGSeq and VarScan are reported in the Supplemental Material.
Processing modes available in VariantMaster
VariantMaster implements several modes of inheritance allowing for missing individuals in the pedigree. Missing individuals will be considered as unaffected by default if not otherwise specified in the configuration file. Dominant, X-linked, recessive homozygous, and compound heterozygous modes can work with NGS data and/or genotypes. However, VariantMaster requires, at minimum, the sequenced variants of at least one affected individual.
De novo mode of inheritance is applied to trios (father, mother, child), and it requires the availability of HTS data for all members of the family.
Tumor-germline comparison works with HTS data only. It is possible to compare several replicates from the same tumor to its related normal sample to identify primary driver mutations or two replicates from the same tumor to isolate late somatic mutations.
Variants from a pool of unrelated individuals are filtered and organized according to their associated gene and their eventual recurrences in the pool.
VariantMaster collects all the variants according to the requested analysis in all the families, tumors, or unrelated individuals and creates a unified record containing the occurrence of the same variant and the normalized (with respect to gene length) mutational load of each single gene.
Technical notes
VariantMaster is written in python2.7 and is released as executable for Linux systems (http://sourceforge.net/projects/variantmaster/). It imports PySam (http://code.google.com/p/pysam) and a customized wrapped version of BEDTools (Quinlan and Hall 2010). Additional details can be found in the Supplemental Material.
Acknowledgments
We thank Ximena Bonilla, Teresa Didonna, and Sam Lukowsky for useful comments and discussions. This study is supported by grants from the SNF (144082), ERC (249968), and ChildCare and Gebert Foundations to S.E.A., and a grant from the Bodossaki Foundation to P.M.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.163832.113.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12: 745–755 [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Fay JC 2009. Identification of deleterious mutations within three human genomes. Genome Res 19: 1553–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Shendure J 2011. Needles in stacks of needles: Finding disease-causal variants in a wealth of genomic data. Nat Rev Genet 12: 628–640 [DOI] [PubMed] [Google Scholar]
- Edery P, Chabrier S, Ceballos-Picot I, Marie S, Vincent MF, Tardieu M 2003. Intrafamilial variability in the phenotypic expression of adenylosuccinate lyase deficiency: A report on three patients. Am J Med Genet A 120A: 185–190 [DOI] [PubMed] [Google Scholar]
- Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K, Kroken M, Mattingsdal M, Egeland T, Stenmark H, et al. 2008. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am J Hum Genet 82: 1003–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurecka A, Tylki-Szymanska A, Zikanova M, Krijt J, Kmoch S 2008. D-ribose therapy in four Polish patients with adenylosuccinate lyase deficiency: Absence of positive effect. J Inherit Metab Dis (Suppl 2) 31: S329–S332 [DOI] [PubMed] [Google Scholar]
- Knocking on the clinic door. [Editorial] 2012. Nat Biotechnol 30: 1009. [DOI] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK 2012. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22: 568–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Chen W, Zhan X, Busonero F, Sanna S, Sidore C, Cucca F, Kang HM, Abecasis GR 2012a. A likelihood-based framework for variant calling and de novo mutation detection in families. PLoS Genet 8: e1002944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MX, Gui HS, Kwan JS, Bao SY, Sham PC 2012b. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res 40: e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makrythanasis P, Antonarakis SE 2012. High-throughput sequencing and rare genetic diseases. Mol Syndromol 3: 197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie S, Race V, Vincent MF, Van den Berghe G 2000. Adenylosuccinate lyase deficiency: From the clinics to molecular biology. Adv Exp Med Biol 486: 79–82 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DJ, Onoufriadis A, Shoemark A, Simpson MA, Zur Lage PI, de Castro SC, Bartoloni L, Gallone G, Petridi S, Woollard WJ, et al. 2013. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am J Hum Genet 93: 346–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev SI, Sotiriou SK, Pateras IS, Santoni F, Sougioultzis S, Edgren H, Almusa H, Robyr D, Guipponi M, Saarela J, et al. 2012. A single-nucleotide substitution mutator phenotype revealed by exome sequencing of human colon adenomas. Cancer Res 72: 6279–6289 [DOI] [PubMed] [Google Scholar]
- Peng G, Fan Y, Palculict TB, Shen P, Ruteshouser EC, Chi AK, Davis RW, Huff V, Scharfe C, Wang W 2013. Rare variant detection using family-based sequencing analysis. Proc Natl Acad Sci 110: 3985–3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race V, Marie S, Vincent MF, Van den Berghe G 2000. Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum Mol Genet 9: 2159–2165 [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D 2010. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7: 575–576 [DOI] [PubMed] [Google Scholar]
- Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O'Meara S, Latimer C, Dicks E, Menzies A, et al. 2009. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 41: 535–543 [DOI] [PMC free article] [PubMed] [Google Scholar]