

Abstract
Ontology organizes and formally conceptualizes information in a knowledge domain with a controlled vocabulary having defined terms and relationships between them. Several ontologies have been used to annotate numerous databases in biology and medicine. Due to their unambiguous nature, ontological annotations facilitate systematic description and data organization, data integration and mining, and pattern recognition and statistics, as well as development of analysis and prediction tools. The Variation Ontology (VariO) was developed to allow the annotation of effects, consequences, and mechanisms of DNA, RNA, and protein variations. Variation types are systematically organized, and a detailed description of effects and mechanisms is possible. VariO is for annotating the variant, not the normal-state features or properties, and requires a reference (e.g., reference sequence, reference-state property, activity, etc.) compared to which the changes are indicated. VariO is versatile and can be used for variations ranging from genomic multiplications to single nucleotide or amino acid changes, whether of genetic or nongenetic origin. VariO annotations are position-specific and can be used for variations in any organism.
The ever increasing production of genetic and other information related to variations demands more efficient data analysis tools and systematics for storage, annotation, search, and mining of data and databases. Several recommendations, best practices, and minimum requirements have been published for locus-specific variation databases (LSDBs), their establishment, maintenance, and curation, and were recently updated (Celli et al. 2012; Vihinen et al. 2012) (Human Mutation virtual issue, “Recommendations and Standards for the Reporting and Databasing of Genetic Variations”; http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)1098-1004/homepage/virtual_issue_recommendatations_and_standards_for_the_reporting_and_databasing_.htm). Systematics is required to handle the exponentially growing data and also to allow for data integration and simultaneous searches in several sources, as well as software development for biomedical informatics and numerous so-called semantic web applications. The “semantic web” means more effective data management because of standardized ways of expressing the relationships between web pages, thereby allowing computers to understand the meaning of the information.
Annotation is a process that adds information to databases by describing key features of the data items. Annotations can be, e.g., free text or more systematic, using controlled vocabularies or ontologies. Ontologies are central for implementation of the semantic web.
Several ontologies and other standardized systems are available to describe genes, proteins, and diseases. The HUGO Gene Nomenclature Committee (HGNC) nomenclature (Seal at al. 2011) provides official gene names, abbreviations, and symbols. Many genes have traditionally had several names, and a name may have meant different genes/proteins in different contexts or papers. Systematic names prevent such problems.
Locus Reference Genomic (LRG) sequence entries (Dalgleish et al. 2010) allow unequivocal mapping of sequence positions. As LRGs will never be changed, the positions at different levels (DNA, RNA, protein) are explicit. The Gene Ontology (GO) (Ashburner et al. 2000) was the first biological ontology and currently (December 2013) provides 40,260 terms for explanation and annotation of cellular components, molecular functions, and biological processes. The Sequence Ontology (SO) (Eilbeck et al. 2005) is used to describe features and properties of biological sequences.
The Variation Ontology (VariO; http://variationontology.org) was developed to annotate effects of variants in different types of databases such as LSDBs, central, variation effect or frequency databases, ethnic/national variation databases, benchmark data sets, and variant management software. It does not explain the properties of the normal state; instead, it describes what is changed in a variant in relation to the reference. VariO can be used for systematic annotation of all kinds of effects, consequences, and mechanisms of variations on DNA, RNA, and protein levels in any organism and includes terms for variations and effects of both genetic and nongenetic origin. Variations of any size and mechanism can be described.
Results and Discussion
VariO contains a total of 384 terms on eight levels. Because the terms can be combined and further modified with variation attributes, annotations can be very specific and detailed. As many VariO terms as possible should be used to describe the effects of a variant to cover all its features.
VariO organization
VariO has four major levels (Fig. 1)—in addition to DNA, RNA, and protein, it also has variation attributes as modifier terms. Each of the major levels—DNA, RNA, and protein—has four main sublevels. The variation type describes the origin and classification of variation without providing the actual nucleotide or amino acid change. HGVS nomenclature provides the systematic naming of variations. VariO explains the variation type so that information can be easily searched. This is an example of how VariO is used together with other systematic approaches without repeating their content. HGVS names for complicated cases are rather difficult to interpret unless one is familiar with the naming conventions. VariO provides the variation type annotations with generally used genetic terms.
Figure 1.

Organization of sublevels in VariO. Examples of terms in the different levels. The three central levels are DNA, RNA, and protein, to which modifier attributes add further versatility. On each major level certain details are shown to illustrate the organization of VariO and types of terms.
Function terms provide annotation for the general function(s) affected by the variation. Structure sublevel terms are for describing affected structural features. Property terms are used for defining diverse features.
VariO terms can be further modified by attributes, increasing the versatility of the ontology without increasing the number of terms. For example, the use of VariO:0289 quantity change terms “VariO:0290 decreased,” “VariO:0291 increased,” “VariO:0292 missing,” and “VariO:0140 not changed,” saved hundreds of terms describing altered properties at several other levels.
Figure 2 indicates how the sublevels contain increasingly more defined terms. For example, VariO:0129 DNA variation type has annotation VariO:0322 DNA variation classification and is of type VariO:0135 DNA chain variation. This can be a VariO:0136 DNA substitution, which is a VariO:0313 transition, more specifically a VariO:0315 purine transition. This kind of description allows very detailed annotation and, e.g., a search for just one kind of variation, several types, different term combinations, etc.
Figure 2.

Variation types at the DNA, RNA, and protein levels. Terms with “is a” relation are indicated by lines with arrowheads, and those with “part of” relation are indicated by a dashed line.
An additional layer of information is brought to the VariO annotation by describing the method(s) and data based on which the annotation is made. Evidence Ontology terms are used for this purpose. ECO contains close to 300 terms for experimental methods as well as for predictions and inference ranging from author statements to experimental evidence. ECO annotations should be provided for specific VariO annotations. They allow the database users to evaluate the annotated cases and their reliability. VariO annotations should be supported by appropriate literature references, when available.
Figure 2 shows how the VariO terms are organized, in this case for the variation type. On each level, the variation type has two subtypes, those for variation origin and variation classification. VariO can be used for annotation of all kinds of variations whether genetic, somatic, or artificial (e.g., genetic engineering). The nongenetic variations include, on the DNA level, epigenetic changes and artificial variants, and variations emerging at the RNA level include decayed, edited, and modified RNA and artificially modified RNA. Variations emerging at the protein level are further divided into those of artificial origin, epigenetic nature, and post-translationally modified protein.
The VariO:0322 DNA variation classification divides into the VariO:0132 chromosomal variation, the VariO:0135 DNA chain variation, and the VariO:0131 genomic variation. Genomic variations are genome-wide, while chromosomal variations include variations in chromosome number (euploidy) and structure. DNA chain variations are further divided into five sublevels, which have still more detailed terms.
Functional terms vary for the different levels and so do structural terms. For example, on the DNA level the structure includes changes to chromatin, chromosome set number, chromosome variations, DNA secondary structural changes, epigenetic DNA modifications, and gene structure variations. On the protein level the terms have subheadings such as dynamics, quaternary structure, tertiary structure, and complex structural variation with increasingly more detailed subterms.
Design principles of VariO
VariO has been built based on certain principles which affect its structure, annotations, and applications. The goal has been to provide a generic ontology that is applicable to all cases and effects of variations. Another key design principle was the simplicity of the ontology structure, which, however, allows detailed description by combining terms and modifying them with additional attributes.
Scope
VariO is for explaining the effects, consequences, and mechanisms of variations. Therefore, VariO does not describe the normal or wild-type situation. Instead, it describes what is changed in relation to it. The reference state for each level needs to be established by the ontology users. For example, all sequence-related terms are used together with reference sequences for DNA, RNA, or protein. LRGs, if available, are recommended for that purpose as they are stable and will never be changed (Dalgleish et al. 2010). Similarly, reference states should be defined at all necessary levels. The reference states should be mentioned and explained in the databases.
Clear hierarchy of terms
Annotations with VariO should be made on as many levels as possible. The three major levels are the three major molecules involved in biology; namely, DNA, RNA, and protein. Each of these is further divided into sublevels common to the major levels. The terms vary from level to level. Variation type provides a general description of the type of the variation, function implies what is the affected function, and the structure sublevel provides the possibility for detailed annotation of diverse structural features. Property terms, for describing diverse characteristics of the variation, is the sublevel that has the largest diversity between the major levels.
Simple terms allow complex annotations when combined
The terms in VariO have been kept simple. Some other ontologies have very extensive and long terms, such as “GO:0016706 oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, 2-oxoglutarate as one donor, and incorporation of one atom each of oxygen into both donors.” In VariO, complex annotations are made by combining several terms together. This provides versatility and facilitates even very complex annotations and consequently powerful computational searches from VariO annotated databases. VariO has a clear structure. Terms on one level, e.g., on the variation type, can be further defined by effects on function or structure. This is possible on all three molecular levels.
The versatility of terms is further expanded by variation attributes that are used as modifiers, e.g., to describe effects on quantity such as increased, decreased, or missing property. Thus, instead of having four separate terms for protein abundance (increased abundance, decreased abundance, no abundance, abundance not changed), it is annotated with a single term: VariO:0052 effect on protein abundance plus the quantity attributes.
Evidence for annotations
An integral part of annotations with VariO is additional information in the form of the method used to obtain the annotated results. The Evidence Ontology (ECO; http://www.evidenceontology.org/) terms are used to indicate the type of method by which the evidence for a certain annotation was obtained. This information makes it possible for the users to estimate the quality and significance of individual annotations. ECO contains terms for experimental methods and, in addition, terms to describe prediction approaches.
Combined use with other systematics approaches
VariO has a very focused use, on purpose. It can and should be used together with other variation-related systematics, such as those listed in the introduction, to capture further characteristics of the variation and the organism/individual having the variant.
Any variation consequence, effect, mechanism
The goal of the VariO is to allow description of any kind of variation effect, consequence, or mechanism, at least to a certain degree. There is a balance between how detailed the terms can be and how many cases there are to annotate with the terms. Certain very specific mechanism descriptions may just be available for a single or only a few cases in the entire literature. VariO aims at covering all effects currently studied. New terms can be added in the future as the need arises.
Any size
The size range of variations is very large, ranging from nucleotide changes to duplications or multiplications of entire genomes. VariO has been designed to cover the full range of sizes. On the DNA level it covers changes at the genome, chromosome, and DNA-chain level. The sizes vary on RNA and protein, as well.
Position-specific
VariO annotations are always position-specific. The concept of position is flexible, including, e.g., a chromosome arm in translocations, or an entire genome in euploidy. We recommend the use of the HGVS nomenclature (den Dunnen and Antonarakis 2000) to indicate the actual position on DNA, RNA, and protein chains; LRG reference sequences or versioned sequence entries for individual genes, transcripts, and proteins; and genome builds (e.g., hg19 or GRCh37) for genomes.
Origin of variation
VariO terms can be used equally well for description of variations of genetic and of nongenetic origin. The nongenetic origin includes on DNA-level epigenetic DNA variation; on RNA-level decayed, edited, and modified RNA; and on protein-level modified amino acids, proteolytically processed proteins, and spliced proteins. Variation can also be of artificial origin, produced, e.g., with protein engineering.
Organism independent
VariO is generic and therefore can be used for any kind of organism, whether having a genome on DNA or RNA, whether the genetic material is genomic or extrachromosomal, whether it contains exons or not, whether living alone or in other cells, etc.
Fulfillment of formal suggestions for ontologies
In addition to these variation-specific design principles, VariO follows the general principles for ontologies put forward by Gruber (1995). These include the following:
Clarity. The developed VariO terms are objective and have full definitions.
Coherence. Special attention was given to make the terms coherent in and between all the levels.
Extendibility. VariO can be easily extended in the future.
Minimal coding bias. The conceptualization of the variation knowledge domain is based on widely used genetic concepts.
Minimal ontological commitment. The extensive conceptual modeling of the variations at different levels made it possible to make VariO generic and thus easily usable for various applications.
Furthermore, VariO fulfills the 5-Star vocabulary requirements (http://bvatant.blogspot.fr/2012/02/is-your-linked-data-vocabulary-5-star_9588.html) and AMOR Manifesto principles (http://knowledgecraver.blogspot.com.es/2013/04/the-amor-manifesto.html) developed based on Tim Berners Lee's 5 Star Linked Open Data rating (http://www.w3.org/DesignIssues/LinkedData.html). The goals of these suggestions are to make data (ontologies) accessible, also in machine readable format including nonproprietary format(s), use open standards, provide metadata, full term descriptions, have a stable Universal Resource Identifier (URI), and link to and reuse other ontologies and vocabularies.
VariO annotation
Examples of VariO annotations are in Table 1 and Supplemental Figures 1–4. Detailed instructions for annotators are in M Vihinen (in prep.). In the first example, a missense variation has an effect on protein structure and binding (Table 1). Based on these data, the DNA and RNA levels contain only variation-type annotations. On the protein level, effects are described also on function, structure, and properties. Note that modifier attributes are used to indicate, e.g., how the protein solubility is affected.
Table 1.
Use of VariO for annotation of a missense variation
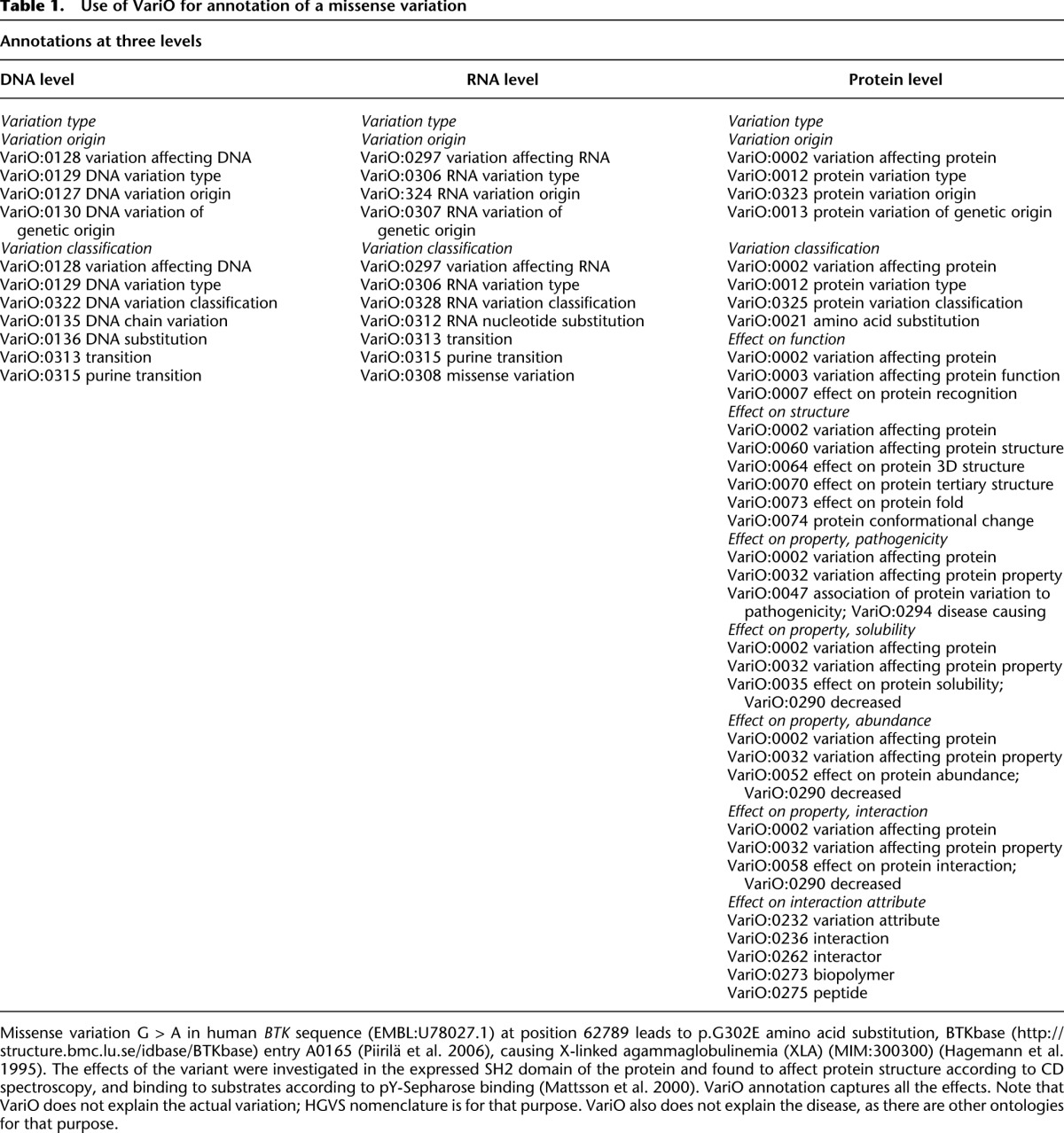
VariO annotations should be as detailed as possible and include all necessary terms. The terms consist of VariO followed by a four-digit code and the name of the term. Always when using the terms, at least the number, e.g., VariO:0123, should be mentioned. The evidence for the terms is indicated with ECO annotations. The annotations are position-specific on all the three major levels (DNA, RNA, protein). The position size can range from a single nucleotide to an entire genome.
The examples are shown for simplicity without the Evidence Codes annotations, which should be together with the VariO annotation. For example, in the case of protein abundance in Table 1, the ECO term ECO:0000046 protein expression level evidence should be used and the term to join effect on protein solubility has evidence ECO:0000112 Western blot evidence. Effects on interaction have a detailed description, and insertion attributes provide details for interaction partners. This exemplifies the principle in VariO that annotations should be as abundant and detailed as possible.
The example in Supplemental Figure 1 is for a case in which a substitution in an exon–intron boundary leads to the insertion of a DNA stretch causing a frameshift and premature termination. The variant has been identified from a patient. The protein was not detectable.
Effects of a variation on a start codon are shown in Supplemental Figure 2. On the RNA level this is a missense variation, but as the protein is not produced due to the initiation codon alteration, the outcome on the protein level is a VariO:0240 missing protein. This site is a hotspot for variation in the BTK gene.
Variations affecting splicing are frequent. The example in Supplemental Figure 3 shows a case in which a canonical splice site is altered, leading to activation of a cryptic splice site in the middle of the exon. This leads to the skipping of 11 nucleotides (nt) from the exon and amphigoric deletion on the amino acid level. The variant appears on the noncoding region of the coding sequence, i.e., in the intron and the cryptic splice site on the coding region.
Effects on protein function and property, especially reaction kinetics and specificity, are indicated in Supplemental Figure 4. The effects of engineered variations at conserved glutamates were studied in Trypanosoma brucei virulence protein oligopeptidase B (Mohd Ismail et al. 2010). The artificial variant was generated with site-directed mutagenesis and is indicated at the DNA level. The RNA and protein variants originate from it. Effects in this variant were noticed in substrate specificity and reaction kinetics (Km, kcat, and their ratio). Note how the VariO quantity term “VariO:0140 not changed” is used to indicate experimental evidence that does not affect a feature. In this way, VariO annotations can indicate also those studied properties that remain unchanged compared with the reference type.
The amount of labor required for making the annotations varies case by case. The annotations can be made relatively quickly with VariOtator once the details are available. The major work is related to finding and mining the details and the methods used for obtaining them. Quite often the database curators have these facts available when entering cases. If it is necessary to search and filter such information from the literature, databases, and other sources, much more time and effort may be needed. Once the annotations are in the database, they will make even very complex data search queries possible.
Availability and browser
In addition to the OBO format, VariO is available as OWL and OWX files at http://variationontology.org. The site has a unique and permanent Uniform Resource Identifier (URI) at MIRIAM registry (Juty et al. 2012) and at OBO. VariO is participating in the OBO Foundry (http://www.obofoundry.org/) project (Smith et al. 2007) and the file is downloadable there as well. VariO terms are also accessible via The National Center for Biomedical Ontology (NCBO; BioPortal http://bioportal.bioontology.org/) and the Ontology Lookup Service (http://www.ebi.ac.uk/ontology-lookup/init.do), where terms can be searched for.
The ontology, terms, definitions, relationships, and cross-references to other ontologies can be visualized with the AmiVariO viewer, which is based on the AmiGO tool (Carbon et al. 2009). The browser can be used to search terms and for visualization of the ontology. The ontology tree can be downloaded in three formats: OBO, RDF-XML, or Graphviz dot for further processing and visualization.
Annotation tool
An annotation tool is available to help curators annotate their databases easily as well as to provide related ECO evidence details for VariO annotations. The current version of the annotation tool, called VariOtator and available on the website, provides variation-type annotations on all three molecular levels based on HGVS nomenclature, which can be generated by the Mutalyzer tool (Wildeman et al. 2008). The tool can at the moment also make function and structure annotations, and the property features will follow. Suggestions for terms can be sent via the e-mail address listed on the website.
Relation to other ontologies
VariO is a unique ontology, but it shares some terms with other systematic representations. Exclusive comparison was made to other ontologies. Cross-references are provided when terms are identical.
The highest number of terms (32) is shared with the Molecular Interaction (MI) ontology (Kerrien et al. 2007). These terms had to be modified to suit the purpose of VariO. Sequence Ontology is a related ontology used to describe features and attributes of biological sequences (Eilbeck et al. 2005), while VariO is for effects and mechanisms of variations including (in addition to sequence) structure, function, and properties. There are altogether 29 common terms with SO—mainly on the DNA and RNA levels—18 and nine, respectively. More than half of them describe DNA structure—16 terms out of the total of 74 such terms in VariO. VariO provides deeper annotations and goes beyond the sequence-based concepts.
Definitions of protein secondary structural elements are from PDBeMotif (Golovin and Henrick 2008). All terms in VariO are manually curated and the reused terms are usually modified to suit the purpose.
Applications
Databases annotated with VariO terms will allow interoperability, collection, and analysis of cases simultaneously from single or multiple databases with simple or even very complex queries. The number of databases and services providing VariO annotations is increasing. There are several applications, including, e.g., generation of benchmark data sets for different effects and mechanisms to test the performance of prediction tools, searching for certain types of variations over several genes or proteins, and the integration of heterogeneous information sources for extensive analysis and interpretation of variations and their effects. VariO is most suitable for annotation of effects of variations in LSDBs, central variation, and sequence databases. It can equally well be applied to specific variation databases—whether devoted to diseases, mechanisms, or effects. One user of VariO annotations is VariBench, a database for benchmark data sets for variation effect analysis and for training and testing of prediction methods (Nair and Vihinen 2013).
Variation-type annotations can be made automatically with the VariOtator tool connected to the sequence variant nomenclature tool Mutalyzer (Wildeman et al. 2008) or from existing HGVS names. These annotations will be available for the thousands of LOVD-based variation databases (Fokkema et al. 2011). We hope that the annotations will be widely used in LSDBs such as IDbases (Piirilä et al. 2006) and also in some central variation databases. Other possible users include, e.g., specific variation effect databases such as ProThem and ProNIT (Kumar et al. 2006), allele frequency databases like FINDbase (van Baal et al. 2007), and ethnic and/or national databases like the Hellenic National Mutation Database (Patrinos et al. 2005).
Once databases have VariO annotations available, queries can be made within and across databases to find cases of interest such as variation types in certain protein structural elements, variants with certain kinds of functional effects, or, for example, disease-causing variants in protein–protein interaction interfaces. The possibilities are limitless, as exemplified by GO annotations, which have already been used in 5000 publications in numerous different ways.
In Figure 3 is shown an example of the use of VariO annotations for data mining and analysis. Wiskott-Aldrich syndrome (WAS) is a rare recessive primary immunodeficiency characterized by a reduced ability to form blood clots due to platelet abnormality. Other features include eczema, thrombocytopenia, and bloody diarrhea. The defective gene WAS codes for Wiskott-Aldrich syndrome protein (also known as WASP), which is a Rho-type GTPase effector that regulates actin filament reorganization. In addition to WAS, the gene and protein are involved in three other conditions (Ochs and Thrasher 2006). X-linked thrombocytopenia (XLT) is a milder disease with less severe symptoms, and intermittent XLT (IXLT) is an even less severe form. Congenital neutropenia (XLN) is caused when the WASP protein is constitutively active.
Figure 3.
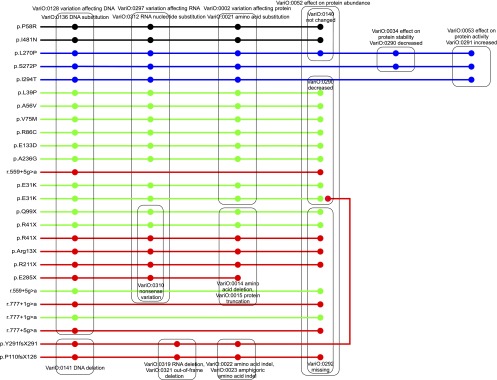
Example of data analysis with VariO annotations. Wiskott-Aldric syndrome protein (WASP) variations are involved in four conditions including Wiskott-Aldrich syndrome (WAS; red), the most severe disease; X-linked thrombocytopenia (XLT; green), a somewhat milder disorder; intermittent XLT (IXLT; black); and congenital neutropenia (XLN; blue). Annotations are shown only for relevant features of variation type, protein abundance, protein stability, and protein activity. The cases are grouped based on variation types to indicate genotype–phenotype correlations. The less severe disorders typically have less drastic changes—mainly amino acid substitutions—while the more severe diseases such as XLT and especially WAS contain protein truncations and splice-site-affecting alterations. The protein abundance follows this trend, being unaffected in XLN and IXLT, while in XLT it is mainly decreased and in WAS mainly missing.
Variations representing the four diseases were collected from the literature and from WASbase (http://structure.bmc.lu.se/idbase/WASbase) and annotated with VariO terms. All the known cases were taken for IXLT (Notarangelo et al. 2002) and XLN (Devriendt et al. 2001; Ancliff et al. 2006; Beel et al. 2009). Since hundreds of cases have been reported in XLT and WAS, a random sample was taken from an article describing 262 patients from 227 families (Jin et al. 2004). In addition to the variation at the DNA, RNA, and protein levels, information about protein abundance, activity, and stability was collected.
Partial annotations for relevant features are shown in Figure 3. The number of features of the diseases can be revealed from this figure. IXLT cases are clustered and are the only ones with protein abundance not affected. In XLN, protein activity is increased and stability is decreased. In both these diseases all the variants lead to amino acid substitutions. The substitutions are also dominant in XLT and WAS, however, they appear together with other kinds of variations including deletions and indels.
In XLT, variants are typically less extensive than in WAS. In WAS, several truncating variations appear with the consequent effect of missing protein. Even the WAS substitutions lead more often to missing than decreased protein abundance, contrary to XLT, which has the the opposite situation. Intron variants are more common in WAS than in XLT. We can draw conclusions about the genotype–phenotype correlations due to WAS variants. In the least severe form, IXLT, patients have normal protein abundance. XLN is characterized by increased protein activity. The majority of the XLT-causing variants are somewhat milder, and are mainly substitutions. These appear also in WAS; however, there are even more drastic variations like protein truncations and splice-site changes. The correlations of variation types to diseases are not complete; there are, e.g., protein truncations in XLT, and the same variant can have different outcomes in different individuals probably due to additional genetic differences and environmental effects. VariO clearly highlights the features of the four diseases.
VariO is compliant with the VarioML variation data exchange format (http://varioml.org/) (Byrne et al. 2012), which is used, e.g., in the Café Variome exchange portal (http://cafevariome.org/) for variation data produced, e.g., in diagnostic laboratories. The use of VariO has been recommended by the Human Variome Project (Kohonen-Corish et al. 2010). Together with other systematic descriptions it will enrich information, e.g., with the HGVS nomenclature and the Human Phenotype Ontology (HPO) (Robinson et al. 2008) annotations to describe the sequence, structure, function, type, and pathogenicity of variations.
Conclusion
VariO can be used to describe basically any kind of variation, whether natural or engineered, in any organism, and for any mechanism. In addition to variations of genetic origin, those of nongenetic origin such as epigenetic modifications can be annotated. VariO is position-specific; however, the concept of position is wide and may even cover a complete chromosome or genome. VariO is used together with the Evidence Codes ontology to describe what evidence and methods have been used to study the case. VariO annotations should be attributed to published information and literature citations, if available.
Methods
VariO is based on extensive conceptual modeling of the nature and features of variations at different levels and on defining terms so that they are organized in a coherent and unambiguous way. The consistency and coherence of the terms at different levels were an important design feature. VariO was constructed with OBO-Edit (Day-Richter et al. 2007), a widely used ontology editor.
VariO terms have detailed definitions. Terms are related by “is a” and “part of” relationships. Apart from a few cases, suitable terms were found in the literature. Only if a term was missing or the databases and literature used ambiguous terms was a new one with a clear and specific definition introduced. For example, a suitable term was missing for deletions, indels, and insertions at the amino acid sequence when the protein sequence is changed after the modification site, usually leading to premature termination. In VariO, these are called “VariO:0017 amphigoric amino acid deletion, VariO:0023 amphigoric indel, or VariO:0019 amphigoric amino acid insertion.” Although “nonsense” would have been a perfect term for this kind of alteration missing the “sense” of the sequence, it could not be used because it is reserved for RNA-level alterations. Thus, new terms were coined.
Software availability
VariO and VariOtator are available at http://variationontology.org.
Acknowledgments
I thank Janet Thornton, Kati Laiho, Jennifer Deegan, Sandra Orchard, Fiona Cunningham, Tomasz Adamusiak, Helen Parkinson, and Karen Eilbeck for valuable discussions. Jouni Väliaho and Gerard Schaafsma are acknowledged for establishing the website and mining term definitions, and Gabriel Teku for drawing figures. The Finnish Academy is acknowledged for financial support. The research leading to these results received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant number 200754 (the GEN2PHEN project).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.157495.113.
Freely available online through the Genome Research Open Access option.
References
- Ancliff PJ, Blundell MP, Cory GO, Calle Y, Worth A, Kempski H, Burns S, Jones GE, Sinclair J, Kinnon C, et al. 2006. Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood 108: 2182–2189 [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene Ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beel K, Cotter MM, Blatny J, Bond J, Lucas G, Green F, Vanduppen V, Leung DW, Rooney S, Smith OP, et al. 2009. A large kindred with X-linked neutropenia with an I294T mutation of the Wiskott-Aldrich syndrome gene. Br J Haematol 144: 120–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne M, Fokkema IAC, Lancaster O, Adamusiak T, Ahonen-Bishopp A, Atlan D, Béroud C, Cornell M, Dalgleish R, Devereau A, et al. 2012. VarioML framework for comprehensive variation data representation and exchange. BMC Bioinformatics 13: 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S 2009. AmiGO: Online access to ontology and annotation data. Bioinformatics 25: 288–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli J, Dalgleish R, Vihinen M, Taschner PEM, den Dunnen JT 2012. Curating gene sequence variant databases (LSDBs). Hum Mutat 33: 291–297 [DOI] [PubMed] [Google Scholar]
- Dalgleish R, Flicek P, Cunningham F, Astashyn A, Tully RE, Proctor G, Chen Y, McLaren WM, Larsson P, Vaughan W, et al. 2010. Locus Reference Genomic sequences: An improved basis for describing human DNA variants. Genome Med 2: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day-Richter J, Harris MA, Haendel M 2007. OBO-Edit—an ontology editor for biologists. Bioinformatics 23: 2198–2200 [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE 2000. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum Mutat 15: 7–12 [DOI] [PubMed] [Google Scholar]
- Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Van Den Oord JJ, Verhoef GE, Boogaerts MA, Fryns JP, You D, et al. 2001. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet 27: 313–317 [DOI] [PubMed] [Google Scholar]
- Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, Ashburner M 2005. The Sequence Ontology: A tool for the unification of genome annotations. Genome Biol 6: R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT 2011. LOVD v.2.0: The next generation in gene variant databases. Hum Mutat 32: 557–563 [DOI] [PubMed] [Google Scholar]
- Golovin A, Henrick K 2008. MSDmotif: Exploring protein sites and motifs. BMC Bioinformatics 9: 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber TR 1995. Towards principles for the design of ontologies used for knowledge sharing. Int J Hum Comput Stud 43: 907–928 [Google Scholar]
- Hagemann TL, Rosen FS, Kwan SP 1995. Characterization of germline mutations of the gene encoding Bruton's tyrosine kinase in families with X-linked agammaglobulinemia. Hum Mutat 5: 296–302 [DOI] [PubMed] [Google Scholar]
- Jin Y, Mazza C, Christie JR, Giliani S, Fiorini M, Mella P, Gandellini F, Stewart DM, Zhu Q, Nelson DL, et al. 2004. Mutations of the Wiskott-Aldrich syndrome protein (WASP): Hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood 104: 4010–4019 [DOI] [PubMed] [Google Scholar]
- Juty N, Le Novère N, Laibe C 2012. Identifiers.org and MIRIAM Registry: Community resources to provide persistent identification. Nucleic Acids Res 40: D580–D586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerrien S, Orchard S, Montecchi-Palazzi L, Aranda B, Quinn AF, Vinod N, Bader GD, Xenarios I, Wojcik J, Sherman D, et al. 2007. Broadening the horizon—level 2.5 of the HUPO-PSI format for molecular interactions. BMC Biol 5: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohonen-Corish M, Al-Aama J, Auerbach A, Axton M, Barach CI, Bernstein I, Béroud C, Burn J, Cunningham F, Cutting G, et al. 2010. How to catch all those mutations—the report of the third Human Variome Project Meeting, UNESCO Paris, May 2010. Hum Mutat 31: 1374–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar MD, Bava KA, Gromiha MM, Parabakaran P, Kitajima K, Uedaira H, Sarai A 2006. ProTherm and ProNIT: Thermodynamic databases for proteins and protein–nucleic acid interactions. Nucleic Acids Res 34: D204–D206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson PT, Lappalainen I, Bäckesjö CM, Brockmann E, Laurén S, Vihinen M, Smith CIE 2000. Six X-linked agammaglobulinemia-causing missense mutations in the Src homology 2 domain of Bruton's tyrosine kinase: Phosphotyrosine-binding and circular dichroism analysis. J Immunol 164: 4170–4177 [DOI] [PubMed] [Google Scholar]
- Mohd Ismail NI, Yuasa T, Yuasa K, Nambu Y, Nisimoto M, Goto M, Matsuki I, Nagahama M, Tsuji A 2010. A critical role for highly conserved Glu610 residue of oligopeptidase B from Trypanosoma brucei in thermal stability. J Biochem 147: 201–211 [DOI] [PubMed] [Google Scholar]
- Nair PS, Vihinen M 2013. VariBench: A benchmark database for variations. Hum Mutat 34: 42–49 [DOI] [PubMed] [Google Scholar]
- Notarangelo LD, Mazza C, Giliani S, D'Aria C, Gandellini F, Ravelli C, Locatelli MG, Nelson DL, Ochs HD, Notarangelo LD 2002. Missense mutations of the WASP gene cause intermittent X-linked thrombocytopenia. Blood 99: 2268–2269 [DOI] [PubMed] [Google Scholar]
- Ochs HD, Thrasher AJ 2006. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol 117: 725–738 [DOI] [PubMed] [Google Scholar]
- Patrinos GP, van Baal S, Petersen MB, Papadakis MN 2005. Hellenic National Mutation database: A prototype database for mutations leading to inherited disorders in the Hellenic population. Hum Mutat 25: 327–333 [DOI] [PubMed] [Google Scholar]
- Piirilä H, Väliaho J, Vihinen M 2006. Immunodeficiency mutation databases (IDbases). Hum Mutat 27: 1200–1208 [DOI] [PubMed] [Google Scholar]
- Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S 2008. The Human Phenotype Ontology: A tool for annotating and analyzing human hereditary disease. Am J Hum Genet 83: 610–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal RL, Gordon SM, Lush MJ, Wright MW, Bruford EA 2011. genenames.org: The HGNC resources in 2011. Nucleic Acids Res 39: D514–D519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B, Ashburner M, Rosse C, Bard J, Bug W, Ceusters W, Goldberg LJ, Eilbeck K, Ireland A, Mungall CJ, et al. 2007. The OBO Foundry: Coordinated evolution of ontologies to support biomedical data integration. Nat Biotechnol 25: 1251–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Baal S, Kaimakis P, Phommarinh M, Koumbi D, Cuppens H, Riccardino F, Macek M Jr, Scriver CR, Patrinos GP 2007. FINDbase: A relational database recording frequencies of genetic defects leading to inherited disorders worldwide. Nucleic Acids Res 35: D690–D695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen M, den Dunnen JT, Dalgleish R, Cotton RGH 2012. Guidelines for establishing locus specific databases. Hum Mutat 33: 298–305 [DOI] [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE 2008. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat 29: 6–13 [DOI] [PubMed] [Google Scholar]