

Abstract
Introduction:
One sober consequence of the current epidemic of diabetes mellitus is that an increasing number of people world-wide will partially or completely lose their sight to diabetic retinopathy. Clinically, the sight-threatening complications of diabetes are diagnosed and treated based on visible retinal lesions (e.g., dot-blot hemorrhages or retinal neovascularization). However, such anatomical microvascular lesions are slow to respond with treatment. Thus, there remains an urgent need for imaging biomarkers that are abnormal before retinal lesions are visibly apparent and are responsive to treatment.
Areas covered:
Here, the development of new MRI methods, such as manganese-enhanced MRI, for evaluating early diabetes-evoked retinal pathophysiology, and its usefulness in guiding new treatments for diabetic retinopathy are reviewed.
Expert opinion:
In diabetic retinopathy, not all important diagnostic and prognostic needs are well served by optical methods. In the absence of gross anatomy changes, critical times when drug intervention is most likely to be successful at reducing vision loss are missed by most light-based methods and thus provide little help in guiding diagnosis and treatment. For example, before clinical symptoms, is there an optimal time to intervene with drug therapy? Is a drug reaching its target? How does one assess optimal drug dose, schedule, and routes? How well do current experimental models mimic the clinical condition? As discussed herein, MRI is as an analytical tool for addressing these unmet needs. Future clinical applications of MRI can be envisioned such as in clinical trials to assess drug treatment efficacy, or as an adjunct approach to refine or clarify a difficult clinical case. New MRI-generated hypotheses about the pathogenesis of diabetic retinopathy and its treatment are discussed. In the coming years, a substantial growth in the development and application of MRI is expected to address relevant question in both the basic sciences and in the clinic.
Keywords: animal models, calcium channels, diabetes, MRI, retinopathy
There remains an urgent need to prevent vision loss from diabetic retinopathy (DR), a common and significant problem for patients with diabetes. The anatomical vascular lesions of DR can appear years after diabetes becomes well-controlled (i.e., blood glucose levels maintained in the normal range) suggesting a persistent and physiologic lesion well before the vascular histopathology is detectable. The visible retinal lesions of DR form the basis for its diagnosis and treatment evaluation. However, such anatomical lesions do not respond quickly with treatment and so clinical trials looking for beneficial drugs typically need to evaluate large numbers of patients over very long periods of time. The expense of such trials has limited the development and testing of new drug therapies for DR. The availability of imaging biomarkers that are sensitive to early diabetes-induced retinal pathophysiology will allow for more rapid evaluation of treatment benefits and thus greatly reduce overall cost of drug trials while improving drug discovery, optimization of treatment, and therapeutic success in DR.
1. Vascular functional biomarkers and DR
Most efforts to develop useful non-invasive imaging biomarkers have focused on the impact of diabetes on retinal vascular physiology and, in particular, retinal blood flow. However, even after decades of research, there is limited consensus as to diabetes’ influence on retinal blood flow [1-4]. To date, most studies of retinal blood flow are usually performed in the absence of a challenge (i.e., a stress to the retinal circulation). However, normally the retinal circulation is always responding to changing neuronal demands, as well as systemic changes in, for example, blood pressure. Thus, it is hard to know how to interpret measurements of blood flow in the minimally challenged retina. For example, if retinal blood flow decreases and autoregulation remains intact, then the retinal circulation will be able to compensate, at least somewhat, for the reduced perfusion. Autoregulation is a measure of how retinal vascular diameter changes to maintain retinal blood flow. However, studies find, regardless of method, patient population, or type of challenge, that before the appearance of diabetic microangiopathy autoregulation is impaired in the diabetic patient [5]. These studies are usually performed by measuring the adaptability (or functionality) of the retinal circulation to a provocation such as flickering light, breathing oxygen, or changing blood pressure [1,6-9]. Furthermore, in experimental DR, drugs which corrected early diabetes-induced autoregulatory dysfunction also corrected later histopathology [9]. Exactly how autoregulatory dysfunction and histopathology are mechanistically connected remains unclear. Nonetheless, as a biomarker of treatment efficacy, correction of diabetes-induced autoregulatory dysfunction is a promising approach. One possible limitation is that in a clinical setting, it might be difficult to maintain quality control of the provocation experienced by the retinal vasculature in each patient. Thus, efforts continue to identify additional functional biomarkers of early diabetes-evoked retinal pathophysiology that also respond to therapy.
2. Neuroretinal function and DR
Because retinal vascular function and neuronal demands are normally tightly coupled, research efforts have moved from investigating retinal vascular pathophysiology induced by diabetes to identifying neuroretinal problems produced by chronic hyperglycemia. Before the appearance of DR, non-vascular retinal dysfunction is clinically and experimentally evident on visual performance and retinal electrophysiology examinations [10,11]. However, these metrics generally do not have the fine spatial resolution needed to map function panretinaliy, a critical need because diabetic lesions do not appear uniformly across the retina [12]. One approach to addressing this problem is multifocal ERG (mfERG), an electrophysiologic method that can evaluate spatially heterogeneous function across central retina [13]. Applications of mfERG in DR have been promising. For example, clinical studies find that diabetes-evoked mfERG defects are spatially predictive, with high sensitivity (88%) and specificity (84%), of where on the retina microangiography will develop up to a year later [13]. Again, exactly how diabetes-evoked vascular and non-vascular problems are linked remains unknown. While there are effective treatments for DR in rodent models, mfERG has limited use in murine and rat due to the very small rodent eyes’ sensitivity to even small amounts of internally reflected light and presence of diabetic cataract in diabetic rats [14,15]. Because few treatments currently exist for patients with diabetes, it is not clear if new treatments that slow progression of DR also correct the earlier neuroretinal dysfunction on mfERG.
3. MRI metrics and technologies for evaluating experimental DR
There are several abnormalities commonly thought of as characteristic of DR in humans which are robustly mimicked in diabetic rodent models including acellular capillary and pericytes ghost formation, impairment in vessel autoregulation, and deterioration of neuroretinal function [5,7,8]. A standard way to evaluate micrography, which is a late-stage indicator, is with trypsin digest [16]. Optical imaging is useful for evaluating retinal blood vessel appearance in the clinic and these methods are translatable rodent models. On the other hand, there are fewer measures of functional retinal blood flow in diabetic rodents, in part because of the small size of the rodent eye and the appearance of cataract in rat models. MRI methods have been developed which can make high-resolution 3D maps of the ocular circulation and its hemodynamic changes in rodents in vivo [17] although the value of such MR microangiography has not yet been evaluated in diabetic models.
Experimental DR also demonstrates early retinal auto-regulatory defects on oxygen-enhanced MRI [9]. When this defect is corrected by normalization of biochemical abnormalities associated with diabetes, the results are predictive of subsequent treatment efficacy [9]. These results – which take advantage of the physiologic accuracy of MRI and thus are independent of MRI – suggest a new diagnostic: evaluating retinal autoregulation in patients with diabetes for prognostically evaluating drug treatment efficacy.
As for assessing neuroretinal function in rodent models, the most common approach involves electrophysiology (e.g., ERG). However, ERG only measures a response from the entire retina making studies of how dysfunction and vascular histopathology are linked difficult to interpret. Here, another imaging tool, manganese-enhanced MRI (MEMRI), is highlighted because it can map retinal function with even higher spatial resolution than mfERG, is not limited by cataract, is readily performed in rodents, and can monitor early diabetes-induced changes in neuroretinal physiology and their response to treatment (this latter point is discussed in detail below) [18-23]. The focus in the remaining review is on MEMRI because it accurately measures retinal calcium channel activity from awake and conscious animals with extremely high spatial resolution while automatically providing co-localization with retinal structure (retinal thickness).
One disadvantage of all MR methods is accessibility because they can not be performed in a doctor’s lab or office. However, most hospitals have MRI on-site. A major advantage of MRI is that often, several of MR methods can be combined to more fully phenotype disease and treatment efficacy in the same eye over time, something that is a major advance for imaging science of the retina.
4. MEMRI basics
The usual MEMRI procedure is straightforward: animals are overnight dark adapted and then injected, while still in the dark, with manganese chloride (MnCl2, ip). After 4 h of additional darkness, animals are anesthetized and gently placed into the MRI machine. A very high spatial resolution spin-lattice relaxation time (T1 or, less ideally, T1-weighted) is then generated from which the extent of manganese accumulated in each retinal layer can be measured. All of the conditions of the MEMRI experiment are carefully considered. Dark adaptation, the most often used condition, represents a retinal ‘stress test’ since the retina uses 50% of its ATP in the dark to maintain outer segment cyclic guanosine monophosphate (cGMP) channels open [24,25]. After 4 h plasma manganese levels have returned to baseline and good retinal uptake has occurred [26]. Doses of manganese (44 – 66 mg/kg) are not toxic to retinal histology, blood retinal barrier integrity, and electrophysiology [23,27,28].
One common variation of this procedure is to light adapt the animal for 20 – 30 min before manganese injection and continue the light exposure during the next 4 h (until placement in the magnet) [23]. Alternatively, one eye can be patched the day before the experiment followed by overnight dark adaptation and light adaptation procedure [29]. This latter condition effectively produces a dark adapted and light adapted eye in the same animal, thus reducing the number of animals studied by 50%. As an aside, patched animals seem to have a lower uptake of manganese in the unpatched or light adapted eye (relative to unpatched light adapted animals) although more work is needed to better understand how patching one eye influences adaptation of the unpatched retina.
Why manganese? First, manganese is a paramagnetic MRI contrast agent and shares a few properties with another commonly used paramagnetic agent, gadolinium. Like gadolinium, manganese linearly alters water spin-lattice relaxation rates (1/T1) at low concentrations. An FDA-approved manganese-based contrast agent, Teslascan, was originally used as a blood pool agent like gadolinium chelates [26]. Importantly, the manganese in Teslascan is taken up by the retina in a stimulus-dependent manner [26]. Clinical evaluation of MEMRI is in progress but no results have yet been published. Nonetheless, the findings reviewed herein are expected to inform future clinical studies.
Unlike gadolinium, manganese is an essential metal that is critical for a mammal’s health. For this reason, manganese can enter cells via a number of ion channels, such as the divalent metal transporter 1 (DMT1) and zinc transporters [30,31]. However, influx through these ion channels does not appear related to tissue function. Critically, manganese also enters cells via voltage-gated calcium channels, such as L-type calcium channels (LTCCs), which are important calcium channels that regulate fundamental neuronal functions, such as neurotransmitter and melantonin release [32-34].
5. MEMRI and LTCCs
In vitro, Mn2+ uptake is increased both by membrane depolarization (opening LTCCs) and the specific LTCC agonist BayK8644 [35,36]. In vivo studies confirm that Mn2+ uptake is inhibited by specific LTCC antagonists verapamil [37], nifedipine [38], and diltiazem [28]. Here, new data are presented that further highlights that normal photoreceptor uptake of manganese (MEMRI) is regulated by LTCC activity based on studies with systemic pretreatment with either nifedipine (NIF, specific LTCC antagonist) and BAYK8644 (BAY, specific LTCC agonist) in dark (patched Long Evans rat eye) and light (unpatched fellow Long Evans rat eye) adaptation (Figure 1) [38]. MEMRI clearly has the robust sensitivity needed to detect the expected results that the antagonist NIF is effective at closing LTCCs in the dark (when photoreceptor membranes are depolarized and LTCCs are open) whereas the agonist BAY is effective at opening LTCC in the light (when photoreceptor membranes are hyperpolarized and LTCCs are closed).
Figure 1.
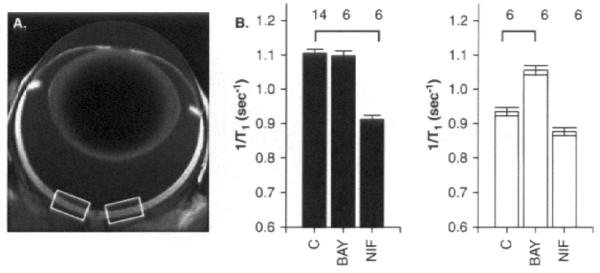
A. Representative MEMRI of an eye of a 200 g Long Evans rat in vivo illustrating central retinal regions-of-interest (white boxes). T1 data acquisition was performed using a 7T Clinscan MRI as previously published [81]. B. Summary of specific LTCC antagonist (nifedipine (NIF), 30 mg/kg dissolved in DMSO) and agonist (BayK8644 (BAY), 2 mg/kg dissolved in saline) on outer retinal manganese uptake in dark adapted (patched, left) and light adapted (unpatched, right) animals [29]. Long Evans rats were patched and prepared as previously described [29,81]. Calcium channel modulators were administered 30 min prior to manganese injection, as in [81]. Non-LTCC antagonist had no effect on retina manganese uptake (data in [38]). Bold black lines indicate a significant (p < 0.05) difference from vehicle control values. Error bars represent standard error of the mean (SEM), and numbers above the bars represent the number of animals studied. No manganese baseline values were 0.6 s−1.
In general, manganese influx can also be modified by other calcium channels, such as transient receptor potential vanilloid (TRPV) [39] and ligand-gated channels in the brain but not apparently in the retina [38,40]. The activity of these calcium channels are closely linked with neuronal activity. Also, Mn2+ efflux from cells is slow, taking days to leave, for example, the retina [26]. Thus, measuring retinal uptake with MEMRI provides an analytical metric of the history of LTCC activity while the animal was awake and freely moving, and thus is a powerful measure assay of a critical modulator of photoreceptor neurotransmitter release, among many other aspects of neuronal activity.
6. MEMRI accurately measures retinal physiology
Initially, sensitivity of MEMRI to normal light/dark adaptation was examined. MEMRI was able to robustly report on the status of photoreceptor LTCCs that are maximally open in the dark and progressively closed with brighter light [23,41]. Also, MEMRI was sensitive to photoreceptor membrane polarization modulation by either visual cycle or sodium-potassium adenosine triphosphatase (Na/K ATPase) activity [21,41]. In contrast to the outer retina, in the inner retina manganese uptake did not change with light and dark adaptation and this likely reflected the roughly equal representation of ON and OFF pathways in the inner retina [23,41]. There were two exceptions to this finding. In much younger mice (postnatal day 7), which do not have functional photoreceptors, inner retinal manganese uptake was significantly greater in the light than in the dark [42]. While initially puzzling, melanopsin was eventually identified as the reason for this difference [42]. In another experiment, an exogenous and intrinisically photosensitive ion channel, called channel rhodopsin, was injected into the inner retina, and again more manganese uptake was found in light than dark exposure [43].
While not part of this application, we note that we have already confirmed that the manganese in an FDA-approved contrast agent (Teslascan) is taken up by the retina in a stimulus-dependant manner [26] and that high resolution MRI of human eyes are possible (Figure 4). Thus, we expect that findings from the proposed research will inform future clinical studies. Together, the above considerations provide strong evidence supporting the physiologic accuracy of MEMRI in the retina.
Figure 4. Representative fine resolution of human eye (courtesy of Drs. S. Lalith Talagala, Daniel Reich and Alan Korestsky (NIH) who also provided much of the following summary).

MRI was performed on a 3.0 T scanner (GE Signa Excite HDx, Waukesha, WI, USA) using a receive-only surface coil (diameter of 40 mm) connected to a head support. The coil was positioned over the right eye, without touching the face. The left eye was patched. The head was slightly tilted to the contra-lateral side. Participants were asked to fixate when necessary on a red tape X positioned on the upper internal surface of the scanner bore. Images were collected using a 2D fast spoiled gradient echo sequence (TR = 16 ms; TE = 3.4 ms; flip angle 25; matrix size 512 × 128; FOV 3.8 × 3.8 cm; slice thickness 2.5 mm; pixel size 75 × 75 μm). 20 separate images, each taking 12 s to acquire while the patient does not blink followed by 5 s of interval where rest and blinking are encouraged, were acquired. These 20 images were acquired on the same location. This cued blinking protocol has been described elsewhere [93]. Following rigid image registration (and removal of images with too much movement artifact), an average image was generated as shown. Top inserts: region of interest of images collected with resolutions of 75 and 58 μm2.
7. MEMRI, ischemia/reperfusion, and a new hypothesis
Previous studies have reported that suppressing calcium ion influx through LTCC is neuroprotective in ischemia/reperfusion studies [44-47]. Thus, the sensitivity of MEMRI to ischemia/reperfusion was tested, an injury that also causes diabetes-like microangiography in non-diabetic rats [48,49]. Using this paradigm, a strikingly significant subnormal uptake of manganese before the appearance of retinal degeneration (24 h of reperfusion) was noted [48]. At 7 days of reperfusion, there was substantial retinal thinning and, surprisingly, manganese uptake had recovered to normal levels [48]. This raised the question: which event (the early subnormal manganese uptake or the later apparent recovery) was linked with retinal histopathology? To begin to address this question, a powerful preconditioning procedure known as repetitive hypoxic preconditioning (RHP) was used because it largely prevents retinal histopathology following ischemia/reperfusion [50]. In control rats (i.e., not exposed to ischemia/reperfusion), RHP treatment alone resulted in subnormal manganese accumulation [48]. Furthermore, in rats that had undergone ischemia/reperfusion, RHP prevented both the recovery in manganese uptake at 7 days of reperfusion as well as retinal degeneration [48]. Thus, the dramatic subnormal manganese uptake found immediately following ischemia/reperfusion appeared neuroprotective while the apparent recovery likely contributed to the retinal morbidity. Intraperitoneal injections of nifedipine (a specific LTCC blocker) protected rats from ischemia-induced retinal degeneration damage [44]. These considerations suggested the hypothesis that therapies which prevent or prolong subnormal manganese uptake will be beneficial against ischemia/reperfusion (and possibly diabetes, see below).
Here, new data are presented which begin to test this hypothesis (Figure 2). Angiotensin-converting enzyme inhibitors (ACEi), such as captopril and enalapril, and angiotensin II subtype 1 receptor antagonist losartan, have all been found to reduce ischemia/reperfusion damage in heart, liver, and lung [51-54]. While the exact mechanism(s) of such protection remains somewhat unclear, there is strong evidence that ACEis, or receptor antagonist, can potentiate/mimic preconditioning, at least in the heart, and this is one reason their clinical benefit is currently being evaluated [53-57]. For the present purposes, it is intriguing that ACEi have also been found to be effective at reducing calcium currents via LTCCs in heart cells and tissue, apparently via a direct interaction with the outer surface of the channel [58-68]. In rat retina, Fukuda et al. reported that either ACEi (captopril) or an angiotensin II type 1 receptor blocker, administered acutely prior to an ischemia–reperfusion insult, attenuated the later histologic damage [69]. Thus, it is possible that captopril, enalapril, or an angiotensin II type 1 receptor antagonist losartan would significantly reduce retinal uptake of manganese in control animals as might be expected if they were inducing a preconditioned-like state (i.e., channel closure). To test this, dark adapted non-diabetic age- and strain-matched controls were compared to rats treated with either captopril (2 mo Lewis rats, ~ 25 mg/kg body weight/day, drinking water [70]) for 5 days before MEMRI study), enalapril (6 mo Sprague–Dawley rats, ~ 6 mg/kg/day, drinking water) for 1 week prior to MEMRI examination, or losartan (2 mo Sprague–Dawley rats, ~ 20 mg/kg/day, drinking water). A suppression of retinal LTCC activity by these drugs in vivo was found for the first time (Figure 2). This antagonism did not seem related to systemic effects on blood pressure (data not shown, manuscript submitted). Although captopril and preconditioning have antioxidant properties, and this could explain some of their beneficial effects, many ACEi are not antioxidants [61,64,65,71-75]. Overall, while the beneficial actions of ACEi and angiotensin II type 1 receptor blockers appear to be multifaceted, the above considerations support the hypothesis that therapies which prevent or extend subnormal manganese uptake effect may also contribute to a beneficial outcome.
Figure 2. ACEi and angiotensin II subtype 1 receptor antagonist suppress retinal manganese uptake, particularly in the outer retina.

Central (−0.4 – 1 mm from the optic nerve head) retinal manganese uptake (i.e., (1/T1)) profiles were compared between non-treated non-diabetic control rats (filled black circles) and non-diabetic control rats (open square treated with (A) captopril (B) enalapril, or (C) losartan. T1 data acquisition was performed using a 7T Clinscan MRI as previously published [81]. Other details are in text. Data were analyzed as previously described and shown here as a function of distance from the retina/non-retina borders, where 0% is the vitreous/retina border and 100% is the retina/choroid border [81]. Regions near borders were not included in the analysis because these regions likely include some signal from outside of the retina. For those areas tested, lines above profiles indicate significant differences in manganese uptake as indicated (p < 0.05, two-tailed test (bold black lines); details on statistical analysis can be found elsewhere) [81].
8. MEMRI, DR, and oxidative stress
Next, MEMRI was used to study experimental DR. Diabetic dark adapted rats and mice, within a couple of months after conversion to chronic hyperglycemia, both demonstrated subnormal retinal manganese uptake [20,22]. Interestingly, at longer durations of diabetes (and at increased age), uptake in diabetic mice retina dramatically recovered to normal [20]. Here, new data are presented (Figure 3) which demonstrate that this dramatic recovery was not limited to diabetic mice and also occurs in 9 mo diabetic rats. Both diabetic mouse and rat results have features that mirror those from ischemia/reperfusion.
Figure 3. Summary of natural history of manganese uptake for dark adapted inner retina (left panel) and outer retina (right panel) of diabetic Sprague–Dawley rats.

Error bars are SEM. The y-axis scale starts at a signal intensity of 50 (arb. units) because this is the pre-manganese baseline level determined from rats not injected with manganese (data not shown). T1 weighted data acquisition was performed using a 4.7T MRI as previously published [41]. Data were analyzed as previously described [41]. Numbers over bars are numbers of animals studied. Details on statistical analysis can be found elsewhere [41].
*p < 0.05 from 100%.
It is established that retinal oxidative stress is a major contributor to the pathogenesis of early diabetes-related changes in retinal microvascularture (i.e., acellular capillaries and pericytes ghosts) [5] and antioxidants are effective at preventing the development of DR in diabetic models. Furthermore, oxidative stress alone alters/suppresses retinal and brain LTCC activity [76,77]. Thus, it is possible that preventative antioxidant therapy could stop the development of the early subnormal retinal uptake seen on MEMRI. In diabetic rats, treatment with a potent antioxidant, α-lipoic acid, prevented subnormal intraretinal uptake [22]. Also, diabetic superoxide dismutase overexpressor mice (SODIOE), unlike diabetic wildtype mice, did not develop subnormal retinal manganese uptake [20]. Consistent with the hypothesis that preventing subnormal uptake is beneficial, treatment with α-lipoic acid or SODIOE prevented later DR [20,22]. Recently, another factor was found to contribute to subnormal manganese uptake in the outer retina. Acute treatment with 11-cis-retinal significantly corrected only outer retinal uptake, although not to control levels [18]. 11-cis-retinal is a key participant of the visual cycle, but also has other concentration-dependent actions such as on gap junctions [78-80]. Studies are on-going to better define the mechanism(s) underlying its beneficial action on impaired outer retinal channel activity.
The apparent recovery of manganese uptake in the diabetic rodents might be due to changes in retinal calcium channels with increased age. To address this question, the relationship between MEMRI and age was characterized in non-diabetic rats [81]. Indeed, a significant age-related supernormal accumulation of manganese was found. This increased uptake was important because it predicted later visual contrast sensitivity declines. A likely explanation for the supernormal uptake was an age-related increase in retinal expression of a drug-insensitive LTCC subtype α1D, but not α1c [81]. As a side note, it is possible that the results from the earlier ischemia/reperfusion study might be a consequence of change in LTCC subtype which contributed to the apparent recovery in retinal manganese uptake after a 1 week of reperfusion in the ischemia model. Interestingly, treatments which suppress manganese uptake in control rodents (e.g., ACEi, preconditioning (see above)) have been found to inhibit or prevent the appearance of DR [70,82-87]. These considerations raise the possibility that the hypothesis that therapies which prevent or prolong subnormal manganese uptake will be beneficial against ischemia/reperfusion may also apply to diabetes. Furthermore, it is possible that alterations in LTCC subtype induced by diabetes contribute to both the apparent recovery in older diabetic rodents and the initial subnormal uptake. These questions are actively being pursued in the lab. In any event, MEMRI appears as a sensitive test of early abnormalities in retinal physiology in diabetic models, as well as treatment efficacy (also see Section 3).
9. MEMRI and retinal neovascularization
A late-stage retinal complication of diabetes that is closely associated with blindness is an abnormal growth of new blood vessels from the retinal circulation into the vitreous (or neovascularization (NV)) which can lead to a retinal detachment. Unfortunately diabetic rodent models do not develop retinal NV [5]. Nonetheless, there are non-diabetic rodent models that do develop retinal NV [88,89]. Of the two generally used models, the rat retinal NV model of John Penn is preferred because it offers dial-in control over the severity and incidence of retinal NV produced and does not involve vaso-obliteration with associated inflammation like other NV models [28,90-92]. When the rat NV model was examined with MEMRI, a striking supernormal uptake of manganese was noted prior to the appearance of retinal NV [28,38]. In fact, during the appearance of NV, different rat strains exhibited dissimilar MEMRI time course and NV regression patterns, with the more prolonged supernormal retinal uptake occurring in retinas with greater NV severity [28,38]. Furthermore, calcium channel blocker treatment administered before the appearance of retinal NV partially inhibited NV severity [38]. In support of these findings, another group has recently reported similar benefits of calcium therapy on retinal NV in the mouse model (ARVO 2013, abstract #5614 - B0137).
10. Conclusion
The above considerations highlight MRI in general and MEMRI in particular as useful biomarker/discovery tools of retinal morbidity and treatment efficacy, and strongly support a link between neuronal dysfunction and the appearance of vascular lesions.
11. Expert opinion
As the world-wide population ages, and the incidence and severity of metabolic disorders such as obesity and diabetes increases, retinal disease continues to be a major public health problem. There is a clear and pressing need for better ways to prevent vision loss and blindness. Imaging is likely to be the best approach for addressing this need in patients and in experimental models by highlighting new biomarkers of disease progression and analytically evaluating drug treatment efficacy. By far, optical imaging remains the primary modality in part because such methods can be performed in a doctor’s office and can time-efficiently screen patients. Yet, moving forward, not all important diagnostic and prognostic needs are well served by optical methods. For example, in the absence of gross anatomy changes, a critical time point when drug intervention is most likely to be successful at reducing vision loss, most light-based methods provide little help in guiding diagnosis and treatment. Thus, urgent questions remain: before clinical symptoms, is there an optimal time to intervene with drug therapy? Is a drug reaching its target? How does one assess optimal drug dose, schedule, and routes? How well do current experimental models mimic the clinical condition?
New imaging methods are clearly needed to address these unmet needs and this has prompted a growing interest in MRI. As discussed herein, MRI is an analytical tool for hypothesis testing in animal models. In other words, MRI is a powerful discovery tool and not well suited for screening. However, future niche clinical applications of MRI can be envisioned such as in clinical trials to assess drug treatment efficacy, or as an adjunct approach to refine or clarify a difficult clinical case.
Here, a new type of MRI examination, MEMRI, is discussed as a particularly powerful, but currently underutilized, high spatial resolution imaging method for analytical assessment of functional retinal cell biology and anatomy in vivo. Applications to date of MEMRI to DR have already advanced understanding of a previously unappreciated role of LTCCs in DR. Such data have motivated a new line of scientific inquiry into the problem of DR and suggest new therapeutic strategies for ameliorating vision loss in this disorder. For example, information derived from the proposed research would motivate the investigation of the efficacy of Cav-specific therapies in treating the long-term vascular histopathologic lesions that are the hallmark of DR. Another focus of future work will be to determine exactly how diabetes impacts the different LTCC subtype channels and leads to oxidative stress via, for example, mitochondrial and NADPH oxidase mechanisms. In addition, it is possible to envision performing future MEMRI studies to non-invasively evaluate retinal LTCC activity in select groups of patients with diabetes treated with subtype-specific LTCC antagonists. Clinical MEMRI studies are nearing reality and are expected to be a major advance for DR and other retinopathies in terms of testing mechanistic hypotheses and evaluating treatment efficacy. Currently, fine resolution images, similar to those in rodents, but in humans without sedation is possible using a cued-blinking procedure (Figure 4) [93]. In the coming years, a substantial growth in the development and application of MRI is expected to address relevant question in both the basic sciences and in the clinic.
Article highlights.
Retinal MRI represents an important advance in imaging of the retina by providing a unique and high spatial resolution combination of co-localized functional and anatomical metrics of retinal physiology and pathophysiology.
MRI of the retina fills an unaddressed need for discovering new biomarkers of disease progression and for evaluating therapeutic benefits before the appearance of gross lesions by reporting on whether or not a treatment was active in the retina, and what combination of dose/timing optimizes therapeutic benefit.
MRI studies have generated new and testable hypotheses with regard to treating DR including the ideas that therapies which correct early subnormal autoregulation will also prevent the develop of retinal histopathology in diabetes, that preventing or prolonging subnormal manganese uptake will be protective against the damaging effects of chronic hyperglycemia on the retina, and that treatments which target photoreceptor L-type voltage gated calcium channels can improve diabetes-induced visual performance impairments.
This box summarizes key points contained in the article
Acknowledgements
Supported by NIH EY018109 (BAB), Juvenile Diabetes Research Foundation (BAB), NIH AG034752 (DB), Wayne State University School of Medicine MD/PhD program (DB), and an unrestricted grant from Research to Prevent Blindness (Kresge Eye Institute).
Footnotes
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Bibliography
- Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Kiss B, Polska E, Dorner G, et al. Retinal blood flow during hyperoxia in humans revisited: concerted results using different measurement techniques. Microvasc Res. 2002;64:75–85. doi: 10.1006/mvre.2002.2402. [DOI] [PubMed] [Google Scholar]
- 2.Higashi S, Clermont AC, Dhir V, Bursell SE. Reversibility of retinal flow abnormalities is disease-duration dependent in diabetic rats. Diabetes. 1998;47:653–9. doi: 10.2337/diabetes.47.4.653. [DOI] [PubMed] [Google Scholar]
- 3.Konno S, Feke GT, Yoshida A, et al. Retinal blood flow changes in type I diabetes – a long-term, follow-up study. Invest Ophthalmol Vis Sci. 1996;37:1140–8. [PubMed] [Google Scholar]
- 4.Bursell SE, Clermont AC, Oren B, King GL. The in vivo effect of endothelins on retinal circulation in nondiabetic and diabetic rats. Invest Ophthalmol Vis Sci. 1995;36:596–607. [PubMed] [Google Scholar]
- 5.Kern TS, Tang J, Berkowitz BA. Validation of structural and functional lesions of diabetic retinopathy in mice. Mol Vis. 2010;16:2121–31. [PMC free article] [PubMed] [Google Scholar]
- •.Discusses key lesions associated with diabetic retinopathy in patients and in experimental models.
- 6.Guan K, Hudson C, Wong T, et al. Retinal hemodynamics in early diabetic macular edema. Diabetes. 2006;55:813–18. doi: 10.2337/diabetes.55.03.06.db05-0937. [DOI] [PubMed] [Google Scholar]
- 7.Lorenzi M, Feke GT, Pitler L, et al. Defective myogenic response to posture change in retinal vessels of well-controlled type 1 diabetic patients with no retinopathy. Invest Ophthalmol Vis Sci. 2010;51:6770–5. doi: 10.1167/iovs.10-5785. [DOI] [PubMed] [Google Scholar]
- •.Discusses the importance of detecting impaired autoregulation before the appearance of diabetic retinopathy.
- 8.Lorenzi M, Feke GT, Cagliero E, et al. Retinal haemodynamics in individuals with well-controlled type 1 diabetes. Diabetologia. 2008;51:361–4. doi: 10.1007/s00125-007-0872-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berkowitz BA, Roberts R. Prognostic MRI biomarkers of treatment efficacy for retinopathy. NMR Biomed. 2008;21:957–67. doi: 10.1002/nbm.1303. [DOI] [PubMed] [Google Scholar]
- •.Review of several different MRI methods for the retina.
- 10.Trick GL, Burde RM, Gordon MO, et al. The relationship between hue discrimination and contrast sensitivity deficits in patients with diabetes mellitus. Ophthalmology. 1988;95:693–8. doi: 10.1016/s0161-6420(88)33125-8. [DOI] [PubMed] [Google Scholar]
- •.Discusses the importance of detecting impaired neuroretinal physiology before the appearance of diabetic retinopathy.
- 11.Jackson G, Barber A. Visual dysfunction associated with diabetic retinopathy. Curr Diab Rep. 2010;10:380–4. doi: 10.1007/s11892-010-0132-4. [DOI] [PubMed] [Google Scholar]
- 12.Tang J, Mohr S, Du YD, Kern TS. Non-uniform distribution of lesions and biochemical abnormalities within the retina of diabetic humans. Curr Eye Res. 2003;27:7–13. doi: 10.1076/ceyr.27.2.7.15455. [DOI] [PubMed] [Google Scholar]
- 13.Adams AJ, Bearse MA., Jr Retinal neuropathy precedes vasculopathy in diabetes: a function-based opportunity for early treatment intervention? Clin Exp Optom. 2012;95:256–65. doi: 10.1111/j.1444-0938.2012.00733.x. [DOI] [PubMed] [Google Scholar]
- •.Discusses the importance of detecting impaired neuroretinal physiology before the appearance of diabetic retinopathy.
- 14.Ball SL, Petry HM. Noninvasive assessment of retinal function in rats using multifocal electroretinography. Invest Ophthalmol Vis Sei. 2000;41:610–17. [PubMed] [Google Scholar]
- 15.Nusinowitz S, Ridder WH, Heckenlively JR. Rod multifocal electroretinograms in mice. Invest Ophthalmol Vis Sei. 1999;40:2848–58. [PubMed] [Google Scholar]
- 16.Cogan DG, Kuwabara T. Comparison of retinal and cerebral vasculature in trypsin digest preparations. Br J Ophthalmol. 1984;68:10–12. doi: 10.1136/bjo.68.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shih YY, Muir ER, Li G, et al. High-resolution 3D MR microangiography of the rat ocular circulation. Radiology. 2012;264:234–41. doi: 10.1148/radiol.12112033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berkowitz BA, Bissig D, Patel P, et al. Acute systemic 11-cis-retinal intervention improves abnormal outer retinal ion channel closure in diabetic mice. Mol Vis. 2012;18:372–6. [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, McClellan ME, Tanito M, et al. Loss of caveolin-1 impairs retinal function due to disturbance of subretinal microenvironment. J Biol Chem. 2012;287:16424–34. doi: 10.1074/jbc.M112.353763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berkowitz BA, Gradianu M, Bissig D, et al. Retinal ion regulation in a mouse model of diabetic retinopathy: natural history and the effect of Cu/Zn superoxide dismutase overexpression. Invest Ophthalmol Vis Sei. 2009;50:2351–8. doi: 10.1167/iovs.08-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •.Discusses the importance of evaluating antioxidative stress using MEMRI.
- 21.Berkowitz BA, Roberts R, Luan H, et al. Manganese-enhanced MRI studies of alterations of intraretinal ion demand in models of ocular injury. Invest Ophthalmol Vis Sei. 2007;48:3796–804. doi: 10.1167/iovs.06-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berkowitz BA, Roberts R, Stemmler A, et al. Impaired apparent ion demand in experimental diabetic retinopathy: correction by lipoic acid. Invest Ophthalmol Vis Sci. 2007;48:4753–8. doi: 10.1167/iovs.07-0433. [DOI] [PubMed] [Google Scholar]
- •.Discusses the importance of evaluating antioxidative stress using MEMRI.
- 23.Berkowitz BA, Roberts R, Goebel DJ, Luan H. Noninvasive and simultaneous imaging of layer-specific retinal functional adaptation by manganese-enhanced MRI. Invest Ophthalmol Vis Sci. 2006;47:2668–74. doi: 10.1167/iovs.05-1588. [DOI] [PubMed] [Google Scholar]
- ••.First description of MEMRI.
- 24.Ames A, III, Li YY, Heher EC, Kimble CR. Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. J Neurosci. 1992;12:840–53. doi: 10.1523/JNEUROSCI.12-03-00840.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linton JD, Holzhausen LC, Babai N, et al. Flow of energy in the outer retina in darkness and in light. PNAS. 2010;107:8599–604. doi: 10.1073/pnas.1002471107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tofts PS, Porchia A, Jin Y, et al. Toward clinical application of manganese-enhanced MRI of retinal function. Brain Res Bull. 2010;81:333–8. doi: 10.1016/j.brainresbull.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •.Discusses the potential application of MEMRI in patients.
- 27.Berkowitz BA, Roberts R, Luan H, et al. Manganese-enhanced MRI studies of alterations of intraretinal ion demand in models of ocular injury. Invest Ophthalmol Vis Sci. 2007;48:3796–804. doi: 10.1167/iovs.06-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berkowitz BA, Roberts R, Penn JS, Gradianu M. High-resolution manganese-enhanced MRI of experimental retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2007;48:4733–40. doi: 10.1167/iovs.06-1516. [DOI] [PubMed] [Google Scholar]
- 29.Bissig D, Berkowitz BA. Same-session functional assessment of rat retina and brain with mangancsc-enhanced MRI. Neuroimage. 2011;58:749–60. doi: 10.1016/j.neuroimage.2011.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Au C, Benedetto A, Aschner M. Manganese transport in eukaryotes: the role of DMT1. Neurotoxicology. 2008;29:569–76. doi: 10.1016/j.neuro.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang CY, Jenkitkasemwong S, Duarte S, et al. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J Biol Chem. 2012;287:34032–43. doi: 10.1074/jbc.M112.367284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmitz Y, Witkovsky P. Dependence of photoreceptor glutamate release on a dihydropyridine-sensitive calcium channel. Neuroscience. 1997;78:1209–16. doi: 10.1016/s0306-4522(96)00678-1. [DOI] [PubMed] [Google Scholar]
- 33.Nachman-Clewner M, Jules R, Townes-Anderson E. L-type calcium channels in the photoreceptor ribbon synapse: localization and role in plasticity. J Comp Neurol. 1999;415:1–16. [PubMed] [Google Scholar]
- 34.Avendano G, Butler BJ, Michael Iuvone P. K+-evoked depolarization induces serotonin N-acetyltransferase activity in photoreceptor-enriched retinal cell cultures. Involvement of calcium influx through l-type channels. Neurochem Int. 1990;17:117–26. doi: 10.1016/0197-0186(90)90075-5. [DOI] [PubMed] [Google Scholar]
- 35.Drapeau P, Nachshen DA. Manganese fluxes and manganese-dependent neurotransmitter release in presynaptic nerve endings isolated from rat brain. J Physiol. 1984;348:493–510. doi: 10.1113/jphysiol.1984.sp015121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlson RO, Masco D, Brooker G, Spiegel S. Endogenous ganglioside GM1 modulates L-type calcium channel activity in N18 neuroblastoma cells. J Neurosci. 1994;14:2272–81. doi: 10.1523/JNEUROSCI.14-04-02272.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cross DJ, Flexman JA, Anzai Y, et al. In vivo manganese MR imaging of calcium influx in spontaneous rat pituitary adenoma. AJNR Am J Neuroradiol. 2007;28:1865–71. doi: 10.3174/ajnr.A0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berkowitz BA, Bissig D, Bergman D, et al. Intraretinal calcium channels and retinal morbidity in experimental retinopathy of prematurity. Mol Vis. 2011;17:2516–26. [PMC free article] [PubMed] [Google Scholar]
- 39.Pritschow B, Lange T, Kasch J, et al. Functional TRPV4 channels are expressed in mouse skeletal muscle and can modulate resting Ca2+ influx and muscle fatigue. Pflugers Arch Eur J Physiol. 2011;461:115–22. doi: 10.1007/s00424-010-0883-4. [DOI] [PubMed] [Google Scholar]
- 40.Itoh K, Sakata M, Watanabe M, et al. The entry of manganese ions into the brain is accelerated by the activation of N-methyl-d-aspartate receptors. Neuroscience. 2008;154:732–40. doi: 10.1016/j.neuroscience.2008.03.080. [DOI] [PubMed] [Google Scholar]
- 41.Berkowitz BA, Roberts R, Oleske DA, et al. Quantitative mapping of ion channel regulation by visual cycle activity in rodent photoreceptors in vivo. Invest Ophthalmol Vis Sci. 2009:1880–5. doi: 10.1167/iovs.08-2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berkowitz BA, Roberts R, Bissig D. Light-dependant intraretinal ion regulation by melanopsin in young awake and free moving mice evaluated with manganese-enhanced MRI. Mol Vis. 2010;16:1776–80. [PMC free article] [PubMed] [Google Scholar]
- 43.Ivanova E, Roberts R, Bissig D, et al. Retinal channelrhodopsin-2-mediated activity in vivo evaluated with manganese-enhanced magnetic resonance imaging. Mol Vis. 2010;16:1059–67. [PMC free article] [PubMed] [Google Scholar]
- 44.Crosson CE, Willis JA, Potter DE. Effect of the calcium antagonist, nifedipine, on ischemic retinal dysfunction. J Ocul Pharmacol. 1990;6:293–9. doi: 10.1089/jop.1990.6.293. [DOI] [PubMed] [Google Scholar]
- 45.Toriu N, Akaike A, Yasuyoshi H, et al. Lomerizine, a Ca2+ channel blocker, reduces glutamate-induced neurotoxicity and ischemia/reperfusion damage in rat retina. Exp Eye Res. 2000;70:475–84. doi: 10.1006/exer.1999.0809. [DOI] [PubMed] [Google Scholar]
- 46.Hirooka K, Kelly MEM, Baldridge WH, Barnes S. Suppressive actions of betaxolol on ionic currents in retinal ganglion cells may explain its neuroprotective effects. Exp Eye Res. 2000;70:611–21. doi: 10.1006/exer.2000.0822. [DOI] [PubMed] [Google Scholar]
- 47.Massote P, Pinheiro A, Fonseca C, et al. Protective Effect of retinal ischemia by blockers of voltage-dependent calcium channels and intracellular calcium stores. Cell Mol Neurobiol. 2008;28:847–56. doi: 10.1007/s10571-007-9243-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berkowitz BA, Gradianu M, Schafer S, et al. Ionic dysregulatory phenotyping of pathologic retinal thinning with manganese-enhanced MRI. Invest Ophthalmol Vis Sci. 2008;49:3178–84. doi: 10.1167/iovs.08-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng L, Gong B, Hatala DA, Kern TS. Retinal Ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest Ophthalmol Vis Sci. 2007;48:361–7. doi: 10.1167/iovs.06-0510. [DOI] [PubMed] [Google Scholar]
- 50.Zhu Y, Zhang Y, Ojwang BA, et al. Long-term tolerance to retinal ischemia by repetitive hypoxic preconditioning: role of HIF-lalpha and heme oxygenase-1. Invest Ophthalmol Vis Sci. 2007;48:1735–43. doi: 10.1167/iovs.06-1037. [DOI] [PubMed] [Google Scholar]
- •.Discusses the importance of a new preconditioning protocol.
- 51.Anthuber M, Farkas S, Rihl M, et al. Angiotensin-converting enzyme inhibition by enalapril: a novel approach to reduce ischemia/reperfusion damage after experimental liver transplantation. Hepatology. 1997;25:648–51. doi: 10.1002/hep.510250326. [DOI] [PubMed] [Google Scholar]
- •.Discusses the importance of ACEi-induced preconditioning.
- 52.Fischer S, MacLean AA, Liu M, et al. Inhibition of angiotensin-converting enzyme by captopril: a novel approach to reduce ischemia-reperfusion injury after lung transplantation. J Thorac Cardiovasc Surg. 2000;120:573–80. doi: 10.1067/mtc.2000.107828. [DOI] [PubMed] [Google Scholar]
- 53.Flynn JD, Akers WS. Effects of the Angiotensin II subtype 1 receptor antagonist losartan on functional recovery of isolated rat hearts undergoing global myocardial ischemia-reperfusion. Pharmacotherapy. 2003;23:1401–10. doi: 10.1592/phco.23.14.1401.31947. [DOI] [PubMed] [Google Scholar]
- 54.Messadi-Laribi E, Griol-Charhbili V, Gaies E, et al. Cardioprotection and kallikreingçôkinin system in acute myocardial ischaemia in mice. Clin Exp Pharmacol Physiol. 2008;35:489–93. doi: 10.1111/j.1440-1681.2008.04902.x. [DOI] [PubMed] [Google Scholar]
- 55.Køber L, Torp-Pedersen C, Carlsen JE, et al. A clinical trial of the angiotensin-convertingg-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 1995;333:1670–6. doi: 10.1056/NEJM199512213332503. [DOI] [PubMed] [Google Scholar]
- 56.Morris SD, Yellon DM. Angiotensin-converting enzyme inhibitors potentiate preconditioning through bradykinin B2 receptor activation in human heart. J Am Coll Cardiol. 1997;29:1599–606. doi: 10.1016/s0735-1097(97)00087-9. [DOI] [PubMed] [Google Scholar]
- 57.Leesar MA, Jneid H, Tang XL, Bolli R. Pretreatment with intracoronary enalaprilat protects human myocardium during percutaneous coronary angioplasty. J Am Coll Cardiol. 2007;49:1607–10. doi: 10.1016/j.jacc.2007.01.060. [DOI] [PubMed] [Google Scholar]
- 58.Alvin Z, Laurence GG, Coleman BR, et al. Regulation of L-type inward calcium channel activity by captopril and angiotensin II via the phosphatidyl inositol 3-kinase pathway in cardiomyocytes from volume-overload hypertrophied rat hearts. Can J Physiol Pharmacol. 2011;89:206–15. doi: 10.1139/Y11-011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •.Discusses the importance of ACEi and LTCC.
- 59.Bryant SM, Ryder KO, Hart G. Effects of captopril on membrane current and contraction in single ventricular myocytes from guinea-pig. Br J Pharmacol. 1991;102:462–6. doi: 10.1111/j.1476-5381.1991.tb12195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J, Zhang L, Qi JH, et al. Effects of captopril and enalaprilat on intracellular Ca2+ content in isolated cardiomyocytes from rats. Zhongguo Yao Li Xue Bao. 1996;17:233–5. [PubMed] [Google Scholar]
- 61.Bartosz M, Kedziora J, Bartosz G. Antioxidant and prooxidant properties of captopril and enalapril. Free Radic Biol Med. 1997;23:729–35. doi: 10.1016/s0891-5849(97)00014-2. [DOI] [PubMed] [Google Scholar]
- 62.de Cavanagh EM, Fraga CG, Ferder L, Inserra F. Enalapril and captopril enhance antioxidant defenses in mouse tissues. Am J Physiol. 1997;272:R514–18. doi: 10.1152/ajpregu.1997.272.2.R514. [DOI] [PubMed] [Google Scholar]
- 63.Scribner AW, Loscalzo J, Napoli C. The effect of angiotensin-converting enzyme inhibition on endothelial function and oxidant stress. Eur J Pharmacol. 2003;482:95–9. doi: 10.1016/j.ejphar.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 64.Sugimoto KI, Tsuruoka S, Fujimura A. Effect of enalapril on diabetic nephropathy in oletf rats: the role of an anti-oxidative action in its protective properties. Clin Exp Pharmacol Physiol. 2001;28:826–30. doi: 10.1046/j.1440-1681.2001.03530.x. [DOI] [PubMed] [Google Scholar]
- 65.Kedziora-Kornatowska K. Effect of angiotensin convertase inhibitors and AT1 angiotensin receptor antagonists on the development of oxidative stress in the kidney of diabetic rats. Clin Chim Acta. 1999;287:19–27. doi: 10.1016/s0009-8981(99)00115-1. [DOI] [PubMed] [Google Scholar]
- 66.Yamasaki T, Kawahara M, Akiyama Y, et al. Effect of enalaprilat, an angiotensin converting enzyme inhibitor, on the membrane potential of cultured neuroblastoma-glioma hybrid NG108-15 cells. No To Shinkei. 1993;45:1039–44. [PubMed] [Google Scholar]
- 67.De Mello WC, Cherry RC, Manivannan S. Electrophysiologic and morphologic abnormalities in the failing heart: effect of enalapril on the electrical properties. J Card Fail. 1997;3:53–61. doi: 10.1016/s1071-9164(97)90008-7. [DOI] [PubMed] [Google Scholar]
- 68.De Mello WC. Electrical activity of the heart and angiotensin-converting enzyme inhibitors on the hyperpolarising action of enalapril. J Hum Hypertens. 2002;16(Suppl 1):S89–92. doi: 10.1038/sj.jhh.1001351. [DOI] [PubMed] [Google Scholar]
- 69.Fukuda K, Hirooka K, Mizote M, et al. Neuroprotection against retinal ischemia-reperfusion injury by blocking the angiotensin II type 1 receptor. Invest Ophthalmol Vis Sci. 2010;51:3629–38. doi: 10.1167/iovs.09-4107. [DOI] [PubMed] [Google Scholar]
- 70.Zhang JZ, Xi X, Gao L, Kern TS. Captopril inhibits capillary degeneration in the early stages of diabetic retinopathy. Curr Eye Res. 2007;32:883–9. doi: 10.1080/02713680701584123. [DOI] [PubMed] [Google Scholar]
- 71.Peng PH, Huang HS, Lee YJ, et al. Novel role for the delta-opioid receptor in hypoxic preconditioning in rat retinas. J Neurochem. 2009;108:741–54. doi: 10.1111/j.1471-4159.2008.05807.x. [DOI] [PubMed] [Google Scholar]
- 72.Benzie IF, Tomlinson B. Antioxidant power of angiotensin-converting enzyme inhibitors in vitro. Br J Clin Pharmacol. 1998;45:168–9. doi: 10.1046/j.1365-2125.1998.00664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De NF, D’Armiento FP, Somma P, et al. Chronic treatment with sulfhydryl angiotensin-converting enzyme inhibitors reduce susceptibility of plasma LDL to in vitro oxidation, formation of oxidation-specific epitopes in the arterial wall, and atherogenesis in apolipoprotein E knockout mice. Int J Cardiol. 2001;81:107–15. doi: 10.1016/s0167-5273(01)00542-3. [DOI] [PubMed] [Google Scholar]
- 74.Goldschmidt JE, Tallarida RJ. Pharmacological evidence that captopril possesses an endothelium-mediated component of vasodilation: effect of sulfhydryl groups on endothelium-derived relaxing factor. J Pharmacol Exp Ther. 1991;257:1136–45. [PubMed] [Google Scholar]
- 75.de Cavanagh EMV, Inserra F, Toblli J, et al. Enalapril attenuates oxidative stress in diabetic rats. Hypertension. 2001;38:1130–6. doi: 10.1161/hy1101.092845. [DOI] [PubMed] [Google Scholar]
- 76.Agostinho P, Duarte CB, Carvalho AP, Oliveira CR. Oxidative stress affects the selective ion permeability of voltage-sensitive Ca2+ channels in cultured retinal cells. Neurosci Res. 1997;27:323–34. doi: 10.1016/s0168-0102(96)01165-0. [DOI] [PubMed] [Google Scholar]
- 77.Shirotani K, Katsura M, Higo A, et al. Suppression of Ca2+ influx through L-type voltage-dependent calcium channels by hydroxyl radical in mouse cerebral cortical neurons. Brain Res Mol Brain Res. 2001;92:12–18. doi: 10.1016/s0169-328x(01)00128-0. [DOI] [PubMed] [Google Scholar]
- ••.Discusses the importance of oxidative stress on LTCC activity.
- 78.Pulukuri S, Sitaramayya A. Retinaldehyde, a potent inhibitor of gap junctional intercellular communication. Cell Commun Adhes. 2004;11:25–33. doi: 10.1080/15419060490471784. [DOI] [PubMed] [Google Scholar]
- 79.Long AC, Bomser JA, Grzybowski DM, Chandler HL. All-trans retinoic acid regulates cx43 expression, gap junction communication and differentiation in primary lens epithelial cells. Curr Eye Res. 2010;35:670–9. doi: 10.3109/02713681003770746. [DOI] [PubMed] [Google Scholar]
- 80.Zhang DQ, McMahon DG. Direct gating by retinoic acid of retinal electrical synapses. Proc Natl Acad Sci USA. 2000;97:14754–9. doi: 10.1073/pnas.010325897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bissig D, Goebel D, Berkowitz BA. Diminished vision in healthy aging is associated with increased retinal l-type voltage gated calcium channel ion influx. PLoS ONE. 2013;8:e56340. doi: 10.1371/journal.pone.0056340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••.Discusses the importance of the calcium hypothesis of aging evaluating in the aging retina.
- 82.Mauer M, Zinman B, Gardiner R, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med. 2009;361:40–51. doi: 10.1056/NEJMoa0808400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••.Discusses the importance of ACEi in DR.
- 83.Salido EM, Dorfman D, Bordone M, et al. Ischemic conditioning protects the rat retina in an experimental model of early type 2 diabetes. Exp Neurol. 2013;240:1–8. doi: 10.1016/j.expneurol.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 84.Fernandez DC, Sande PH, Chianelli MS, et al. Induction of ischemic tolerance protects the retina from diabetic retinopathy. Am J Pathol. 2011;178:2264–74. doi: 10.1016/j.ajpath.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •.Discusses the importance of preconditioning in DR.
- 85.Zheng Z, Chen H, Ke G, et al. Protective effect of perindopril on diabetic retinopathy is associated with decreased vascular endothelial growth factor-to-pigment epithelium-derived factor ratio: involvement of a mitochondria-reactive oxygen species pathway. Diabetes. 2009;58:954–64. doi: 10.2337/db07-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ebrahimian TG, Tamarat R, Clergue M, et al. Dual effect of angiotensin-converting enzyme inhibition on angiogenesis in type 1 diabetic mice. Arterioscler Thromb Vasc Biol. 2005;25:65–70. doi: 10.1161/01.ATV.0000149377.90852.d8. [DOI] [PubMed] [Google Scholar]
- 87.Zheng Z, Chen H, Xu X, et al. Effects of angiotensin-converting enzyme inhibitors and beta-adrenergic blockers on retinal vascular endothelial growth factor expression in rat diabetic retinopathy. Exp Eye Res. 2007;84:745–52. doi: 10.1016/j.exer.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 88.Barnett J, Yanni S, Penn J. The development of the rat model of retinopathy of prematurity. Doc Ophthalmol. 2010;120:3–12. doi: 10.1007/s10633-009-9180-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••.First description of the rat model of ROP.
- 89.Smith LE, Wesolowski E, McLellan A, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–11. [PubMed] [Google Scholar]
- •.First description of the mouse model of OIR.
- 90.Berkowitz BA, Penn JS. Abnormal panretinal response pattern to carbogen inhalation in experimental retinopathy of prematurity. Invest Ophthalmol Vis Sci. 1998;39:840–5. [PubMed] [Google Scholar]
- 91.Penn JS, Henry MM, Wall PT, Tolman BL. The range of PaO2 variation determines the severity of oxygen-induced retinopathy in newborn rats. Invest Ophthalmol Vis Sci. 1995;36:2063–70. [PubMed] [Google Scholar]
- 92.Penn JS, Henry MM, Tolman BL. Exposure to alternating hypoxia and hyperoxia causes severe proliferative retinopathy in the newborn rat. Pediatr Res. 1994;36:724–31. doi: 10.1203/00006450-199412000-00007. [DOI] [PubMed] [Google Scholar]
- 93.Berkowitz BA, McDonald C, Ito Y, et al. Measuring the human retinal oxygenation response to a hyperoxic challenge using MRI: eliminating blinking artifacts and demonstrating proof of concept. Magn Reson Med. 2001;46:412–16. doi: 10.1002/mrm.1206. [DOI] [PubMed] [Google Scholar]
- ••.First description of cued-blinking.