

Abstract
Purpose
Merkel cell carcinoma (MCC) is an aggressive cutaneous neuroendocrine tumor, often metastatic at presentation, for which current chemotherapeutic regimens are largely ineffective. As its pathogenesis is still unknown, we hypothesized that deregulation of signaling pathways commonly activated in cancer may contribute to MCC tumorigenesis and may provide insights into targeted therapy approaches for this malignancy.
Experimental Design
We retrospectively profiled 60 primary MCC samples using a SNaPshot-based tumor genotyping assay to screen for common mutations in 13 cancer genes.
Results
We identified mutations in 9 (15%) MCC primary tumors, including mutations in TP53 (3/60) and activating mutations in the PIK3CA gene (6/60). Sanger sequencing of the primary MCC tumors detected one additional PIK3CA mutation (R19K) that had not been previously described in cancer. Merkel cell polyoma virus (MCPyV) was detected in 38 (66%) MCC cases and patients with MCPyV-positive cancers showed a trend toward better survival. With one exception, the presence of MCPyV and of activating mutations in PIK3CA appeared mutually exclusive. We observed that signaling through the PI3K/pAKT pathway was active in one MCPyV-positive and in all MCPyV-negative MCC cell lines, as evidenced by AKT phosphorylation. Importantly, the presence of a PIK3CA activating mutation was associated with sensitivity to treatment with ZST474, a specific PI3K inhibitor and to NVP-BEZ235, a dual PI3K-mTOR inhibitor, targeted agents under active clinical development.
Conclusions
PI3K pathway activation may drive tumorigenesis in a subset of MCC and screening these tumors for PIK3CA mutations could help identify patients who may respond to treatment with PI3K pathway inhibitors.
INTRODUCTION
Merkel cell carcinoma (MCC), originally defined as a “trabecular carcinoma of the skin”, is an aggressive neuroendocrine skin tumor first described almost forty years ago (1). Although initially believed to derive from Merkel cells, the slowly adapting mechanoreceptors located in the basal cell layers of the epidermis and the only cutaneous cells containing electron-dense neurosecretory granules (2), the histogenesis of MCC is still disputed with some evidence pointing to a pluripotent stem cell origin (3, 4).
MCC is quite rare, with an estimated incidence of three new cases per million people per year in the United States (5). The incidence of MCC has been on the rise (6), and increases with age, exposure to ultraviolet radiation and with immunosuppression, being up to 15 times higher in patients with HIV, organ transplants and hematological malignancies such as multiple myeloma, non-Hodgkin’s lymphomas and chronic lymphocytic leukemias (7, 8). The most common primary site of presentation is the skin of the head and neck, followed by the skin of the upper extremities, lower extremities, and trunk. Interestingly, albeit to a much lower extent (<1%), MCC may also arise in extracutaneous sites including the parotid gland, submandibular glands, the nasal cavity and the lymph nodes (9).
MCC tends to present as a firm, flesh-colored, red or blue-livid painless nodule, which is fast growing and can rapidly spread to regional lymph nodes and to distant sites (10, 11). The extent of disease at presentation is a strong predictor of survival, with a 10-yr survival rate of 70% for patients with localized disease, which sharply drops to 20% if the patients present with distant metastases. (9)
Histological presentation of MCC typically includes a dermal or subcutaneous centered mass of monotonous round blue tumor cells, often with a trabecular pattern, a scant eosinophilic cytoplasmic rim, round nuclei with finely granular “salt and pepper” chromatin and multiple nucleoli. Numerous apoptotic and mitotic figures are also common. Helpful features in reaching a diagnosis include: the expression of neuroendocrine markers, a characteristic paranuclear staining in a dot-like pattern of cytokeratin 20 (CK-20), and the absence of thyroid transcription factor –1 (TTF1) expression (12, 13).
While the contribution of established oncogenes and tumor suppressors to the pathogenesis of MCC remains unknown, a new polyomavirus has been recently identified in about 80% of these tumors (14, 15). Merkel cell polyomavirus (MCPyV) appears to be a common infection in humans (16, 17) and evidence of a possible mechanistic role for MCPyV in MCC tumorigenesis is starting to emerge. First, MCPyV DNA was shown to integrate into the genome of MCC tumors in a clonal pattern, strongly suggesting that viral infection precedes clonal expansion of the cancer cells (14). Although integrated MCPyV cannot replicate, due to the presence of prematurely truncating mutations in the viral large T antigen (LT) DNA, the retinoblastoma (RB) binding motif of LT is spared and both LT and small T antigen (ST) viral sequences are significantly expressed in MCC (18, 19). Furthermore, knockdown experiments targeting the viral T antigens result in cell cycle arrest and death of MCPyV-positive MCC cell lines, suggesting that T antigen expression may play a role in the oncogenesis of MCPyV-infected MCC (20, 21).
Unfortunately, despite optimal early management, many MCC metastasize and at that stage treatment options are very limited and typically fail. Thus, new mechanistic and therapeutic insights would be very valuable. In an attempt to identify novel oncogenic pathways involved in MCC tumorigenesis and with the hope of uncovering therapeutically-relevant mutations that could help improve management of MCC patients, we retrospectively profiled 60 primary tumors using the previously described SNaPshot tumor genotyping assay. This test screens for hotspot mutations across 13 cancer genes, and has been used at our institution for over two years to guide therapy for patients with solid tumors (22).
MATERIALS AND METHODS
Patient specimens
This study was conducted according to the principles expressed in the Declaration of Helsinki. Work on human tissue was performed on excess archival human material after obtaining approval from the Massachusetts General Hospital (MGH) Institutional Review Board (protocol reference: 2008-P-002165; MGH). We collected a retrospective cohort of patients with a diagnosis of MCC, who had undergone surgery at MGH between January 1995 and September 2010. Sixty patients were identified, for whom formalin-fixed paraffin embedded (FFPE) tumor specimens suitable for molecular analysis could be obtained. The patients’ medical records were reviewed to collect demographic data and relevant information regarding clinical history, presentation, management and outcome. Stage at diagnosis was determined according the American Joint Committee on Cancer (AJCC) guidelines (23).
Cell lines
We established a permanent cell line, MGH-mcc1, derived from one of the primary tumors in our cohort (MCC-12). Briefly, fresh tumor (isolated within 1 hr after surgery) was isolated from a left axillary dissection and washed with 50ml of wash buffer (DMEM + 2%FBS + gentamicin and fungizone). Tissues were minced using sterile crossed blades to produce small (<1×1 mm) pieces under sterile conditions. Tumor pieces were left to grow in 6-well plates with 17% FBS+ RPMI+ L-glutamine+ antibiotic/antimycotic. After approximately 4-6 weeks, single colonies were subcloned and pooled to generate the MGH-mcc1 cell line. Human MCC cell lines MCC13, MCC26, MKL-1, MKL-2, UISO-mcc1 had been previously described (18, 24-27). All MCC cell lines were maintained in RPMI medium 1640 supplemented with 10% FBS.
MCPyV detection by PCR
Testing for the presence of Merkel cell polyoma virus was carried out using two previously described sets of primers: MCVPS1 (28) and MCPyV (29), both of which yield short PCR products (109 bp and 195 bp, respectively) being well suited for testing poor quality DNA derived from formalin-fixed paraffin-embedded tissue. In short, 20 ng of tumor DNA were subject to PCR amplification using the primers described below and the Platinum Taq kit (Invitrogen, Carlsbad, CA, USA) for 40 cycles of: denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 68 °C for 1 min. To ensure that a negative signal was not an artifact related to DNA integrity, in addition to the MCVPS1 (Forward, 5’-TCAGCGTCCCAGGCTTCAGA-3’; Reverse 5’-TGGTGGTCTCCTCTCTTGCTACTG-3’), and MCPyV (Forward, 5’-ACTTGGGAAAGTTTTGACTGGTGGCAA-3’; Reverse 5’- GGGCCTCGTCAACCTAGATGGGAAAG-3’), primer sets, which detect viral-specific sequences in MCPyV-positive tumors (28, 29), we set up a parallel amplification reaction to detect KRAS exon 2 (Forward, 5’-TCATTATTTTTATTATAAGGCCTGCTG -3’; Reverse 5’-AGAATGGTCCTGCACCAGTAA-3’), which yielded a 185 bp fragment for all tested samples. Due to the lack of sufficient tumor material for two cases, MCPyV analysis was performed on 58 out of the 60 MCC specimens.
Tumor mutational analysis
Histological examination of hematoxylin and eosin-stained slides derived from FFPE was performed by a pathologist and assessed for the presence of tumor. Available tumor tissue was manually micro-dissected, or cored from the paraffin block using a 1.5 mm dermal punch. Total nucleic acid was extracted from FFPE material using a modified FormaPure System (Agencourt Bioscience Corporation, Beverly, MA). Mutational analysis for a panel of 13 cancer genes, which included BRAF, EGFR, KRAS, NRAS, PIK3CA and TP53 (Supp. Table S1), was performed as previously described (22). This analysis used a recently developed tumor genotyping protocol based on the SNaPshot multiplex platform (Applied Biosystems) that efficiently detects multiple mutations in tumor DNA extracted from FFPE tissue (22).
Sequence analysis of mutational hotspots mapping within the PIK3CA adaptor-binding, helical and kinase domains was performed by direct sequencing of M13-tagged PCR products generated using the following primer pairs: PIK3CA exon 1, 5′- TGTAAAACGACGGCCAGTTGCTTTGGGACAACCATACA -3′ (forward) and 5′- CAGGAAACAGCTATGACCTTTTAGAAAGGGACAACAGTTAAGC -3′ (reverse); exon 9, 5′-TGTAAAACGACGGCCAGTCTGTGAATCCAGAGGGGAAA-3′ (forward) and 5′- CAGGAAACAGCTATGACCCATGCTGAGATCAGCCAAAT-3′ (reverse); exon 20, 5′- TGTAAAACGACGGCCAGTCATTTGCTCCAAACTGACCA -3′ (forward) and 5′- CAGGAAACAGCTATGACCTGTGGAATCCAGAGTGAGCTT -3′ (reverse). Thermocycling was performed at 95 °C for 8 min, followed by 45 cycles of 95 °C for 20 s, annealing at 58 °C (exon 1 and 20) or at 68 °C (exon 9) for 30 s and 72°C for 1 min, and one last cycle of 72 °C for 3 min. Sequencing was performed utilizing the incorporated M13 primer tag, as previously described (22).
EGFR and HER2 gene copy number were assessed by fluorescence in situ hybridization (FISH) as previously reported (30).
Immunoblotting
Cells were prepared and lysed as previously described (31). Proteins were resolved using the NuPAGE® Novex® Midi Gel system on 4-12% Bis-Tris Gels (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose membranes. Immunoblotting was performed using the following antibodies according to the manufacturers’ recommendations: total Akt (Santa Cruz); phospho-AKT (308) and phospho-AKT (473) (both from Cell Signaling) and actin (Sigma-Aldrich).
Flow Cytometry
FACS analysis was performed on a BD LSR III (Becton Dickenson). For apoptosis measurements, experiments were conducted as previously described (31). The annexin Cy5 was from Biosource International (Camarillo, CA). Experiments were carried out in triplicate and error bars represent standard deviations.
Statistical analysis
Fisher’s exact test and Wilcoxon rank-sum test were used to assess the association of MCPyV or PIK3CA genotype status with baseline patient characteristics. Overall survival (OS) was calculated from the date of initial diagnosis of MCC to the date of death and was censored at the date of last follow-up for patients who were alive. OS was estimated by the Kaplan-Meier method, and the difference between MCPyV groups was assessed using proportional hazards regression to adjust for the independent effects of stage and nodal status. The data analysis was computed by SAS 9.2 (SAS Institute, Cary, NC), and all p-values were based on a two-sided hypothesis.
RESULTS
Merkel cell carcinoma cohort characteristics
To study the genetic etiology of MCC, we identified surgical pathology specimens from patients who underwent resection of their tumors at the Massachusetts General Hospital from 1995 to 2010. We identified sixty cases of MCC with sufficient tissue for molecular analysis.
In our cohort the median age at disease presentation was 73 years (range 44-94), 38 (63%) patients were male, and 8 (13%) were immunocompromised due to HIV infection, solid organ transplant or leukemia (CLL or CML) (Table 1). The most common site of presentation was the head and neck (45%) followed by the extremities (35%), with only 5 (8%) tumors presenting in the trunk. In 7 (12%) cases the primary tumor site was unknown, as these patients first presented with nodules in the lymph nodes or with disseminated metastatic disease. Approximately half (28) of the patients presented with either stage I (25%) or stage II (22%) disease, 23 (38%) patients presented with stage III and a minority (15%) had stage IV disease. Disease staging was determined according to the AJCC guidelines (23).
Table 1.
MCC patient baseline characteristics.
| All Patients n=60 |
PIK3CA Mutational Status | MCPyV Status* | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Positive n=7 | Negative n=53 | P value | Positive n=38 | Negative n=20 | P value | ||
| No. (%) | No. (%) | No. (%) | No. (%) | No. (%) | |||
| Age, yrs | 0.326 | 0.615 | |||||
| Median | 73 | 68 | 77 | 73.5 | 71 | ||
| Range | 44-94 | 59-89 | 44-94 | 44-90 | 52-94 | ||
|
| |||||||
| Sex | 0.040 | <0.001 | |||||
| Male | 38 (63) | 7 (100) | 31 (58) | 18 (47) | 19 (95) | ||
| Female | 22 (37) | 0 (0) | 22 (42) | 20 (53) | 1 (5) | ||
|
| |||||||
| Immunosuppression | 1.000 | 1.000 | |||||
| Yes | 8 (13) | 1 (14) | 7 (13) | 5 (13) | 3 (15) | ||
| No | 52 (87) | 6 (86) | 46 (87) | 33 (87) | 17 (85) | ||
|
| |||||||
| Stage | 0.176# | 0.016## | |||||
| I | 15 (25) | 0 (0) | 15 (28) | 12 (32) | 3 (15) | ||
| II | 13 (22) | 2 (29) | 11 (21) | 9 (24) | 4 (20) | ||
| III | 23 (38) | 3 (43) | 20 (38) | 15 (39) | 7 (35) | ||
| IV | 9 (15) | 2 (29) | 7 (13) | 2 (5) | 6 (30) | ||
|
| |||||||
| Lymph Nodes | 0.432 | 0.174 | |||||
| Involved | 32 (53) | 5 (71) | 27 (51) | 17 (45) | 13 (65) | ||
| None | 28 (47) | 2 (29) | 26 (49) | 21 (55) | 7 (35) | ||
|
| |||||||
| Primary Tumor | 0.748 | 0.111 | |||||
| Head & Neck | 27 (45) | 3 (43) | 24 (45) | 15 (39) | 11 (55) | ||
| Extremity | 21 (35) | 1 (14) | 20 (38) | 17 (45) | 3 (15) | ||
| Trunk | 5 (8) | 0 (0) | 5 (9) | 3 (8) | 2 (10) | ||
| unknown | 7 (12) | 3 (43) | 4 (8) | 3 (8) | 4 (20) | ||
|
| |||||||
| Margins | 1.000 | 0.530 | |||||
| Negative | 22 (37) | 3 (43) | 19 (36) | 16 (42) | 6 (30) | ||
| Close | 12 (20) | 2 (29) | 10 (19) | 8 (21) | 4 (20) | ||
| Positive | 11 (18) | 1 (14) | 10 (19) | 6 (16) | 5 (25) | ||
| unknown | 15 (25) | 1 (14) | 14 (26) | 8 (21) | 5 (25) | ||
due to limited amount of tumor, we were unable to determine MCPyV status for two cases
Stage I vs. II-IV;
Stage I-III vs. IV
Out of the 60 MCC specimens available for molecular analysis, 40 (67%) were primary tumors and 20 (33%) were recurrences. Median tumor size was 2.2 cm (range 0.5-12.5). Representative images of the MCC cases are illustrated in Figure 1 and include the classical neuroendocrine morphology (Fig. 1A) and the typical, although non-specific, immunohistochemistry with cytoplasmic positivity for synaptophysin (Fig. 1B) and perinuclear dot-like positivity for CK20 (Fig. 1C). Additional histopathological features are outlined in Table 2 and included an unusual spindle shape morphology observed in only a subset of cases (5 out of 60, 8%) (Fig. 1D), tumor cell necrosis identified in 23 (38%) cases (Fig. 1E) and the presence of highly prominent nucleoli in 18 (30%) specimens (Fig. 1F).
Figure 1. Histology and immunohistochemistry of MCC.
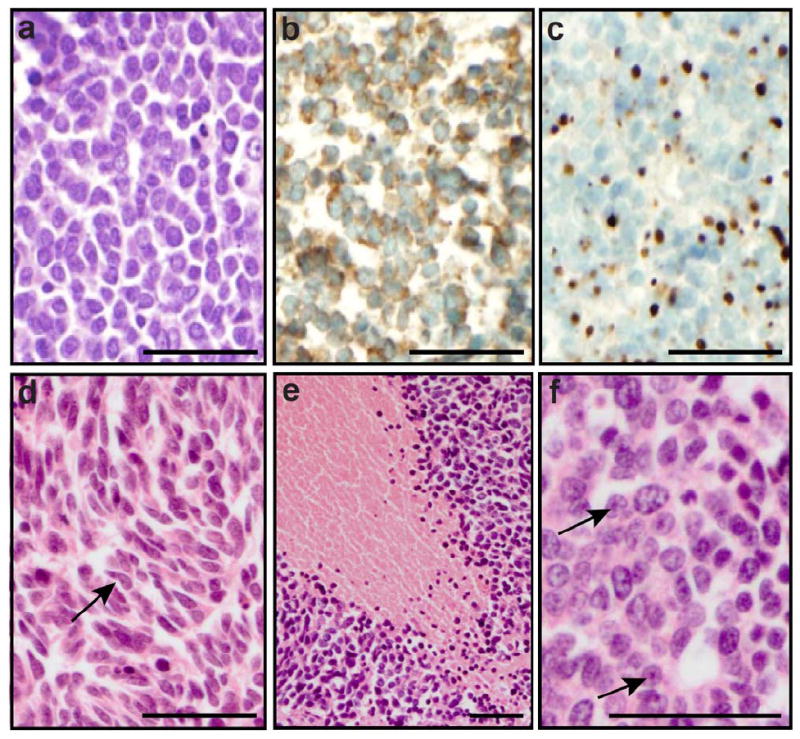
(A) H&E staining illustrating a population of monomorphic, non-cohesive round cells with a high nuclear to cytoplasmic ratio and finely granular “salt and pepper” chromatin. (B) Positive staining for the neuroendocrine marker synaptophysin. (C) cytokeratin 20 dot-like perinuclear staining. Additional features observed in some tumors: (D) spindle morphology (arrows), (E) large zone of necrosis, and (F) prominent nucleoli (arrows). Scale bar: 50 micron.
Table 2.
Pathology of MCC cases.
| All Patients n=60 |
PIK3CA Mutational Status | MCPyV Status* | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Positive n=7 | Negative n=53 | P value | Positive n=38 | Negative n=20 | P value | ||
| No. (%) | No. (%) | No. (%) | No. (%) | No. (%) | |||
| Specimen size, cm | <0.001 | 0.120 | |||||
| Median | 2.2 | 8.0 | 2.0 | 2.00 | 3.65 | ||
| Range | 0.5-12.5 | 3.5-12.5 | 0.5-8.0 | 0.5-8.0 | 0.5-12.5 | ||
|
| |||||||
| Specimen size | 0.070 | 0.239 | |||||
| < 2cm | 21 (35) | 0 (0) | 21 (40) | 16 (42) | 5 (25) | ||
| 2-12.5cm | 31 (52) | 6 (86) | 25 (47) | 18 (47) | 13 (65) | ||
| unknown | 8 (13) | 1 (14) | 7 (13) | 4 (11) | 2 (10) | ||
|
| |||||||
| % Necrosis | 0.053 | 0.010 | |||||
| Median | 0% | 20% | 0% | 0% | 5% | ||
| Range | 0-95% | 0-60% | 0-95% | 0-95% | 0-60% | ||
|
| |||||||
| Necrosis | 0.095 | 0.004 | |||||
| 0% | 37 (62) | 2 (29) | 35 (66) | 29 (76) | 7 (35) | ||
| 5-95% | 23 (38) | 5 (71) | 18 (34) | 9 (24) | 13 (65) | ||
|
| |||||||
| Nucleoli visible | 0.663 | 0.373 | |||||
| Negative | 42 (70) | 6 (86) | 36 (68) | 28 (74) | 12 (60) | ||
| Positive | 18 (30) | 1 (14) | 17 (32) | 10 (26) | 8 (40) | ||
|
| |||||||
| Spindle Shape | 0.099 | 0.044 | |||||
| Negative | 55 (92) | 5 (71) | 50 (94) | 37 (97) | 16 (80) | ||
| Positive/Pleomor. | 5 (8) | 2 (29) | 3 (6) | 1 (3) | 4 (20) | ||
|
| |||||||
| Vascular Invasion | 0.101 | 0.195 | |||||
| Absent | 31 (52) | 1 (14) | 30 (32) | 23 (61) | 7 (35) | ||
| Present | 8 (13) | 2 (29) | 6 (57) | 4 (11) | 4 (20) | ||
| unknown | 21 (35) | 4 (57) | 17 (11) | 11 (29) | 9 (45) | ||
due to limited amount of tumor, we were unable to determine MCPyV status for two cases
The presence of Merkel cell polyoma virus correlated with better survival
To determine the prevalence of Merkel cell polyoma virus (MCPyV) in our cohort, we used PCR-based protocols and two sets of primers that had been previously optimized to detect viral load in nucleic acid extracted from archival material (28, 29). We detected MCPyV in 38 (66%) of 58 tumors (Supp. Table S2). Patients with MCPyV-positive MCC were significantly more likely than MCPyV-negative patients to be female (50% versus 5%, p<0.001) and to have a lower stage disease at presentation (I-III vs. IV) (95% vs. 70%, p=0.016) (Table 1).
MCPyV-positive tumors were significantly less likely than MCPyV-negative MCC to show evidence of pleomorphic spindle-cell morphology (3% vs. 20%, p=0.044) or to be associated with necrosis (24% vs 65%, p=0.004) (Table 2). Stratification of the overall patient population according to viral status revealed a statistically significant improvement in the overall survival (OS, p=0.023) and recurrence-free survival (RFS, p=0.027) of MCPyV-positive patients. After adjusting for the independent effects of stage and nodal status, patients with MCPyV-positive tumors showed a trend toward longer OS (p=0.065), when compared to patients with no detectable virus (Fig. 2, Supp. Table S3).
Figure 2. Trend toward improved survival in MCPyV-positive MCC patients.
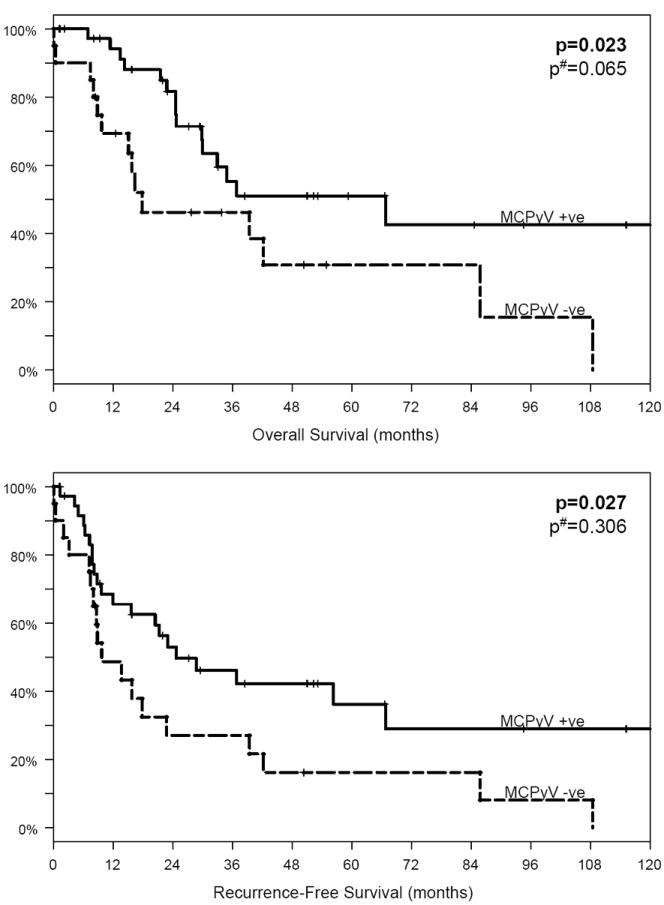
(A) Overall survival and (B) recurrence-free survival depicted according to MCPyV status, for all disease stages. Upon adjustment for the independent effects of disease stage and nodal status, statistical analysis revealed that patients with MCPyV-positive tumors showed a trend toward a longer overall survival (p=0.065) when compared to patients with no detectable virus. Values in bold indicate statistical significance. MCPyV+ (solid line) and MCPyV- (dashed line) patients (28, 29).
# adjusted for stage and nodal status
SNaPshot genotyping detected PIK3CA activating mutations in MCC
SNaPshot mutational profiling of the 60 specimens identified point mutations in 9 (15%) cases (Supp. Table S4). Three (5%) tumors harbored mutations in the TP53 tumor suppressor gene (R248P, R248W and R273H). Interestingly, 6 (10%) MCC had activating mutations in the p110 alpha subunit of the phosphatidylinositol 3-kinase (PIK3CA) gene (Fig. 3 and Supp. Table S4). Five tumors harbored mutations located within the helical domain of PIK3CA, including three cases with the E542K substitution, and one case each with the E545K and the Q546K activating mutations. The sixth case carried the H1047L somatic mutation located within the kinase domain of PIK3CA.
Figure 3. Detection of PIK3CA activating mutations in MCC.

Mutational profiling was performed by SNaPshot analysis (left panels) and confirmed by Sanger sequencing (right panels). Nucleic acid extracted from each MCC archival specimen was run in parallel with a genomic DNA control, as indicated. The arrows point to the mutant alleles: PIK3CA Q546K (c.1636C>A) in tumor MCC-12 (A), PIK3CA E545K (c.1633G>A) in MCC-13 (B), and PIK3CA E542K (c.1624G>A) for MCC-20 (C). The assayed loci for each panel are as follows: (A) 1.PIK3CA 3140, 2.CTNNB1 101, 3.BRAF 1798, 4.NRAS 37, 5.PIK3CA 1636, 6.APC 4348 and 7.APC 3340; (B) 1.KRAS 34, 2.EGFR 2235_49del F, 3.EGFR 2369, 4.NRAS 181, 5.PIK3CA 1633, 6.CTNNB1 94 and 7.CTNNB1 121; and (C) 1.EGFR 2236_50del F, 2.EGFR 2573, 3.CTNNB1 133, 4.PIK3CA 1624 and 5.NRAS 35.
All of the variants detected by SNaPshot were confirmed by conventional Sanger sequencing, which was also used to screen MCC for the presence of additional mutations within the PIK3CA gene. This effort identified one extra variant located within the adaptor binding domain of PI3KCA. This mutation (R19K) has not been previously reported in the literature, and arises from a guanine (G) to adenosine (A) change at nucleotide position 56, resulting in an amino acid substitution of the arginine (R) at codon 19 with a lysine (K). The clinical significance of this variant is currently unknown.
In our cohort, PIK3CA mutations occurred in MCC tumors that presented exclusively in male patients (p=0.040). Patients with a PIK3CA-mutant MCC presented with stage II-IV disease and the location of their primary tumors included the head and neck (3 cases), the lower extremity (1 case), or was undetermined due to disease presentation either in the lymph nodes (2 cases) or as disseminated metastases (1 case) (Table 1).
Three of the PIK3CA-mutant MCC cases retrieved for molecular profiling were primary tumors and the remaining four tumors were recurrences for which a primary specimen was unavailable for testing. PIK3CA-mutant tumors were significantly larger than their mutation-negative counterparts (median 8 cm vs. 2 cm, p<0.001). In addition, and in contrast to MCPyV-positive tumors, PIK3CA mutations occurred more frequently in tumors with visible necrosis (p=0.095) and with pleomorphic or spindle shape cell morphology (p=0.099), but the association between these features and tumor genotype did not reach strict statistical significance due to limited power given the small number of mutant cases (Table 2). These clinicopathological characteristics were suggestive of a more aggressive tumor behavior and interestingly, with the exception of the H1047L kinase domain mutation, PIK3CA-mutant tumors had a negative MCPyV status. However, we found no association between the presence of a PI3KCA mutation and MCC patient overall survival (Supp. Table S3, p=0.628).
The epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor 2 (HER2/ERBB2) genes encode for receptor tyrosine kinases that are often overexpressed in human cancer and that can signal through activation of the PI3K/pAKT pathway. This prompted us to screen our MCC cohort for EGFR and HER2 gene amplification, using FISH analysis. We did not detect EGFR amplification in any of the tumors, but one sample showed HER2 gene amplification. This particular case (MCC-16, Supp. Table S4) was a local recurrence with a highly unusual histology of very atypical and anaplastic morphologic features, which were most likely secondary to the fact that that the primary tumor had been treated with radiation therapy. Unfortunately the primary tumor was not available for genetic testing, and we could not determine whether HER2 amplification arose after radiation, or was present in the original tumor.
For the majority of mutation-positive MCC cases (9 out of 11), we were able to obtain matching normal tissue from the same patient and confirm that these were somatic mutations, unique to the tumor (Supp. Table S4).
Sensitivity of PIK3CA-mutant MCC cells to PI3K pathway inhibitors
We had access to six primary MCC cell lines, five of which (MCC13, MCC26, UISO-mcc1, MKL-1 and MKL-2) had been previously described (18, 24-27). Molecular analysis of these five cell lines revealed that none of them harbored PIK3CA mutations, and identified three that were positive for MCPyV (MKL-1, MKL-2 and UISO-mcc1; Supp. Table S2). The sixth cell line (MGH-mcc1) was derived from tumor MCC-12, which had been resected from one of the patients in our cohort. Consistent with the molecular profile of the original tumor (Suppl. Table S4), the MGH-mcc1 cell line carried an activating mutation in the helical domain of PI3KCA (Q546K) and was negative for MCPyV.
To evaluate PI3K pathway activation in MCC cell lines, we measured AKT phosphorylation at two residues, threonine 308 and serine 473, which are downstream targets of PI3K. AKT phosphorylation could not be detected by immunoblot in two of the three MCPyV-positive cell lines, MKL-1 and MKL-2. By contrast, the remaining four cell lines yielded robust phospho-AKT signals, suggesting strong activation of the PI3K/pAKT pathway in a substantial fraction of MCC cell lines (Fig 4A).
Figure 4. Sensitivity of MCC cell lines to PI3K pathway inhibitors.
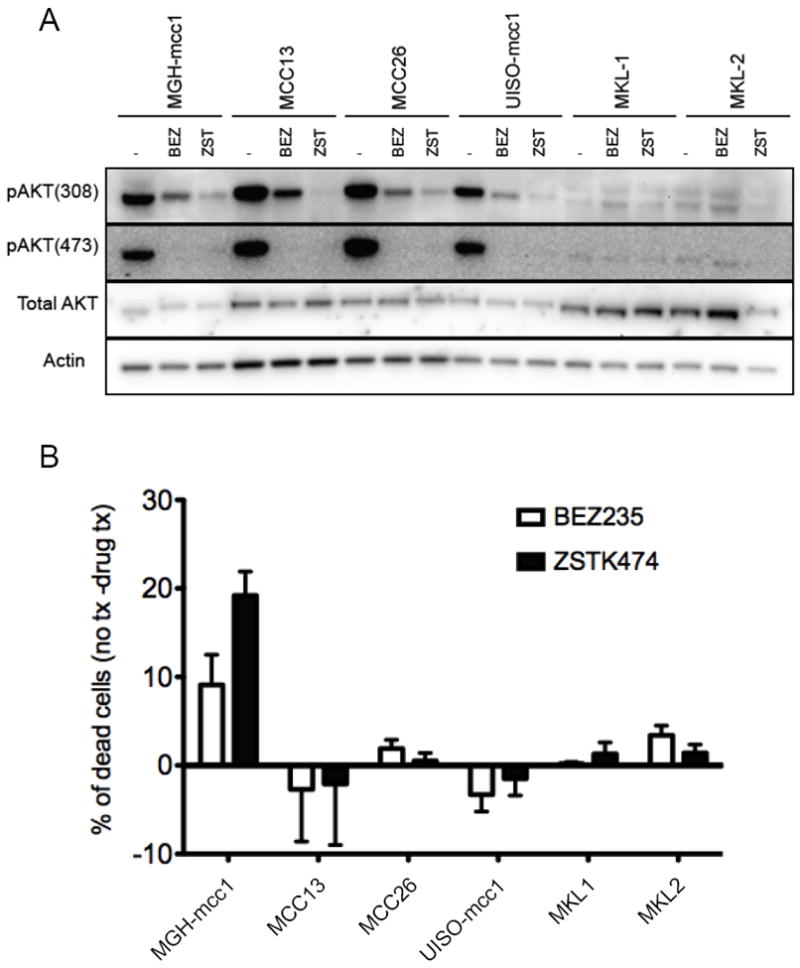
(A) PI3K pathway activation in MCC cell lines. MGH-mcc1 (PIK3CA-mutant), MCC13, MCC26, UISO-mcc1 (MCPyV+), MKL-1 (MCPyV+), and MKL-2 (MCPyV+) cells were treated with vehicle control (-), with 200nM of the dual PI3K/mTORC inhibitor, NVP-BEZ235, or with 1mM of the pure PI3K inhibitor, ZSTK474, for 6 hours. Lysates were prepared and subject to immunoblotting with the indicated antibodies. Blots were stripped and probed with α-Actin to ensure proper loading. (B) Targeted PI3K and PI3K-mTOR inhibition reduced cell viability in PIK3CA-mutant MCC. The cell lines were treated as described above for 72 hours. The cells were then stained with propidium iodide (PI) and Annexin V and analyzed by FACS. Because we found that plasma membranes of healthy MKL-1 and MKL-2 cells displayed constitutive binding to Annexin V, these cells were instead permeabilized and stained with only propidium iodide, and sub-G1 cells were quantified and scored as dead by FACS, as previously described (31).
We treated the six MCC cell lines with two targeted agents under active clinical development: ZSTK474 (32), a PI3K inhibitor, and NVP-BEZ235 (33, 34), a dual inhibitor of PI3K and the mammalian target of rapamycin (mTOR). Exposure to these drugs reduced AKT-phosphorylation in all four MCC cell lines that exhibited high baseline levels of PI3K signaling (Fig. 4A). In addition, the PI3KCA mutant cell line (MGH-mcc1) was particularly sensitive to inhibition of the PI3K/pAKT pathway, as evidenced by an increase in apoptosis (Fig. 4B).
DISCUSSION
We genotyped 60 patient samples of Merkel cell carcinoma with the hope of identifying mutations in oncogenes involved in the pathogenesis of this aggressive cancer that could serve as potential targets for small molecule inhibitors.
The clinical and pathological characteristics of our MCC patient cohort (Table 1) were comparable to those described in previously published studies: the median age at disease presentation was 73 years, the male to female ratio was approximately 2:1, immunodeficiency was present in about 13% of patients, and the head and neck region was the most common location of disease at presentation. The median overall survival was lower in our patient population, where we observed approximately 50% OS at 3 yrs (Suppl. Table S3) compared to 67% at 3 yrs or 45 to 65% at 5 yrs in prior reports (9, 12, 35). This difference is most likely due to the fact that our institution is a tertiary care center that serves as a reference center for Merkel cell carcinoma. Thus, the majority of MCC patients referred to our service have more advanced and aggressive disease, presenting with a higher disease stage (53% stage III-IV) than in other reports where the majority of patients present with stage I or II disease (9, 12, 36).
Since its discovery in 2008 (14), MCPyV has been detected in the majority of MCC, and only occasionally in other cutaneous and hematological malignancies (15, 37, 38). We detected MCPyV in our cohort at a frequency of 66% (38/58 patients), which is well within the wide range of 24-100% reported in the literature (35). We observed that MCPyV+ cases showed a trend towards a better prognosis and prolonged survival, consistent with previous reports (36, 39, 40, 41). A similarly favorable prognosis for tumors containing viral DNA has also been noted for human papillomavirus (HPV)-positive oropharyngeal squamous cell carcinomas and for Epstein-Barr virus (EBV)-positive leiomyosarcomas (42, 43). A possible explanation, at least for MCC, might be that, in the absence of MCPyV, tumorigenesis occurs through more complex or more directly oncogenic genetic abnormalities like the activation of the PI3K/pAKT pathway. An alternative explanation might be that the immune system of patients whose tumors contain an oncogenic virus, although likely compromised and permissive to a viral infection, might retain some anti-tumor activity.
Historically, it appears that MCPyV positive cases are mostly indistinguishable from MCPyV negative cases, in terms of histological and clinical features(41). Only a few authors noticed some modest differences between the two groups, including lighter, more abundant cytoplasm and more irregular or polygonal nuclei in virus negative cases (35, 44) and a higher likelihood of presentation on the extremities (41) for virus positive cases. In our cohort, most patients with virus positive MCC were female (p<0.001) with lower stage disease at presentation (p=0.016). In addition, we noticed a significant correlation between the presence of MCPyV and less aggressive histological features, like very little or no necrosis (p=0.004), and the absence of spindle cell or unusual tumor morphology (p=0.044).
To date, molecular genetic analysis of MCC has been predominantly limited to TP53, revealing mutations in 14 to 28% of cases, mostly through sequencing of the most common hotspots for TP53 mutations on exons 5 to 9 (45, 46). In an effort to better understand the molecular pathogenesis of MCC, we screened our sample cohort for point mutations in 13 cancer genes (Suppl. Table S1) and detected mutations in 15% of the samples: 5% (3/60) of patients had mutations in TP53 and 10% (6/60) had mutations in PI3KCA.
Using FISH, we detected HER2 amplification in one unusually pleomorphic, locally recurrent MCC that developed after irradiation of the primary tumor. Since the primary sample was not available for molecular testing, we do not know whether HER2 amplification was a therapy-related event or if, alternatively, it could possibly characterize a yet unrecognized, rare subtype of MCC.
The number of TP53 gene mutations detected in our cohort (5%) was lower than the previously reported frequencies and likely an underestimate, reflecting the fact that single base–extension genotyping assays such as SNaPshot, while ideal for oncogenes, are not practical approaches to capture the wide range of mutations typical of tumor suppressors. Interestingly, while other authors identified an inverse correlation between the presence of MCPyV and TP53 mutations, two of our three TP53-mutant cases (R248P and R273H) were also positive for MCPyV (Suppl. Table S4).
This is the first time, to our knowledge, that activating mutations in PIK3CA, which encodes for the p110 alpha subunit of PI3K, have been detected in MCC. Activation of the PI3K/pAKT pathway has been observed in a large fraction of human cancers, including colorectal, breast, head and neck, lung, brain, and gynecological tumors, with mutation frequencies as high as 40% in breast cancers and 75% in head and neck squamous cell carcinomas (47). The majority of mutations so far identified map to two hotspots located within the helical and the kinase domains of PI3K. The mutant PIK3CA proteins have higher kinase activity than their wild type counterparts and can transform chicken embryo fibroblasts and NIH-3T3 cells, supporting the hypothesis that these are “driver” mutations with an active role in tumorigenesis. Therefore, detection of activating PIK3CA mutations in a subset of MCC is highly suggestive of an oncogenic role of the PI3K/pAKT pathway in MCC.
Using conventional sequencing, we detected one novel mutation in the first exon of PIK3CA, at position c.56G>A (p.R19K). This mutation maps to the adaptor-binding domain located at the extreme N-terminal region of PIK3CA, which is responsible for binding to p85alpha (48), one of the regulatory subunits of PI3K. Whether this previously unreported mutant could also act as a “driver” is not known, but the R19K substitution was a somatic mutation (absent in normal tissue from the same patient) and involves two positively charged and structurally similar amino acid residues at a position that has been conserved through evolution, including in zebra fish, frogs, chicken, and mammals.
Remarkably, some of the clinical and pathological features of the PIK3CA-mutant MCC are suggestive of a more aggressive tumor behavior, including a significantly larger tumor size (>2 cm), more unusual histopathology, tendency towards a more advanced disease stage at presentation and, with one exception, absence of MCPyV. However, we could not demonstrate a statistically significant correlation with worse survival, perhaps due to the relatively small number of PIK3CA-mutant cases. If our survival estimates were found to be reproducible in larger cohorts, it appears that PIK3CA-mutant MCC might have a worse prognosis. Moreover, our observation suggests that a subset of MCC patients could potentially benefit from PI3K/pAKT inhibitors, which was corroborated by our cell culture experiments.
We made several attempts to investigate PI3K/pAKT pathway activation in FFPE tumor tissue using immunohistochemistry with phospho-AKT antibodies. Unfortunately, we were unable to obtain convincing results and decided to turn instead to the MCC cell lines that were available to us. Out of six primary MCC cell lines, three were positive for MCPyV and two of these showed no activation of the PI3K/pAKT pathway on immunoblot analysis. The remaining three cell lines were negative for MCPyV and showed a strong activation of the PI3K/pAKT pathway. These results and the apparent frequent absence of MCPyV in PIK3CA-mutant MCC (at least in our cohort) suggest that genetically distinct subtypes of Merkel cell carcinoma may rely on alternative oncogenic mechanisms for tumorigenesis, which could also explain the different outcomes of patients with MCPyV-positive versus MCPyV-negative tumors. There is in fact increasing evidence for an etiological role of MCPyV in MCC, such as the clonal integration of the virus in the host genome; the constant expression of small (ST) and large (LT) T antigens; the necessity of the virus for cell growth, survival and the transformed phenotype (in cell transformation experiments); and the almost omnipresent truncation in the 3’ of LT with loss of replication properties, but persistence of the Rb binding domain (35).
MCPyV integration and PI3K activation are not mutually exclusive occurrences, as observed for the MCC-54 primary tumor sample and for the UISO-mcc1 cell line (Supp. Table S2), suggesting independent and possibly cooperative mechanisms of tumorigenesis that may impact prognosis as well as response to future therapeutic regimens, including potential combinations of anti-viral and PI3K targeted agents.
The presence of phosphorylated AKT in four out of six MCC cell lines suggests the existence of alternative mechanisms for activation of the PI3K/pAKT pathway, besides point mutations in PIK3CA. Accordingly, inactivating mutations, deletion, or silencing of the PTEN gene, which encodes for a tumor suppressor that dephosphorylates PIP3 and downregulates PI3K/pAKT signaling (49), have been previously reported in MCC (26, 50, 51).
Finally, our experiments suggest that PI3K/mTOR inhibitors may be an effective treatment approach for the subset of MCC patients with PIK3CA-mutant tumors. These small molecule inhibitors have shown promising anti-tumor activity in the context of PI3K /pAKT pathway activation and an expanding number of alternative PI3K/mTOR inhibitors are undergoing clinical trials for the treatment of a variety of solid tumors (30, 31, 50-53).
In conclusion, our study shows for the first time that a subset of MCC cases harbor activating mutations in PIK3CA, which confer sensitivity to PI3K/mTOR inhibitors in vitro. Our findings suggest that targeted inhibition of the PI3K /pAKT pathway could be explored as a possible therapeutic option for MCC patients with PI3KCA-mutant tumors.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Tumor genotyping is becoming increasingly important for cancer treatment and diagnosis, as more targeted agents are developed and available for clinical use. While patient management strategies are rapidly changing for the most common and well studied malignancies, rare tumor types tend to lag behind as their molecular drivers await elucidation. We used a tumor genotyping assay focused on clinically actionable targets to profile Merkel cell carcinoma (MCC), a rare and very aggressive neuroendocrine malignancy. We detected activating mutations in the PIK3CA gene in a subset of MCC tumors, most of which were not infected with the Merkel cell polyoma virus (MCPyV). Remarkably, in MCC cell lines, the presence of a PIK3CA mutation conferred sensitivity to PI3K inhibitors. Our findings suggest that PI3K pathway activation may drive tumorigenesis in a subset of MCC and that screening for PIK3CA mutations could help identify MCC patients who may potentially benefit from treatment with PI3K pathway inhibitors.
Acknowledgments
Financial support: P30 CA06516 (BYY)
Footnotes
Conflict of interest: DDS and AJI filed a patent application for the SNaPshot methods used in this study. DDS, AJI and DRB are consultants for Bio-Reference Laboratories, Inc. JAE receives consulting and research funding from Novartis.
References
- 1.Toker C. Trabecular carcinoma of the skin. Archives of dermatology. 1972;105:107–10. [PubMed] [Google Scholar]
- 2.Gould VE, Moll R, Moll I, Lee I, Franke WW. Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Laboratory investigation; a journal of technical methods and pathology. 1985;52:334–53. [PubMed] [Google Scholar]
- 3.Walsh NM. Primary neuroendocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol. 2001;32:680–9. doi: 10.1053/hupa.2001.25904. [DOI] [PubMed] [Google Scholar]
- 4.Smith PD, Patterson JW. Merkel cell carcinoma (neuroendocrine carcinoma of the skin) Am J Clin Pathol. 2001;115(Suppl):S68–78. doi: 10.1309/MVXR-8BM1-GPR7-P5NP. [DOI] [PubMed] [Google Scholar]
- 5.TRMCCG. Merkel cell carcinoma: recent progress and current priorities on etiology, pathogenesis, and clinical management. J Clin Oncol. 2009;27:4021–6. doi: 10.1200/JCO.2009.22.6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodgson NC. Merkel cell carcinoma: changing incidence trends. Journal of surgical oncology. 2005;89:1–4. doi: 10.1002/jso.20167. [DOI] [PubMed] [Google Scholar]
- 7.Lunder EJ, Stern RS. Merkel-cell carcinomas in patients treated with methoxsalen and ultraviolet A radiation. The New England journal of medicine. 1998;339:1247–8. doi: 10.1056/NEJM199810223391715. [DOI] [PubMed] [Google Scholar]
- 8.Howard RA, Dores GM, Curtis RE, Anderson WF, Travis LB. Merkel cell carcinoma and multiple primary cancers. Cancer Epidemiol Biomarkers Prev. 2006;15:1545–9. doi: 10.1158/1055-9965.EPI-05-0895. [DOI] [PubMed] [Google Scholar]
- 9.Albores-Saavedra J, Batich K, Chable-Montero F, Sagy N, Schwartz AM, Henson DE. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. Journal of cutaneous pathology. 2009 doi: 10.1111/j.1600-0560.2009.01370.x. [DOI] [PubMed] [Google Scholar]
- 10.Heath M, Jaimes N, Lemos B, Mostaghimi A, Wang LC, Penas PF, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. Journal of the American Academy of Dermatology. 2008;58:375–81. doi: 10.1016/j.jaad.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poulsen M. Merkel cell carcinoma of skin: diagnosis and management strategies. Drugs & aging. 2005;22:219–29. doi: 10.2165/00002512-200522030-00004. [DOI] [PubMed] [Google Scholar]
- 12.Wong HH, Wang J. Merkel cell carcinoma. Archives of pathology & laboratory medicine. 2010;134:1711–6. doi: 10.5858/2009-0165-RSR2.1. [DOI] [PubMed] [Google Scholar]
- 13.Hanly AJ, Elgart GW, Jorda M, Smith J, Nadji M. Analysis of thyroid transcription factor-1 and cytokeratin 20 separates merkel cell carcinoma from small cell carcinoma of lung. Journal of cutaneous pathology. 2000;27:118–20. doi: 10.1034/j.1600-0560.2000.027003118.x. [DOI] [PubMed] [Google Scholar]
- 14.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science (New York, NY) 2008;319:1096–100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kassem A, Schopflin A, Diaz C, Weyers W, Stickeler E, Werner M, et al. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer research. 2008;68:5009–13. doi: 10.1158/0008-5472.CAN-08-0949. [DOI] [PubMed] [Google Scholar]
- 16.Tolstov YL, Pastrana DV, Feng H, Becker JC, Jenkins FJ, Moschos S, et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. International journal of cancer. 2009;125:1250–6. doi: 10.1002/ijc.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A. 2008;105:16272–7. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sastre-Garau X, Peter M, Avril MF, Laude H, Couturier J, Rozenberg F, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218:48–56. doi: 10.1002/path.2532. [DOI] [PubMed] [Google Scholar]
- 20.Houben R, Shuda M, Weinkam R, Schrama D, Feng H, Chang Y, et al. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol. 2010;84:7064–72. doi: 10.1128/JVI.02400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houben R, Adam C, Baeurle A, Hesbacher S, Grimm J, Angermeyer S, et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. International journal of cancer. 2011 doi: 10.1002/ijc.26076. [DOI] [PubMed] [Google Scholar]
- 22.Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO molecular medicine. 2010;2:146–58. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, editors. AJCC cancer staging manual. 7. Chapter 30. New York, NY: Springer; 2010. Merkel cell carcinoma. [Google Scholar]
- 24.Leonard JH, Dash P, Holland P, Kearsley JH, Bell JR. Characterisation of four Merkel cell carcinoma adherent cell lines. International journal of cancer. 1995;60:100–7. doi: 10.1002/ijc.2910600115. [DOI] [PubMed] [Google Scholar]
- 25.Ronan SG, Green AD, Shilkaitis A, Huang TS, Das Gupta TK. Merkel cell carcinoma: in vitro and in vivo characteristics of a new cell line. J Am Acad Dermatol. 1993;29:715–22. doi: 10.1016/0190-9622(93)70236-m. [DOI] [PubMed] [Google Scholar]
- 26.Van Gele M, Leonard JH, Van Roy N, Van Limbergen H, Van Belle S, Cocquyt V, et al. Combined karyotyping, CGH and M-FISH analysis allows detailed characterization of unidentified chromosomal rearrangements in Merkel cell carcinoma. International journal of cancer. 2002;101:137–45. doi: 10.1002/ijc.10591. [DOI] [PubMed] [Google Scholar]
- 27.Rosen ST, Gould VE, Salwen HR, Herst CV, Le Beau MM, Lee I, et al. Establishment and characterization of a neuroendocrine skin carcinoma cell line. Lab Invest. 1987;56:302–12. [PubMed] [Google Scholar]
- 28.Duncavage EJ, Zehnbauer BA, Pfeifer JD. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516–21. doi: 10.1038/modpathol.2009.3. [DOI] [PubMed] [Google Scholar]
- 29.Helmbold P, Lahtz C, Enk A, Herrmann-Trost P, Marsch W, Kutzner H, et al. Frequent occurrence of RASSF1A promoter hypermethylation and Merkel cell polyomavirus in Merkel cell carcinoma. Molecular carcinogenesis. 2009;48:903–9. doi: 10.1002/mc.20540. [DOI] [PubMed] [Google Scholar]
- 30.Growdon WB, Boisvert SL, Akhavanfard S, Oliva E, Dias-Santagata DC, Kojiro S, et al. Decreased survival in EGFR gene amplified vulvar carcinoma. Gynecol Oncol. 2008;111:289–97. doi: 10.1016/j.ygyno.2008.07.038. [DOI] [PubMed] [Google Scholar]
- 31.Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A. 2009;106:19503–8. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yaguchi S, Fukui Y, Koshimizu I, Yoshimi H, Matsuno T, Gouda H, et al. Antitumor activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor. J Natl Cancer Inst. 2006;98:545–56. doi: 10.1093/jnci/djj133. [DOI] [PubMed] [Google Scholar]
- 33.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Molecular cancer therapeutics. 2008;7:1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 34.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer research. 2008;68:8022–30. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 35.Kuwamoto S, Higaki H, Kanai K, Iwasaki T, Sano H, Nagata K, et al. Association of Merkel cell polyomavirus infection with morphologic differences in Merkel cell carcinoma. Hum Pathol. 2011;42:632–40. doi: 10.1016/j.humpath.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 36.Laude HC, Jonchere B, Maubec E, Carlotti A, Marinho E, Couturaud B, et al. Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dworkin AM, Tseng SY, Allain DC, Iwenofu OH, Peters SB, Toland AE. Merkel cell polyomavirus in cutaneous squamous cell carcinoma of immunocompetent individuals. J Invest Dermatol. 2009;129:2868–74. doi: 10.1038/jid.2009.183. [DOI] [PubMed] [Google Scholar]
- 38.Teman CJ, Tripp SR, Perkins SL, Duncavage EJ. Merkel cell polyomavirus (MCPyV) in chronic lymphocytic leukemia/small lymphocytic lymphoma. Leuk Res. 2011;35:689–92. doi: 10.1016/j.leukres.2011.01.032. [DOI] [PubMed] [Google Scholar]
- 39.Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. Journal of the National Cancer Institute. 2009;101:938–45. doi: 10.1093/jnci/djp139. [DOI] [PubMed] [Google Scholar]
- 40.Bhatia K, Goedert JJ, Modali R, Preiss L, Ayers LW. Immunological detection of viral large T antigen identifies a subset of Merkel cell carcinoma tumors with higher viral abundance and better clinical outcome. International journal of cancer. 2010;127:1493–6. doi: 10.1002/ijc.25136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeCaprio JA. Does detection of Merkel cell polyomavirus in Merkel cell carcinoma provide prognostic information? J Natl Cancer Inst. 2009;101:905–7. doi: 10.1093/jnci/djp162. [DOI] [PubMed] [Google Scholar]
- 42.Deyrup AT, Lee VK, Hill CE, Cheuk W, Toh HC, Kesavan S, et al. Epstein-Barr virus-associated smooth muscle tumors are distinctive mesenchymal tumors reflecting multiple infection events: a clinicopathologic and molecular analysis of 29 tumors from 19 patients. The American journal of surgical pathology. 2006;30:75–82. doi: 10.1097/01.pas.0000178088.69394.7b. [DOI] [PubMed] [Google Scholar]
- 43.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. The New England journal of medicine. 363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katano H, Ito H, Suzuki Y, Nakamura T, Sato Y, Tsuji T, et al. Detection of Merkel cell polyomavirus in Merkel cell carcinoma and Kaposi’s sarcoma. J Med Virol. 2009;81:1951–8. doi: 10.1002/jmv.21608. [DOI] [PubMed] [Google Scholar]
- 45.Lassacher A, Heitzer E, Kerl H, Wolf P. p14ARF hypermethylation is common but INK4a-ARF locus or p53 mutations are rare in Merkel cell carcinoma. J Invest Dermatol. 2008;128:1788–96. doi: 10.1038/sj.jid.5701256. [DOI] [PubMed] [Google Scholar]
- 46.Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in merkel cell carcinoma. Clin Cancer Res. 2011;17:4806–13. doi: 10.1158/1078-0432.CCR-10-3363. [DOI] [PubMed] [Google Scholar]
- 47.Qiu W, Schonleben F, Li X, Ho DJ, Close LG, Manolidis S, et al. PIK3CA mutations in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:1441–6. doi: 10.1158/1078-0432.CCR-05-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106:16996–7001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vazquez F, Devreotes P. Regulation of PTEN function as a PIP3 gatekeeper through membrane interaction. Cell Cycle. 2006;5:1523–7. doi: 10.4161/cc.5.14.3005. [DOI] [PubMed] [Google Scholar]
- 50.Van Gele M, Leonard JH, Van Roy N, Cook AL, De Paepe A, Speleman F. Frequent allelic loss at 10q23 but low incidence of PTEN mutations in Merkel cell carcinoma. International journal of cancer. 2001;92:409–13. doi: 10.1002/ijc.1209. [DOI] [PubMed] [Google Scholar]
- 51.Fernandez-Figueras MT, Puig L, Musulen E, Gilaberte M, Lerma E, Serrano S, et al. Expression profiles associated with aggressive behavior in Merkel cell carcinoma. Mod Pathol. 2007;20:90–101. doi: 10.1038/modpathol.3800717. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature reviews. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 53.Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, et al. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther. 2009;8:2204–10. doi: 10.1158/1535-7163.MCT-09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–83. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.