

Abstract
The environmental toxin and carcinogen 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin) binds and activates the transcription factor aryl hydrocarbon receptor (AHR), inducing CYP1 family cytochrome P450 enzymes. CYP1A2 and its avian ortholog CYP1A5 are highly active arachidonic acid epoxygenases. Epoxygenases metabolize arachidonic acid to four regioisomeric epoxyeicosatrienoic acids (EETs) and selected monohydroxyeicosatetraenoic acids (HETEs). EETs can be further metabolized by epoxide hydrolases to dihydroxyeicosatrienoic acids (DHETs). As P450–arachidonic acid metabolites affect vasoregulation, responses to ischemia, inflammation, and metabolic disorders, identification of their production in vivo is needed to understand their contribution to biologic effects of TCDD and other AHR activators. Here we report use of an acetonitrile-based extraction procedure that markedly increased the yield of arachidonic acid products by lipidomic analysis over a standard solid-phase extraction protocol. We show that TCDD increased all four EETs (5,6-, 8,9-, 11,12-, and 14,15-), their corresponding DHETs, and 18- and 20-HETE in liver in vivo and increased 5,6-EET, the four DHETs, and 18-HETE in heart, in a chick embryo model. As the chick embryo heart lacks arachidonic acid–metabolizing activity, the latter findings suggest that arachidonic acid metabolites may travel from their site of production to a distal organ, i.e., heart. To determine if the TCDD–arachidonic acid–metabolite profile could be altered pharmacologically, chick embryos were treated with TCDD and the soluble epoxide hydrolase inhibitor 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA). Cotreatment with AUDA increased hepatic EET-to-DHET ratios, indicating that the in vivo profile of P450–arachidonic acid metabolites can be modified for potential therapeutic intervention.
Introduction
In addition to metabolizing xenobiotics, cytochrome P450 enzymes (P450s) can metabolize endogenous substrates including arachidonic acid (Rifkind, 2006). P450s exhibiting arachidonic acid epoxygenase activity include 1A2 (avian 1A5) (Gilday et al., 1996; Rifkind, 2006), 1B1 (Choudhary et al., 2004), 2J2 (Wu et al., 1996), 2B (avian 2H) (Nakai et al., 1992), 2C8, 2C9 (Daikh et al., 1994; Rifkind et al., 1995; Rifkind, 2006), and 2C19 (Bylund et al., 1998). Some of these epoxygenases can be transcriptionally induced. For example, CYP1A arachidonic acid epoxygenases are increased by aryl hydrocarbon receptor (AHR) activators, including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), benzo(a)pyrene, β-naphthoflavone, and tryptophan photoproducts. CYP2B (avian 2H) arachidonic acid epoxygenases are induced by phenobarbital, an activator of different nuclear receptors, i.e., constitutive androstane receptor (CAR) and the pregnane X receptor (PXR) (Nakai et al., 1992; Gilday et al., 1996; Gannon et al., 2000; Willson and Kliewer, 2002; Diani-Moore et al., 2006a; Rifkind, 2006). Imidazole-based drugs (i.e., vorozole, omeprazole, benzylimidazole) increase arachidonic acid metabolism by mixed induction of CYP1 and CYP2 family P450s (Diani-Moore et al., 2006b). Significantly, the different P450 epoxygenases all produce the same arachidonic acid products: four regioisomeric epoxides, 5,6-, 8,9-, 11,12-, and 14,15-epoxyeicosatetraenoic acids, and terminal hydroxyeicosatetraenoic acids (HETEs), i.e., 16-19 HETE (Capdevila et al., 1981), although the relative amounts of the products can vary for different P450s.
Transcriptional induction of CYP1A arachidonic acid epoxygenases by AHR activation occurs in livers of birds and fish as well as mammals (Nakai et al., 1992; Rifkind et al., 1995; Schlezinger et al., 1998; Gannon et al., 2000; Diani-Moore et al., 2006a; Bui et al., 2012), albeit with species differences in the relative production of different P450 arachidonic acid metabolites (Capdevila, Karara et al., 1990; Lee and Riddick, 2012). In vivo accumulation of arachidonic acid metabolites after TCDD treatment has been reported in liver of the marine fish Stenotomus chrysops and of mice (Schlezinger et al., 1998; Bui et al., 2012), supporting a biologic role for P450-generated arachidonic acid metabolites.
In these studies, we have used the chick embryo near hatching, a well established model for studying TCDD toxicity and P450-mediated arachidonic acid epoxygenation (Rifkind et al., 1990), to ask whether we could improve detection of arachidonic acid epoxygenase products increased in vivo in liver by TCDD treatment using a new extraction method. Further, as eicosatrienoic acids (EETs) and dihydroxyeicosatrienoic acids (DHETs) can affect cardiac function, and TCDD exerts cardiac toxicity in both the chick embryo and mammalian species (Canga et al., 1988; Canga et al., 1993; Walker and Catron, 2000), we asked whether TCDD treatment can alter the content of P450–arachidonic acid products in heart notwithstanding the lack of cardiac TCDD-induced arachidonic acid epoxygenases (Gannon et al., 2000). We also tested the hypothesis that the in vivo profile of P450-mediated arachidonic acid products after exposure to TCDD could be modified pharmacologically.
We demonstrate here, using an improved acetonitrile-based procedure for extracting P450-arachidonic acid metabolites, that TCDD treatment increases EETs and DHETs in liver and increases 5,6-EET and DHETs in heart. We also report that treatment of chick embryos with the epoxide hydrolase inhibitor 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA) (Morisseau et al., 2002) alters in vivo hepatic P450-dependent arachidonic acid metabolites, indicating that the TCDD-induced arachidonic acid metabolite profile in vivo can be modulated for potential therapeutic control.
Materials and Methods
Treatments.
TCDD (1 nmol per egg) or equal amounts of the solvent dioxane (Kanetoshi et al., 1992) were injected in ovo in 14-day-old chick embryo into the fluids surrounding the embryo. Treatments were for 24 hours unless specified. For the experiments involving the soluble epoxide hydrolase inhibitor 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA; Cayman Chemical Company, Ann Arbor MI), AUDA was dissolved in dimethylsulfoxide (20 mg/ml) and 100 μl (2 mg AUDA) were injected into the egg together with TCDD at 1 nmol per egg for 6 hours.
Solid-Phase Extraction of Arachidonic Acid Metabolites.
We initially examined a solid-phase method to extract arachidonic acid metabolites (Powell, 1982) from liver homogenates. Liver samples were homogenized in KPO4 buffer (1:1; w/v). Ethanol (175 μl) and acetic acid (25 μl) were added to 1-ml samples of homogenates, which were then vortexed and centrifuged at 1500 rpm for 3 minutes. Supernatants containing arachidonic acid metabolites were loaded on disposable extraction columns (Bakerbond spe Octadecyl (C18); J T Baker, Phillipsburg, NJ) previously washed with 5 ml of ethanol followed by 15 ml of dH2O. After sample loading, columns were washed four times with 5 ml dH2O. Arachidonic acid metabolites were eluted with 5 ml of ethyl acetate containing 0.005% butylated hydroxytoluene (BHT). The organic (upper) phases of the eluates containing arachidonic acid metabolites were transferred into new glass tubes. One ml of the ethyl-acetate/BHT mixture was added to the aqueous phase, passed through the column, and the upper organic phase was combined with the organic phase for the first eluate. This procedure was repeated to enhance metabolite recovery. The combined organic phases were dried under N2 gas and the pellets were stored at –80°C. The pellets were resuspended in 25 μl of 35% acetonitrile/0.1% acetic acid/65% dH2O and used for liquid chromatography–tandem mass spectroscopy (LC-MS/MS) analysis.
Acetonitrile Extraction of Arachidonic Acid Metabolites.
Livers and hearts from 17-day-old chick embryos were homogenized in acetonitrile (1:2; w/v) and sonicated (5 seconds, 4 times). Four hundred μl of the liver homogenates or the total volumes of the heart homogenates were transferred to glass tubes. 11,12 EETd11 (10 ng) and 11,12 DHETd11 (1 ng) were added as internal standards to each tube. After adding 2 ml of acetonitrile, the homogenates were incubated overnight at –80°C. Then samples were sonicated (5 seconds, 4 times), vortexed for 5 seconds, transferred to fresh tubes, and centrifuged for 10 minutes at 2000 rpm. The supernatants were transferred to glass conical tubes and dried under N2 gas. Dried pellets were resuspended in 50 μl of methanol, transferred to 1.5-ml Eppendorf tubes, and centrifuged for 5 minutes to remove particulates. A 5-μl aliquot of each sample was used for LC-MS/MS analysis.
LC-MS/MS Analysis.
LC-MS/MS analysis was performed using an Agilent 6460 Triple Quad LC/MS system (Agilent Technologies, Palo Alto, CA), containing an Agilent 1290 Infinity HPLC for compound separation and a JetStream ESI source for ion generation. Analysis was performed in the multiple reaction monitoring (MRM) mode. Compounds were separated on an Agilent SB-Aq C18 2.0 × 150 mm, 1.8-μm column at a flow rate of 400 μl/min. Mobile phase A was water with 0.1% formic acid and mobile phase B, acetonitrile with 0.1% formic acid. The LC elution gradient was: 0–2 minutes, 30% B; 2–12 minutes to 65% B; 12–12.5 minutes to 95% B; 12.5–14.5 minutes, 95% B; 14.5–15 minutes to 30% B; 15–20 minutes, 30% B. Heated N2 gas (12 l/ min at 400°C) was used as the sheath gas to facilitate evaporation of solvent from the ionization chamber. MRM analysis was performed in the negative ion mode. In the initial phase of these studies, compounds of interest were monitored using two different mass transitions and the one with the optimal specificity and signal was chosen. The selected parameters for MRM transitions from parent to product ions for the analysis of metabolites in the biologic samples (Supplemental Fig. 1) were as follows: From m/z 337 to m/z 207 (14,15-DHET), to 167 (11,12-DHET), to 127 (8,9-DHET), and to 145 (5,6-DHET); from m/z 319 to m/z 245 (20-HETE), to 261 (18-HETE), to 219 (14,15-EET), to 167 (11,12-EET), to 69 (8,9-EET), and to 191 (5,6-EET); from m/z 348 to m/z 167 (11,12-DHETd11); from m/z 330 to m/z 167 (11,12-EETd11). The MRM data were processed using Agilent Mass Hunter Quantitative Analysis Software. Fifteen-point standard curves containing the following standards (Cayman Chemical Company) were used for quantitation: (±)5(6)-epoxy-8Z,11Z,14Z-eicosatrienoic acid; (±)8(9)-epoxy-5Z,11Z,14Z-eicosatrienoic acid; (±)11(12)-epoxy-5Z,8Z,14Z-eicosatrienoic acid; (±)14(15)-epoxy-5Z,8Z,11Z-eicosatrienoic acid; (±)5,6-dihydroxy-8Z,11Z,14Z-eicosatrienoic acid; (±)8,9-dihydroxy-5Z,11Z,14Z-eicosatrienoic acid; (±)11,12-dihydroxy-5Z,8Z,14Z-eicosatrienoic acid; (±)14,15-dihydroxy-5Z,8Z,11Z-eicosatrienoic acid; (±)18-hydroxy-5Z,8Z,11Z,14Z-eicosatetraenoic acid, and 20-hydroxy-5Z,8Z,11Z,14Z-eicosatetraenoic acid. Concentrations of the standards ranged from 0.3 ng/ml to 5000 ng/ml for EETs and 0.12 ng/ml to 2000 ng/ml for DHETs and HETEs. Successive dilutions (1:2) were made in methanol. All samples and dilutions of the standards were spiked with deuterated standards: (±)11(12)-epoxy-5Z,8Z,14Z-eicosatrienoic-16,16,17,17,18,18,19,19,20,20,20 acid (11,12-EETd11, 200 ng/ml) and (±)11,12-dihydroxy-5Z,11Z,14Z-eicosatrienoic-16,16,17,17,18,18,19,19,20,20,20-d11 acid (11,12-DHETd11, 20 ng/ml). Metabolite concentrations were determined by reference to the standards. Signal-to-noise ratios ≥ 3 were required for a peak to be considered identifiable and ≥ 10 to be quantifiable.
cDNA Preparation and Quantitative Polymerase Chain Reaction.
Total RNA was extracted from samples of chick embryo liver or heart using RNA STAT-60 (Tel-Test “B” Inc., Friendswood, TX) following the manufacturer’s directions. cDNAs were prepared as follows: 1 μg of total RNA, 4 μl of qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD), and water (final volume of 20 μl) were mixed and incubated for 5 minutes at 25°C, 40 minutes at 42°C, 5 minutes at 85°C. The cDNA was diluted 1:5 with water before using it for real-time polymerase chain reaction (PCR). For PCR, 2 μl of cDNA, 10 μl of PerfeCTa SYBR Green FastMix (Quanta Biosciences), 1 μl of each primer (10 μM), and 6 μl of nuclease-free water. Primers and annealing temperatures (in parenthesis) were as follows: for CYP1A4, 5′-tgctcgcactgaaggaatca-3′ and 5′- ctgggcctcttattgctttg-3′ (56°C); for CYP1A5, 5′-cttcatgcccttcaccatc-3′ and 5′-caggccaaaagtcatcac-3′ (55°C); for CYP1B1, 5′-tggctttcctgtacgaatcc-3′ and 5′-tctgggttggaccatttagc-3′ (55°C); for EPHX1, 5′-gatgacagcaagaaaaatgc-3′ and 5′-gaagtctttgaggagggaaa-3′(53°C); for EPHX2, 5′-gatgctggcttccgtgttat-3′ and 5′-tccaggaaaatggtcaggtc-3′(55°C); for CYP4V, 5′-tgtgttgccaaacacctcat-3′and 5′-aactgcaatgggcaagaatc-3′(53°C); for CYP4B, 5′-accccacactgagaaccttg-3′ and 5′-tggaggagaacgaggctaaa-3′ (56°C); for CYP2C45, 5′-gattgaccgggtagtaggac-3′ and 5′-tgaagtggatgtctttggtc-3′ (53°C); for CYP2J2, 5′-acatcatctgctctgtcacc-3′ and 5′-tcttcttgcacacaaacctt-3′(53°C); for CYP2H1, 5′-ccatgggttatttgttttgtgtt-3′and 5′-gctgctgccttgttcgttc-3′ (53°C); for 18s ribosomal RNA, 5′-gaccataaacgatgccgact-3′ and 5′-agacaaatcgctccaccaac-3′ (55°C). Denaturation and elongation temperatures were 95°C and 60°C, respectively. An Eppendorf Mastercycler epgradient machine was used for 40 cycles of amplification. The fold-changes in mRNA were calculated using the standard 2−ΔΔCt method (Livak and Schmittgen, 2001) with 18S mRNA serving as a control for normalization.
Arachidonic Acid Metabolism Assay in Vitro.
Livers and hearts from chick embryos treated for 24 hours with TCDD or solvent (controls) were homogenized in three volumes of 0.1 M KPO4, pH 7.4, and stored at 80°C. Arachidonic acid metabolism was assayed as previously described (Capdevila, Falck et al., 1990; Rifkind et al., 1994; Gannon et al., 2000). Reaction mixtures (total volume 0.25 ml) containing 1 or 3 mg wet weight of liver or heart homogenate, 30 μM [1-14C] arachidonic acid (48 mCi/mmol) (Perkin Elmer, Torrance, CA), and 10 mM MgCl2. After preincubation for 2 minutes at 37°C, reactions were started with a mixture of 1 mM NADPH, 10 mM isocitric acid, 0.2 IU of isocitric dehydrogenase/ml, and incubated for 10 minutes at 37°C. Addition of 0.1 ml of acetic acid was followed by two extractions, each with 3 ml of ethyl acetate containing 0.005% BHT. The organic phases were combined, dried under N2, and resuspended in 0.11 ml of 50% acetonitrile in water containing 0.1% acetic acid. Products in 0.05 ml were resolved by reverse-phase high-performance liquid chromatography (HPLC) using a Vydac C18 column (Vydac, Hesperia, CA) (90 Å, 5-μm particle size, 4.6 × 250 mm), on a linear gradient from 50 to 100% acetonitrile in water containing 0.1% acetic acid, at 1 ml per minute for 40 minutes. Radioactivity was measured using a Flo-One Beta Model S radioactivity flow monitor (Packard Instrument Company, Downers Grove, IL). Products were identified by reference to HPLC retention times of pure standards (Rifkind et al., 1994).
Statistics.
P values for differences between group means were obtained using unpaired t tests (GraphPad Prism 5; San Diego, CA). P values ≤ 0.05 were accepted as statistically significant.
Results
Arachidonic Acid Metabolite Production in Vitro.
The HPLC chromatograms in Fig. 1 demonstrate as previously reported (Gannon et al., 2000) that TCDD treatment of chick embryos in ovo enhances arachidonic acid metabolite production by homogenates of liver (Fig. 1A), but not by homogenates from heart (Fig. 1B) even at three times the protein concentration used for the liver assay. 20-HETE was the only arachidonic acid metabolite detected for liver homogenates from chick embryos treated with solvent alone (controls) while the homogenates from livers of TCDD-treated chick embryos exhibited increased formation of EETs and their epoxide hydrolase products, DHETs, as well as several hydroxyeicosatetraenoic acids, HETEs. TCDD decreased 20-HETE formation. In contrast, heart homogenates from the controls or from TCDD-treated chick embryo (Fig. 1B) did not generate any arachidonic acid metabolites.
Fig. 1.
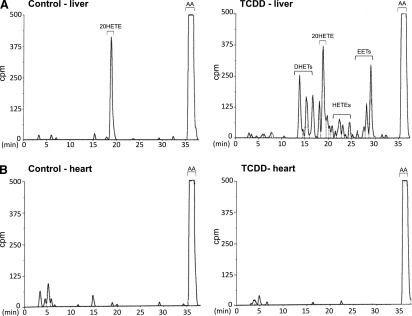
Arachidonic acid metabolite production in vitro by homogenates of chick embryo liver and heart. Representative reverse-phase HPLC chromatograms showing arachidonic acid metabolite production in vitro by 1 mg of liver homogenate (A) and 3 mg of heart homogenate (B) per reaction mixture, after treatment of chick embryos in ovo with TCDD (1 nmol per egg for 24 hours), or for the same amounts of homogenate from solvent controls. Arachidonic acid metabolism was assayed as described in Materials and Methods. Peaks between 0 and 10 minutes are non-P450-mediated polar products. AA, arachidonic acid. Retention times (min) are shown on the abscissas.
Extraction of EETs, DHETs, and HETEs from Liver and Heart; Comparison of Acetonitrile and Solid-Phase Extraction.
We first conducted preliminary experiments on liver samples from TCDD-treated chick embryos, using a standard solid-phase extraction protocol with C18 columns (see Materials and Methods) (Powell, 1982; Nithipatikom et al., 2003). Recoveries of most eicosanoid metabolites of interest were low or undetectable (data not shown). To enhance the recovery of P450-mediated arachidonic acid products, we developed an acetonitrile-based extraction procedure and compared the results with those obtained with C18 extraction. P450 arachidonic acid products were measured by LC-MS/MS. Table 1 shows that use of the solid-phase extraction method permitted measurement of 8,9- and 11,12-EETs and 8,9-, 11,12-, and 14,15-DHETs but not of 5,6- or 14,15-EET or 5,6-DHET in liver after TCDD-treatment. The acetonitrile extraction procedure, in contrast, permitted measurement of all four EET regioisomers and their DHET products. Moreover, product yields were substantially greater for acetonitrile extraction than for solid phase extraction. Mean levels of 11,12-EETd11 in samples spiked with1 ng of 11,12-EETd11 were 4-fold higher for samples extracted in acetonitrile compared with C18 extraction, in agreement with recoveries shown for detectable EETs in Table 1.
TABLE 1.
Comparison of solid-phase (C18 column) and acetonitrile extraction for LC-MS/MS detection of arachidonic acid metabolites from liver of a chick embryo treated for 24 hours with TCDD as described in Materials and Methods
Means ± S.E. (n = 3 replicates) are shown in the table.
| Method | EETs | DHETS | 20-HETE | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 5,6- | 8,9- | 11,12- | 14,15- | 5,6- | 8,9- | 11,12- | 14,15- | ||
| ng/g liver | |||||||||
| C18 column | n.d. | 2 ± 0.8 | 2.4 ± 1.4 | n.d. | n.d. | 2.7 ± 0.9 | 4 ± 0.4 | 7 ± 2 | 4.4 ± 1 |
| Acetonitrile | 195 ± 106 | 9 ± 3 | 10 ± 1.7 | 4.2 ± 0.5 | 5.6 ± 0.5 | 39 ± 7 | 133 ± 11 | 288 ± 37 | 35 ± 4 |
n.d., not detected.
To determine the extent to which EETs were recovered from liver samples after extraction in acetonitrile, we added 10 ng of 11,12-EETd11 to liver samples followed by acetonitrile extraction and compared the values for peak areas for the extracted 11,12-EETd11 to those for replicates of the same amount of 11,12-EETd11 not added to liver samples or extracted (taken as the 100% value). The results (Supplemental Fig. 2) show that acetonitrile extraction of liver samples permitted 100% recovery of the deuterated standard.
Further, to be sure that the DHETs measured in tissues had been produced in vivo and not during sample manipulation, we added deuterated 11,12-EET (11,12-EETd11) to samples and measured its conversion to 11,12-DHETd11. When livers were homogenized in phosphate-buffered saline followed by acetonitrile extraction of arachidonic acid products, high levels of 11,12-DHETd11 were produced (data not shown), but 11,12-DHETd11 formation was barely detectable when tissues were both homogenized and extracted in acetonitrile (e.g., mean peak areas for 11,12-DHETd11 were 3200 versus 32, respectively) indicating that homogenization in acetonitrile prevented EET breakdown during sample preparation. In addition we examined the effects of various compounds that are commonly added to tissues before homogenization for their potential to prevent alteration of biologic products during sample preparation. Neither AUDA, an inhibitor of soluble epoxide hydrolase (Morisseau et al., 2002), miconazole, an inhibitor of cytochrome P450 (Cunningham et al., 2007), nor the hydrogen peroxide scavenger catalase altered the results when added to acetonitrile before tissue homogenization (data not shown), providing assurance that the metabolites extracted and measured were formed in the physiologic environment and not during sample processing. Accordingly, for all subsequent studies, acetonitrile was used for tissue homogenization and arachidonic acid metabolite extraction, without other additions.
TCDD Increases Arachidonic Acid Metabolites in Liver and Heart.
Using the acetonitrile method we then sought to examine the effect of TCDD on P450-mediated arachidonic acid metabolites in vivo, focusing on liver and heart, both of which are targets of TCDD toxicity. LC-MS/MS results for arachidonic acid metabolite profiles of livers and hearts from 14-day-old chick embryos treated for 24 hours with TCDD (1 nmol/egg) or dioxane (solvent controls) are shown in Fig. 2. Low levels of 5,6-EET and 8,9-, 11,12-, and 14,15-DHETs were detected in the control livers. TCDD treatment increased the liver content of all four EETs and DHETs (Fig. 2A). TCDD produced the highest levels for 5,6-EET and 14,15-DHET (increased from 23 ± 6 in controls to 399 ± 22 and from 31 ± 8 to 292 ± 22 ng/g liver, respectively) followed by the 11,12-DHET (from 3 ± 1 to 95 ± 9 ng/g liver). TCDD also increased in vivo levels of hepatic 18- and 20-HETE. Arachidonic acid metabolites were also detected in the heart although at much lower levels than in liver. 8,9-, 11,12-, 14,15- DHET and 18- and 20-HETE were detected in the control hearts (Fig. 2B), but EETs were not detected. After TCDD treatment 5,6-EET became detectable (21 ± 4 ng/g heart). A peak for 11,12-EET could be detected but not quantified (signal-to-noise < 10). Levels of 8,9-, 11,12-, 14,15- DHETs and 18-HETE were also increased, particularly the 14,15-DHET (from 3.7 ± 0.8 to 42 ± 6 ng/g heart).
Fig. 2.

Arachidonic acid metabolite production in vivo in chick embryo liver and heart. Arachidonic acid metabolites were extracted from livers and hearts of chick embryos treated for 24 hours with TCDD (1 nmol/egg) or solvent (controls) as described in Materials and Methods. Means ± S.E. for arachidonic acid–metabolite levels from liver (A) and from heart (B) are plotted. n = 5 individual livers or hearts for each treatment group. **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001; n.s., not significant.
Effects of TCDD on mRNA Levels of Enzymes That Catalyze P450-Dependent Arachidonic Acid Metabolism.
We had previously shown that TCDD increased CYP1A5 in liver but not in heart (Gannon et al., 2000), and that CYP1A5 and 2H were the major arachidonic acid epoxygenases in chick embryo liver microsomes after TCDD or phenobarbital treatment, respectively (Kanetoshi et al., 1992; Nakai et al., 1992). To determine whether TCDD affected mRNA levels of P450 epoxygenases other than CYP1A5 we examined effects of TCDD on mRNA levels of chick orthologs of known mammalian P450 epoxygenases in liver and heart (Fig. 3).
Fig. 3.

Effects of TCDD on mRNA levels of enzymes known to affect EETs, DHETs, or HETEs. Mean quantitative real-time PCR results (± S.E.) for total RNA extracted from livers (A) and hearts (B) from chick embryos treated for 24 hours with TCDD or solvent (controls). PCR conditions and specific primers for CYP1A4, CYP1A5, CYP1B1, CYP2C45, CYP2J2, CYP2H1, CYP4B, CYP4V, EPHX1, EPHX2 are described in Materials and Methods Fold changes in mRNAs for genes in livers and hearts of TCDD-treated chick embryos and controls (solvent-treated) are calculated and compared with the values for each gene in control liver for which each mRNA is set at “1” (n = 5 individual organs per treatment group, with three replicates for each organ). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001; n.s., not significant.
mRNAs for all of the P450s tested were measurable in control liver extracts. TCDD increased hepatic mRNA levels for CYP1A4 (a positive control for TCDD activation of the AHR) and CYP1A5, but not for other P450s with recognized arachidonic acid epoxygenase activity. Rather, TCDD diminished mRNA levels for CYP1B1- and CYP2-related epoxygenases (CYP2J2 and CYP2H1). TCDD did not affect mRNA for CYP2C45, the chicken ortholog of mammalian CYP2C19. TCDD suppressed mRNAs for CYP4V and CYP4B, avian orthologs for CYP4 enzymes, generators of 20-HETE (Kroetz and Xu, 2005; Rifkind, 2006). Both soluble and microsomal epoxide hydrolases were expressed in liver and heart. mRNA for the microsomal epoxide hydrolase (EPHX1) was not significantly affected by TCDD, while TCDD suppressed the mRNA for the soluble epoxide hydrolase (EPHX2). In heart, TCDD treatment increased only CYP1A4 but not 1A5 mRNAs and did not affect mRNAs for P450s CYP2J2 or CYP2C45 or the epoxide hydrolases. mRNA for CYP1B1 was increased 2-fold by TCDD and was also the only transcript whose level was differently affected by TCDD in liver versus heart. mRNAs for CYP2H1, 4B or 4V were undetected in both control and TCDD-treated heart extracts.
Cotreatment with TCDD and the Soluble Epoxide Hydrolase Inhibitor AUDA Alters the Hepatic Arachidonic Acid Metabolite Profile.
To learn whether the P450–arachidonic acid profile elicited by TCDD could be pharmacologically modified, we examined the effect of cotreatment of chick embryos with TCDD and the epoxide hydrolase inhibitor AUDA. A short treatment time of 6 hours was used because AUDA is rapidly metabolized (Hwang et al., 2007). Table 2 shows that cotreatment of chick embryo with TCDD and AUDA increased levels of all four EET regioisomers in liver compared with TCDD alone (by 1.9-, 7.2-, 10.5-, and 2.3-fold for 5,6-, 8,9-, 11,12-, and 14,15-EET, respectively) and increased ratios of EETs to DHETs. These findings are consistent with reports that AUDA inhibits DHET formation (Schmelzer et al., 2005). EET levels in the heart were below the limit of detection, precluding assessment of AUDA on cardiac EET/DHET ratios.
TABLE 2.
Effects of the soluble epoxide hydrolase inhibitor AUDA on EETs and DHETs in liver after treatment with TCDD or TCDD + AUDA for 6 hours
For EETs, P values (T+AUDA vs. T alone) are given in parentheses.a n = 3 individual livers for each treatment group. T, TCDD.
| EETs | Ratio EET/DHET | Fold Change in the Ratio EET/DHET T + AUDA vs. T | |||
|---|---|---|---|---|---|
| T | T + AUDA | T | T + AUDA | ||
| mean ng/g liver ± S.E. | mean ± S.E. | ||||
| 5,6- | 76.6 ± 13.5 | 141.8 ± 32.2 (P = 0.06) | 24.1 ± 4.3 | 75 ± 17 | 3.1 (P = 0.044) |
| 8,9- | 2.0 ± 0.1 | 14.4 ± 5.6 (P = 0.04) | 0.3 ± 0.01 | 3.8 ± 1.5 | 12.7 (P = 0.07) |
| 11,12- | 1.2 ± 0.8 | 12.6 ± 4.4 (P = 0.03) | 0.09 ± 0.06 | 0.9 ± 0.3 | 10 (P = 0.046) |
| 14,15- | 10.4 ± 0.2 | 24.1 ± 4.2 (P = 0.03) | 0.4 ± 0.01 | 0.8 ± 0.1 | 2 (P = 0.04) |
Control values for 5,6- and 14,15-EETs were 30.4 ± 0.8 and 9.6 ± 0.2, respectively, and for 8,9- and 11,12-EETs were not detectable.
Discussion
Using an acetonitrile extraction procedure for P450-dependent arachidonic acid products we report here that: 1) TCDD treatment in ovo increases the liver content in vivo of P450 epoxygenase products, including all four EET regioisomers and their DHET metabolites and of 18-HETE, and in the heart, although to a lesser extent, of 5,6-EET, and 8,9-, 11,12-, and 14,15-DHET; 2) The soluble epoxide hydrolase inhibitor AUDA modified the arachidonic acid profile produced in vivo in liver by TCDD treatment, increasing EET levels and EET to DHET ratios as compared with TCDD alone; and 3) TCDD increased levels of 20-HETE, a CYP4 product, in liver and heart.
The first finding answers affirmatively a recurring question in pharmacology: Is the ability of an agent to enhance microsomal enzyme activity as assayed in vitro reflected in a “real world” increase in product formation in vivo? We show here that enhanced microsomal enzyme production of P450 arachidonic acid products by AHR activation (i.e., by TCDD) is reflected in increased levels of arachidonic acid products in liver in vivo, consistent with previous findings (Schlezinger et al., 1998; Bui et al., 2012), and that this occurs widely among species, including avians as well as mammals.
We expect that our increased detection of EETs and DHETs in chick embryo liver and heart compared with prior reports (Schlezinger et al., 1998; Bui et al., 2012) primarily reflects the use of acetonitrile-based extraction, which greatly enhanced metabolite recovery, although species differences may also be a contributing factor. Thus TCDD increased EET production less in mouse than chick embryo liver microsomes (Lee et al., 1998), and along with other AHR agonists, failed to increase arachidonic acid epoxygenation in rat liver (Lee et al., 1998; Aboutabl et al., 2009). In that context, it may be of interest to note that human CYP1A2, like avian CYP1A5, is a highly active arachidonic acid epoxygenase (Rifkind et al., 1995), suggesting that the chick embryo may be more representative than the mouse of human TCDD-induced arachidonic acid epoxygenase activity.
The small amount of EETs and DHETs detected in control liver could reflect the low levels of expression of constitutive P450 epoxygenases (Fig. 3). The evidence that among the EETs, TCDD increased the 5,6 EET most, and among the DHETs, the 5,6 DHET least and the 14,15-DHET most, is consistent with reports that soluble epoxide hydrolase has greatest substrate preference for the 14,15- EET and least for the 5,6-EET (Chacos et al., 1983; Yu et al., 2000).
Our finding that P450 arachidonic acid products also accumulate in the heart after TCDD treatment is especially noteworthy because heart homogenates from TCDD-treated chick embryos lacked the ability to generate arachidonic acid epoxides, as assayed in vitro (see Fig. 1). Avian TCDD-induced CYP1A5 can entirely account for increased hepatic P450 epoxygenase activity by TCDD treatment, as immunoinhibition of CYP1A5 completely abolished TCDD-induced hepatic microsomal P450 arachidonic acid–metabolizing activity (Kanetoshi et al., 1992), but CYP1A5 cannot account for the increase in arachidonic acid metabolites in heart as TCDD does not induce CYP1A5 in chick embryo heart (Gannon et al., 2000) (see Fig. 3). It seems unlikely that the small increase in heart of mRNA for CYP1B1 (Fig. 3), a P450 reported to have some arachidonic acid epoxygenase activity (Choudhary et al., 2004), could explain the observed increase by TCDD in cardiac arachidonic acid products in vivo (Fig. 2), since the amount of CYP1B1 in heart homogenates was insufficient to generate any P450 arachidonic acid products (as shown in Fig. 1). It seems plausible, therefore, that the P450 arachidonic acid products detected in vivo in heart after TCDD treatment reflect metabolites circulating to heart from distal sites of their production, i.e., liver or kidney, organs in which TCDD increases both CYP1A5 and arachidonic acid epoxygenase activity (Gannon et al., 2000). That suggestion is consistent with reports that arachidonic acid epoxygenase products are found in plasma (Karara et al., 1992; Jiang et al., 2013) and can be increased after TCDD treatment (Bui et al., 2012). Circulation of P450 arachidonic acid products from one organ to another would imply further that EETs have endocrine as well as previously recognized autocrine and paracrine actions (Spector, 2009; Jiang et al., 2013). DHETs in heart could reflect circulation from a different organ, or conversion of EETs in situ in the heart, by resident cardiac epoxide hydrolases (Fig. 3B).
The second major finding, the ability of cotreatment with TCDD and the well established soluble epoxide hydrolase inhibitor AUDA to increase EET/DHET ratios in liver is of interest in several respects. First, although soluble epoxide hydrolase inhibitors have been reported to alter the arachidonic acid product composition in plasma (Revermann et al., 2010; Panigrahy et al., 2013), this is the first evidence of which we are aware for soluble epoxide hydrolase inhibition altering the endogenous P450 arachidonic acid metabolite profile in vivo in liver. The evidence that the in vivo arachidonic acid metabolite profile is susceptible to pharmacologic alteration implies that soluble epoxide hydrolase inhibition could be used for therapeutic modification of organ arachidonic acid composition in vivo. Notably, EETs have been shown to be beneficial by enhancing vascular flow, ameliorating inflammatory responses, cardiac reperfusion injury, and metabolic disorders, while DHETs have been associated with adverse effects (i.e., hypertension, inflammation) (Roman, 2002; Imig and Hammock, 2009; Morisseau and Hammock, 2013). The therapeutic use of soluble epoxide hydrolase inhibitors has attracted increasing attention not only for cardiovascular benefits (Morisseau and Hammock, 2013) but also for treatment of hepatic metabolic disturbances (Liu et al., 2012).
The third finding reported here, an increase by TCDD treatment in the hepatic content of 20-HETE, was unexpected because 20-HETE is a prototypic product of CYP4 enzymes (Kroetz and Xu, 2005; Rifkind, 2006), which are not increased by TCDD (see Fig. 3). Interestingly, Bui et al. (2012) also reported that TCDD increased 20-HETE in vivo in mouse liver. The finding also seems to contradict the substantial evidence that AHR activation by diverse agents, including TCDD, β-naphthoflavone, planar PCBs, and imidazole drugs, suppresses hepatic 20-HETE formation by microsomes in in vitro experiments in many species, including chick embryo, mouse, scup, rats, and guinea pigs (Huang and Gibson, 1991; Nakai et al., 1992; Lee et al., 1998; Schlezinger et al., 1998; Diani-Moore et al., 2006b) (also see Fig. 1). Moreover, as shown in Fig. 3, TCDD suppresses CYP4 mRNA levels. Some possible explanations for the increase in 20-HETE levels in the setting of suppressed CYP4 mRNA include TCDD enhancement of 20-HETE release from tissues and suppression of 20-HETE metabolism by β-oxidation. The latter would also be consistent with the energy-suppressing effects of the TCDD wasting syndrome. 20-HETE can have adverse effects, for example, abetting hypertension and arteriolar vasoconstriction (Roman et al., 2000; Kroetz and Xu, 2005; Hama-Tomioka et al., 2009). Our finding that 20-HETE is detected in control hearts, although not produced by heart homogenates (see Fig. 1) (consistent with the lack of CYP4B or CYP4V mRNA expression in heart, Fig. 3B), suggests that 20-HETE, like some other P450 arachidonic acid metabolites, may derive from another organ, possibly from blood vessels, major sites of 20-HETE production (Kroetz and Xu, 2005).
Supplementary Material
Acknowledgments
The authors thank Dr. Kasem Nithipatikom, Medical College of Wisconsin, for suggesting the use of acetonitrile to extract arachidonic acid metabolites and Dr. Ruba Saba Deeb, Weill Cornell Medical College, for assistance in the initial phases of this project. TCDD was provided by the National Cancer Institute Chemical Carcinogen Standards Repository, operated under contract by Midwest Research Institute, Kansas City, Missouri, NO2-CB-666000.
Abbreviations
- AHR
aryl hydrocarbon receptor
- AUDA
12-(3-adamantan-1-yl-ureido)-dodecanoic acid
- BHT
butylated hydroxytoluene
- DHETs
dihydroxyeicosatrienoic acids
- EETs
eicosatrienoic acids
- HETEs
monohydroxyeicosatetraenoic acids
- HPLC
high-performance liquid chromatography
- LC-MS/MS
liquid chromatography–tandem mass spectroscopy
- MRM
multiple reaction monitoring
- P450
cytochrome P450
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
Authorship Contributions
Participated in research design: Diani-Moore, Ma, Rifkind.
Conducted experiments, Diani-Moore, Ma.
Contributed new reagents or analytic tools: Gross.
Performed data analysis: Diani-Moore, Ma, Rifkind.
Wrote and contributed to the writing of the manuscript: Diani-Moore, Ma, Rifkind.
Footnotes
This work was supported by the National Institutes of Health [Grant R01 ES-03606] (to A.B.R), [Grants R37 HL-087062, P20 HL-113444] (to S.S.G.); and a gift from the Winston Foundation (to A.B.R).
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Aboutabl ME, Zordoky BN, El-Kadi AO. (2009) 3-methylcholanthrene and benzo(a)pyrene modulate cardiac cytochrome P450 gene expression and arachidonic acid metabolism in male Sprague Dawley rats. Br J Pharmacol 158:1808–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui P, Solaimani P, Wu X, Hankinson O. (2012) 2,3,7,8-Tetrachlorodibenzo-p-dioxin treatment alters eicosanoid levels in several organs of the mouse in an aryl hydrocarbon receptor-dependent fashion. Toxicol Appl Pharmacol 259:143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylund J, Ericsson J, Oliw EH. (1998) Analysis of cytochrome P450 metabolites of arachidonic and linoleic acids by liquid chromatography-mass spectrometry with ion trap MS. Anal Biochem 265:55–68 [DOI] [PubMed] [Google Scholar]
- Canga L, Levi R, Rifkind AB. (1988) Heart as a target organ in 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity: decreased beta-adrenergic responsiveness and evidence of increased intracellular calcium. Proc Natl Acad Sci USA 85:905–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canga L, Paroli L, Blanck TJJ, Silver RB, Rifkind AB. (1993) 2,3,7,8-tetrachlorodibenzo-p-dioxin increases cardiac myocyte intracellular calcium and progressively impairs ventricular contractile responses to isoproterenol and to calcium in chick embryo hearts. Mol Pharmacol 44:1142–1151 [PubMed] [Google Scholar]
- Capdevila J, Chacos N, Werringloer J, Prough RA, Estabrook RW. (1981) Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proc Natl Acad Sci USA 78:5362–5366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila JH, Falck JR, Dishman E, Karara A. (1990) Cytochrome P-450 arachidonate oxygenase. Methods Enzymol 187:385–394 [DOI] [PubMed] [Google Scholar]
- Capdevila JH, Karara A, Waxman DJ, Martin MV, Falck JR, Guenguerich FP. (1990) Cytochrome P-450 enzyme-specific control of the regio- and enantiofacial selectivity of the microsomal arachidonic acid epoxygenase. J Biol Chem 265:10865–10871 [PubMed] [Google Scholar]
- Chacos N, Capdevila J, Falck JR, Manna S, Martin-Wixtrom C, Gill SS, Hammock BD, Estabrook RW. (1983) The reaction of arachidonic acid epoxides (epoxyeicosatrienoic acids) with a cytosolic epoxide hydrolase. Arch Biochem Biophys 223:639–648 [DOI] [PubMed] [Google Scholar]
- Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB. (2004) Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1b1. Drug Metab Dispos 32:840–847 [DOI] [PubMed] [Google Scholar]
- Cunningham FX, Jr, Lee H, Gantt E. (2007) Carotenoid biosynthesis in the primitive red alga Cyanidioschyzon merolae. Eukaryot Cell 6:533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikh BE, Lasker JM, Raucy JL, Koop DR. (1994) Regio- and stereoselective epoxidation of arachidonic acid by human cytochromes P450 2C8 and 2C9. J Pharmacol Exp Ther 271:1427–1433 [PubMed] [Google Scholar]
- Diani-Moore S, Labitzke E, Brown R, Garvin A, Wong L, Rifkind AB. (2006a) Sunlight generates multiple tryptophan photoproducts eliciting high efficacy CYP1A induction in chick hepatocytes and in vivo. Toxicol Sci 90:96–110 [DOI] [PubMed] [Google Scholar]
- Diani-Moore S, Papachristou F, Labitzke E, Rifkind AB. (2006b) Induction of CYP1A and cyp2-mediated arachidonic acid epoxygenation and suppression of 20-hydroxyeicosatetraenoic acid by imidazole derivatives including the aromatase inhibitor vorozole. Drug Metab Dispos 34:1376–1385 [DOI] [PubMed] [Google Scholar]
- Gannon M, Gilday D, Rifkind AB. (2000) TCDD induces CYP1A4 and CYP1A5 in chick liver and kidney and only CYP1A4, an enzyme lacking arachidonic acid epoxygenase activity, in myocardium and vascular endothelium. Toxicol Appl Pharmacol 164:24–37 [DOI] [PubMed] [Google Scholar]
- Gilday D, Gannon M, Yutzey K, Bader D, Rifkind AB. (1996) Molecular cloning and expression of two novel avian cytochrome P450 1A enzymes induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem 271:33054–33059 [DOI] [PubMed] [Google Scholar]
- Hama-Tomioka K, Kinoshita H, Azma T, Nakahata K, Matsuda N, Hatakeyama N, Kikuchi H, Hatano Y. (2009) The role of 20-hydroxyeicosatetraenoic acid in cerebral arteriolar constriction and the inhibitory effect of propofol. Anesth Analg 109:1935–1942 [DOI] [PubMed] [Google Scholar]
- Huang S, Gibson GG. (1991) Differential induction of cytochromes P450 and cytochrome P450-dependent arachidonic acid metabolism by 3,4,5,3′,4′-pentachlorobiphenyl in the rat and the guinea pig. Toxicol Appl Pharmacol 108:86–95 [DOI] [PubMed] [Google Scholar]
- Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. (2007) Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem 50:3825–3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Hammock BD. (2009) Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 8:794–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, McGiff JC, Fava C, Amen G, Nesta E, Zanconato G, Quilley J, Minuz P. (2013) Maternal and fetal epoxyeicosatrienoic acids in normotensive and preeclamptic pregnancies. Am J Hypertens 26:271–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanetoshi A, Ward AM, May BK, Rifkind AB. (1992) Immunochemical identity of the 2,3,7,8-tetrachlorodibenzo-p-dioxin- and β-naphthoflavone-induced cytochrome P-450 arachidonic acid epoxygenases in chick embryo liver: distinction from the ω-hydroxylase and the phenobarbital-induced epoxygenase. Mol Pharmacol 42:1020–1026 [PubMed] [Google Scholar]
- Karara A, Wei S, Spady D, Swift L, Capdevila JH, Falck JR. (1992) Arachidonic acid epoxygenase: structural characterization and quantification of epoxyeicosatrienoates in plasma. Biochem Biophys Res Commun 182:1320–1325 [DOI] [PubMed] [Google Scholar]
- Kroetz DL, Xu F. (2005) Regulation and inhibition of arachidonic acid ω-hydroxylases and 20-HETE formation. Annu Rev Pharmacol Toxicol 45:413–438 [DOI] [PubMed] [Google Scholar]
- Lee C, Riddick DS. (2012) Aryl hydrocarbon receptor-dependence of dioxin’s effects on constitutive mouse hepatic cytochromes P450 and growth hormone signaling components. Can J Physiol Pharmacol 90:1354–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CA, Lawrence BP, Kerkvliet NI, Rifkind AB. (1998) 2,3,7,8-Tetrachlorodibenzo-p-dioxin induction of cytochrome P450-dependent arachidonic acid metabolism in mouse liver microsomes: evidence for species-specific differences in responses. Toxicol Appl Pharmacol 153:1–11 [DOI] [PubMed] [Google Scholar]
- Liu Y, Dang H, Li D, Pang W, Hammock BD, Zhu Y. (2012) Inhibition of soluble epoxide hydrolase attenuates high-fat-diet-induced hepatic steatosis by reduced systemic inflammatory status in mice. PLoS ONE 7:e39165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) Method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. (2002) Structural refinement of inhibitors of urea-based soluble epoxide hydrolases. Biochem Pharmacol 63:1599–1608 [DOI] [PubMed] [Google Scholar]
- Morisseau C, Hammock BD. (2013) Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol 53:37–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai K, Ward AM, Gannon M, Rifkind AB. (1992) β-naphthoflavone induction of a cytochrome P-450 arachidonic acid epoxygenase in chick embryo liver distinct from the aryl hydrocarbon hydroxylase and from phenobarbital-induced arachidonate epoxygenase. J Biol Chem 267:19503–19512 [PubMed] [Google Scholar]
- Nithipatikom K, Laabs ND, Isbell MA, Campbell WB. (2003) Liquid chromatographic-mass spectrometric determination of cyclooxygenase metabolites of arachidonic acid in cultured cells. J Chromatogr B Analyt Technol Biomed Life Sci 785:135–145 [DOI] [PubMed] [Google Scholar]
- Panigrahy D, Kalish BT, Huang S, Bielenberg DR, Le HD, Yang J, Edin ML, Lee CR, Benny O, Mudge DK, et al. (2013) Epoxyeicosanoids promote organ and tissue regeneration. Proc Natl Acad Sci USA 110:13528–13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell WS. (1982) Rapid extraction of arachidonic acid metabolites from biological samples using octadecylsilyl silica. Methods Enzymol 86:467–477 [DOI] [PubMed] [Google Scholar]
- Revermann M, Schloss M, Barbosa-Sicard E, Mieth A, Liebner S, Morisseau C, Geisslinger G, Schermuly RT, Fleming I, Hammock BD, et al. (2010) Soluble epoxide hydrolase deficiency attenuates neointima formation in the femoral cuff model of hyperlipidemic mice. Arterioscler Thromb Vasc Biol 30:909–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkind AB. (2006) CYP1A in TCDD toxicity and in physiology-with particular reference to CYP dependent arachidonic acid metabolism and other endogenous substrates. Drug Metab Rev 38:291–335 [DOI] [PubMed] [Google Scholar]
- Rifkind AB, Gannon M, Gross SS. (1990) Arachidonic acid metabolism by dioxin-induced cytochrome P-450: a new hypothesis on the role of P-450 in dioxin toxicity. Biochem Biophys Res Commun 172:1180–1188 [DOI] [PubMed] [Google Scholar]
- Rifkind AB, Kanetoshi A, Orlinick J, Capdevila JH, Lee C. (1994) Purification and biochemical characterization of two major cytochrome P-450 isoforms induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in chick embryo liver. J Biol Chem 269:3387–3396 [PubMed] [Google Scholar]
- Rifkind AB, Lee C, Chang TKH, Waxman DJ. (1995) Arachidonic acid metabolism by human cytochrome P450s 2C8, 2C9, 2E1, and 1A2: regioselective oxygenation and evidence for a role for CYP2C enzymes in arachidonic acid epoxygenation in human liver microsomes. Arch Biochem Biophys 320:380–389 [DOI] [PubMed] [Google Scholar]
- Roman RJ. (2002) P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82:131–185 [DOI] [PubMed] [Google Scholar]
- Roman RJ, Maier KG, Sun CW, Harder DR, Alonso-Galicia M. (2000) Renal and cardiovascular actions of 20-hydroxyeicosatetraenoic acid and epoxyeicosatrienoic acids. Clin Exp Pharmacol Physiol 27:855–865 [DOI] [PubMed] [Google Scholar]
- Schlezinger JJ, Parker C, Zeldin DC, Stegeman JJ. (1998) Arachidonic acid metabolism in the marine fish Stenotomus chrysops (Scup) and the effects of cytochrome P450 1A inducers. Arch Biochem Biophys 353:265–275 [DOI] [PubMed] [Google Scholar]
- Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. (2005) Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci USA 102:9772–9777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA. (2009) Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res 50 (Suppl):S52–S56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MK, Catron TF. (2000) Characterization of cardiotoxicity induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related chemicals during early chick embryo development. Toxicol Appl Pharmacol 167:210–221 [DOI] [PubMed] [Google Scholar]
- Willson TM, Kliewer SA. (2002) PXR, CAR and drug metabolism. Nat Rev Drug Discov 1:259–266 [DOI] [PubMed] [Google Scholar]
- Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC. (1996) Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J Biol Chem 271:3460–3468 [DOI] [PubMed] [Google Scholar]
- Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, et al. (2000) Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res 87:992–998 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.