

Abstract
Hyperglycemia and endothelial dysfunction are associated with hypertension, but the specific causality and genetic underpinning are unclear. Caveolin-1 (cav-1) is a plasmalemmal anchoring protein and modulator of vascular function and glucose homeostasis. Cav-1 gene variants are associated with reduced insulin sensitivity in hypertensive individuals, and cav-1−/− mice show endothelial dysfunction, hyperglycemia, and increased blood pressure (BP). On the other hand, insulin-sensitizing therapy with metformin may inadequately control hyperglycemia while affecting the vascular outcome in certain patients with diabetes. To test whether the pressor and vascular changes in cav-1 deficiency states are related to hyperglycemia and to assess the vascular mechanisms of metformin under these conditions, wild-type (WT) and cav-1−/− mice were treated with either placebo or metformin (400 mg/kg daily for 21 days). BP and fasting blood glucose were in cav-1−/− > WT and did not change with metformin. Phenylephrine (Phe)- and KCl-induced aortic contraction was in cav-1−/− < WT; endothelium removal, the nitric-oxide synthase (NOS) blocker l-NAME (Nω-nitro-l-arginine methyl ester), or soluble guanylate cyclase (sGC) inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) enhanced Phe contraction, and metformin blunted this effect. Acetylcholine-induced relaxation was in cav-1−/− > WT, abolished by endothelium removal, l-NAME or ODQ, and reduced with metformin. Nitric oxide donor sodium nitroprusside was more potent in inducing relaxation in cav-1−/− than in WT, and metformin reversed this effect. Aortic eNOS, AMPK, and sGC were in cav-1−/− > WT, and metformin decreased total and phosphorylated eNOS and AMPK in cav-1−/−. Thus, metformin inhibits both vascular contraction and NO-cGMP-dependent relaxation but does not affect BP or blood glucose in cav-1−/− mice, suggesting dissociation of hyperglycemia from altered vascular function in cav-1-deficiency states.
Introduction
Hypertension is a multifaceted disorder that involves abnormalities in the vascular, renal, nervous, and endocrine systems (Williams, 1994; Hall et al., 1996; Lohmeier et al., 2005). Vascular dysfunction occurs in many forms of hypertension and involves changes in different regulatory proteins and signaling pathways. Caveolin-1 (cav-1) is an anchoring protein in plasma membrane caveolae of many cell types, including vascular smooth muscle (VSM) (Ishizaka et al., 1998) and endothelial cells (Feron et al., 1998; Minshall et al., 2003). In endothelial cells, cav-1 anchors endothelial nitric oxide (NO) synthase (eNOS) in plasma membrane caveolae, which limits its translocation and phosphoactivation (Feron et al., 1998; Batova et al., 2006) and thereby reduces its capacity to generate NO and activate the NO-cGMP vasorelaxation pathway. Endothelial cell dysfunction and abnormalities in NO-cGMP are important factors in hypertension.
Metabolic disorders such as obesity, hyperglycemia, and diabetes mellitus are often detected in association with hypertension (Brands and Hopkins, 1996). Abnormal vascular function may play a role in obesity-related hypertension (Lastra et al., 2010). Interestingly, cav-1 may modulate the metabolic pathways in adipose cells. Indeed, cav-1 expression in human adipose tissue is altered in obesity-associated type 2 diabetes (Catalan et al., 2008). In addition, our studies in humans have shown that cav-1 gene variants are associated with reduced insulin sensitivity in hypertensive, but not normotensive, individuals (Pojoga et al., 2011). Furthermore, cav-1-deficient mice show hyperglycemia, adipose tissue defects in insulin receptor expression and mitochondrial function (Asterholm et al., 2012), as well as dramatic changes in vascular function and blood pressure (BP) (Pojoga et al., 2010, 2011). Although cav-1 has been linked to regulation of both vascular and glycemic control mechanisms, the relation between hyperglycemia and altered vascular function and BP in cav-1 deficiency states is poorly defined. For example, whereas insulin-sensitizing compounds such as metformin are the first-line therapy for type 2 diabetes and glycemic control in prediabetic states and metformin improves hyperglycemia in several experimental models (Heishi et al., 2006; Matsumoto et al., 2008), certain patients with diabetes show inadequate glycemic control with metformin monotherapy (Bailey et al., 2013). On the other hand, metformin may have pleiotropic vascular effects that are independent of its effects on glycemic control. For instance, in women with polycystic ovary syndrome characterized by hyperinsulinemia, hyperandrogenism, dyslipidemia, dysadipocytokinemia, and central obesity, metformin improves cardiovascular outcomes while having minimal effects on glucose levels (Agarwal et al., 2010). Interestingly, some of the cellular actions of metformin require cav-1 (Salani et al., 2012). Thus, it is conceivable that in cav-1-deficiency states metformin may not affect glycemic control; however, whether it could still exert vascular actions under these conditions is unclear.
The purpose of this study was to determine whether the pressor changes and altered vascular function associated with cav-1 deficiency are related to or independent from hyperglycemia. By use of cav-1 knockout (cav-1−/−) mice, which show hyperglycemia and altered vascular reactivity (Cohen et al., 2003; Pojoga et al., 2010, 2011; Asterholm et al., 2012) and metformin treatment at a dose that improved hyperglycemia in other models (Heishi et al., 2006; Matsumoto et al., 2008), we hypothesized that if the vascular changes in cav-1 deficiency states are related to hyperglycemia, then metformin improvement of hyperglycemia should be paralleled with improved vascular function. Conversely, if the vascular changes in cav-1 deficiency states are independent of hyperglycemia and metformin changes vascular outcome despite inadequate glycemic control as shown clinically in certain patients with diabetes (Bailey et al., 2013), metformin then should affect vascular function without modifying hyperglycemia. We used WT and cav-1−/− mice chronically treated with placebo or metformin to test whether metformin affects glycemic control, BP, and vascular contraction and relaxation mechanisms and whether these effects are different in cav-1 deficiency states. We also tested whether the metformin-induced changes in vascular function involve changes in the NO-cGMP pathway.
Materials and Methods
Animals
Cav-1−/− (strain 004585) and genetically matched WT (strain 101045) male mice (16 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME). The genotypes were confirmed by polymerase chain reaction. Animals were housed in 12-hour/12-hour light/dark cycle at an ambient temperature of 22 ± 1°C and were maintained on ad libitum rodent chow and tap water. After 3 days of acclimatization, mice were treated for 21 days with placebo or metformin (400 mg/kg daily in drinking water), a dose reported to improve hyperglycemia in other models (Heishi et al., 2006; Matsumoto et al., 2008). All procedures were approved by the Animal Care and Use Committee at Harvard Medical School.
Blood Pressure
Systolic BP was measured in conscious mice using tail-cuff plethysmography (CODA; Kent Scientific, Torrington, CT) (Pojoga et al., 2010). Mice were kept warm at 30°C for 10 minutes and allowed to rest quietly before BP measurement. BP measurements were taken in a quiet room, and mice were kept calm and handled by the same person.
Fasting Blood Glucose
Animals were fasted overnight. The next morning at 7:00 AM, blood samples were collected via tail vein, and glucose levels were determined using a Freestyle glucometer (Abbott Laboratories, North Chicago, IL).
Plasma Renin Activity
Blood was collected in purple-top BD Microtainer tubes (EDTA). The plasma was separated by centrifugation, and plasma renin activity (PRA) levels were determined in duplicate using a GammaCoat RIA kit (DiaSorin Inc., Stillwater, MN).
Tissue Preparation
Mice were killed with isoflurane overdose, and the thoracic and abdominal aortas were rapidly excised. The thoracic aorta was cleaned of connective tissue and cut into 3-mm-wide rings. The rest of thoracic aorta and all abdominal aortas were placed in liquid nitrogen for protein analysis.
Isometric Contraction and Relaxation
Aortic segments were suspended between two tungsten wire hooks, one hook was fixed at the bottom of tissue bath, and the other hook was connected to Grass force transducer (FT03; Astro-Med, Inc, West Warwick, RI). Aortic segments were stretched under 0.5-g resting tension for 45 minutes in temperature-controlled tissue bath, filled with 50 ml of Krebs’ solution continuously bubbled with 95% O2, 5% CO2 at 37°C. Changes in isometric contraction were recorded on Grass polygraph (Model 7D, Astro-Med).
A control aortic contraction to 96 mM KCl was first elicited followed by rinsing in Krebs’ 3 × 10 minutes. Aortic segments were then stimulated with phenylephrine (Phe, 10−9−10−5 M), concentration-contraction curves were constructed, and maximal Phe contraction was measured. Individual Phe concentration-response curves were further analyzed using a nonlinear regression curve (best-fit sigmoidal dose-response curve; Sigmaplot, La Jolla, CA), and the effective concentration that produced half maximal contraction (ED50) was measured and presented as pED50 (−log M). In other experiments, vessels were precontracted with Phe (10−5 M), acetylcholine (ACh, 10−9−10−5 M) was added, and the percent of relaxation of Phe contraction was measured. Parallel contraction and relaxation experiments were performed in aortic rings pretreated with the NOS inhibitor Nω-nitro-l-arginine methyl ester (l-NAME, 3 × 10−4 M) or guanylate cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 10−5 M) for 10 minutes. In some experiments, the endothelium was removed by scraping the vessel interior around the tip of fine forceps, and the responses to Phe and ACh were compared with those in endothelium-intact vessels. The relaxation of Phe-precontracted aortic rings to the exogenous NO donor sodium nitroprusside (SNP) was also measured. Individual ACh and SNP concentration-relaxation curves were further analyzed using a nonlinear regression best-fit sigmoidal dose-response curve, and ACh and SNP ED50 values were calculated.
Western Blots
Aortic tissues were homogenized in RIPA buffer (Santa Cruz Biotechnology, Santa Cruz, CA). Protein extracts (40 μg) were combined with an equal volume of 2× Laemmli loading buffer, boiled for 5 minutes, and size-fractionated by electrophoresis on 7.5% SDS-polyacrylamide gels. Proteins were transferred to a nitrocellulose membrane by electroblotting. Membranes were incubated with 5% nonfat dried milk in Tris-buffered saline-Tween (USB Corporation, Santa Clara, CA) for 1 hour and then incubated overnight at 4°C with the primary antibody anti-eNOS (1:2500) (BD Transduction Laboratories, Franklin Lakes, NJ), anti-peNOS (1:1000), anti AMP-activated protein kinase (AMPK) (1:400), anti-pAMPK (1:400) (Cell Signaling Technology, Danvers, MA) and antisoluble guanylate cyclase (sGC) (1:200) (Cayman Chemical, Ann Arbor, MI). Membranes were then washed, incubated with peroxidase-conjugated secondary antibody, and analyzed using enhanced chemiluminescence (PerkinElmer Life and Analytical Sciences, Waltham, MA). Blots were then reprobed for β-actin (1:20,000; Sigma-Aldrich), immunoreactive bands were quantitated by optical densitometry, and the results were normalized to β-actin to correct for loading.
Solutions and Drugs
Krebs’ solution contained (in mM): NaCl 120, KCl 5.9, NaHCO3 25, NaH2PO4 1.2, dextrose 11.5, CaCl2 2.5, MgCl2 1.2, at pH 7.4, and 96 mM KCl was prepared as Krebs’ with equimolar substitution of NaCl with KCl. Stock solutions of Phe, ACh, SNP, and l-NAME (10−1 M; Sigma-Aldrich) were prepared in distilled water. Stock solution of ODQ (10−1 M; EMD Bioscience, Darmstadt, Germany) was prepared in DMSO. Final concentration of DMSO in solution was <0.1%. All other chemicals were of reagent grade or better.
Statistical Analysis
Data were presented as means ± S.E.M., with n value representing the number of mice. Data were first analyzed using one-way analysis of variance for comparison of different groups (cav-1 status vs. metformin treatment). When a statistical difference was observed, the data were further analyzed using Student-Newman-Keuls post hoc test for multiple comparisons. Also, pairwise comparisons were performed when a significant effect was noted in analysis of variance, and unpaired Student’s t test was used for comparison of two means. Differences were considered significant if P < 0.05. In all studies, experiments were performed blinded to animal genotype and treatment.
Results
Cav-1−/− mice had lower body weights and higher BP and fasting blood glucose levels compared with WT mice (Fig. 1, A, B, and C). Metformin treatment did not significantly affect body weight, BP, or blood glucose levels in cav-1−/− or WT. PRA levels were not significantly different in cav-1−/− versus WT, and metformin did not modify PRA levels (Fig. 1D).
Fig. 1.
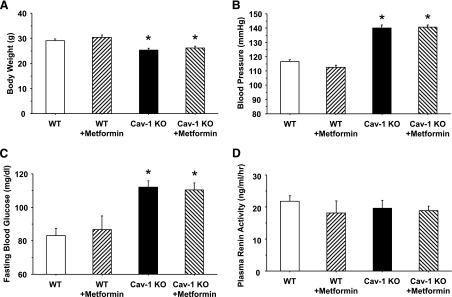
Body weight (A), blood pressure (B), fasting blood glucose (C), and plasma renin activity (PRA) (D) in WT and cav-1−/− mice with or without metformin treatment. Data represent means ± S.E.M. of measurements in six to nine mice. *Measurements in cav-1−/− are significantly different (P < 0.05) from corresponding measurements in WT.
In endothelium-intact aortic rings, Phe caused concentration-dependent contraction that reached a maximum at 10−5 M. Phe contraction was less in cav-1−/− than WT and in cav-1−/−+metformin than WT + metformin. Metformin caused insignificant reduction of Phe contraction in WT and had no effect in cav-1−/− mice (Fig. 2A; Table 1).
Fig. 2.
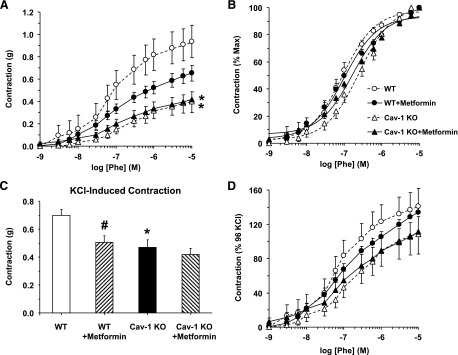
Phe- and KCl-induced contraction in aortic segments of WT and cav-1−/− mice with or without metformin treatment. Endothelium-intact aortic rings were stimulated with increasing concentrations of Phe, and the contractile response was measured and presented in grams (A) or as percentage of maximum Phe contraction (B). Vascular contraction to KCl (95 mM KCl) was also measured (C), and the contractile response to Phe was presented as % of KCl contraction (D). Data represent means ± S.E.M. of measurements in 9–17 mice. *Measurements in cav-1−/− are significantly different (P < 0.05) from corresponding measurements in WT mice. #Measurements in metformin-treated mice are significantly different (P < 0.05) from corresponding measurements in mice of the same genotype without metformin treatment.
TABLE 1.
Phenylephrine (Phe) and potassium chloride (KCl) contraction, and acetylcholine (Ach) and sodium nitroprusside (SNP) relaxation in aortic rings of wild-type (WT) and caveolin-1 (cav-1)−/− mice with and without metformin treatment
Data represent means ± S.E.M. of cumulative measurements in aortic segments of 9 to 17 mice.
| WT |
+ Metformin |
Cav-1−/− |
+ Metformin |
|
|---|---|---|---|---|
| Phe max (10−5 M) | ||||
| Contraction (g) | 0.94 ± 0.15 | 0.65 ± 0.07 | 0.40 ± 0.10* | 0.42 ± 0.07* |
| + l-NAME (3 × 10−4 M) | 1.21 ± 0.14 | 0.64 ± 0.11# | 0.69 ± 0.10* | 0.64 ± 0.10 |
| +ODQ (10−5 M) | 1.10 ± 0.16 | 1.31 ± 0.17† | 0.89 ± 0.13† | 0.76 ± 0.13† |
| − Endo | 1.05 ± 0.19 | 0.58 ± 0.08# | 0.72 ± 0.11† | 0.54 ± 0.07 |
| Phe pED50 (-log M) | 7.11 ± 0.18 | 7.00 ± 0.10 | 6.66 ± 0.13 | 7.47 ± 0.10*# |
| +l-NAME | 7.36 ± 0.17 | 7.36 ± 0.14 | 6.91 ± 0.18 | 7.03 ± 0.14 |
| +ODQ | 7.43 ± 0.14 | 7.53 ± 0.10† | 7.05 ± 0.10*† | 7.10 ± 0.09* |
| − Endo | 7.36 ± 0.16 | 7.36 ± 0.13† | 6.94 ± 0.13 | 7.07 ± 0.11 |
| KCl contraction (g) | 0.70 ± 0.04 | 0.51 ± 0.05# | 0.47 ± 0.05* | 0.42 ± 0.04 |
| -Endo | 0.65 ± 0.06 | 0.42 ± 0.06# | 0.37 ± 0.04* | 0.45 ± 0.06 |
| Phe max contraction | ||||
| (% 96 mM KCl) | 141.18 ± 20.46 | 133.83 ± 7.39 | 108.43 ± 23.30 | 111.46 ± 17.93 |
| +l-NAME | 186.21 ± 22.88 | 176.26 ± 22.35 | 206.67 ± 38.31† | 156.58 ± 21.08 |
| +ODQ | 166.76 ± 20.95 | 202.41 ± 8.24† | 188.93 ± 19.56† | 187.41 ± 27.87† |
| - Endo | 161.98 ± 19.75 | 145.55 ± 15.54 | 223.32 ± 42.08† | 131.62 ± 15.59# |
| ACh (10−5 M) % relaxation | 23.51 ± 5.33 | 24.36 ± 5.63 | 85.20 ± 8.15* | 64.36 ± 5.74*# |
| pED50 (-log M) | 6.32 ± 0.14 | 6.23 ± 0.13 | 7.45 ± 0.27* | 6.70 ± 0.11*# |
| SNP (10−8 M) % relaxation | 57.66 ± 4.60 | 53.03 ± 7.59 | 73.75 ± 4.93* | 50.08 ± 5.11# |
| pED50 (−log M) | 8.12 ± 0.09 | 8.08 ± 0.13 | 8.52 ± 0.09* | 7.98 ± 0.15# |
Measurements in cav-1−/− are significantly different (P < 0.05) from corresponding measurements in WT.
Measurements in metformin-treated mice are significantly different (P < 0.05) from corresponding measurements in mice without metformin treatment.
Measurements in l-NAME or ODQ-treated or endothelium-denuded arteries are significantly different from corresponding measurements in control nontreated arteries.
To test whether the reduced contraction in cav-1−/− versus WT is due to changes in α-adrenergic receptor sensitivity, Phe contraction was presented as the percent of maximum, and Phe ED50 was calculated. Although the Phe concentration-response curve showed a shift to the right in cav-1−/− (Fig. 2B), the Phe ED50 was not significantly different between cav-1−/− and WT (Table 1). Although the Phe ED50 was not significantly different between WT + metformin and WT, Phe was more potent in cav-1−/− + metformin than cav-1−/− (Fig. 2B; Table 1).
To test whether the changes in aortic contraction in cav-1−/− are specific to a particular agonist or receptor, we measured contraction to 96 mM KCl. KCl causes membrane depolarization and stimulates Ca2+ entry through voltage-gated channels via receptor-independent mechanisms (Murphy and Khalil, 2000). KCl contraction was reduced in cav-1−/− versus WT, and metformin treatment reduced KCl contraction in WT, but not cav-1−/− (Fig. 2C; Table 1).
Relative to maximal KCl contraction, the maximal Phe contraction was ∼1.4-fold in WT, but it was almost similar to KCl contraction in cav-1−/− (Fig. 2D; Table 1). Assuming that KCl contraction is due mainly to Ca2+ entry through voltage-gated channels (Murphy and Khalil, 2000), the greater maximal Phe contraction relative to maximal KCl in WT could be due to additional activation of other Ca2+ channels (Lee et al., 2001; Park et al., 2008) or Ca2+-sensitization pathways (Salamanca and Khalil, 2005; Dimopoulos et al., 2007), and these contraction enhancement mechanisms are inactive or obscured in cav-1−/−. Metformin did not significantly affect Phe contraction relative to KCl in WT or cav-1−/− (Fig. 2D; Table 1).
To test the role of endothelium in the altered vascular responses, the magnitude of Phe contraction was enhanced in endothelium-denuded compared with intact aortas, particularly in cav-1−/−, but not in WT, WT + metformin, or cav-1−/− + metformin (Fig. 3A; Table 1). To test for a potential role of endothelium-derived NO in the reduced aortic contraction in cav-1−/−, the NOS inhibitor l-NAME caused insignificant enhancement of Phe contraction in cav-1−/− and no significant change in Phe contraction in WT, WT + metformin or cav-1−/− + metformin. Pretreatment with the guanylate cyclase inhibitor ODQ minimally affected Phe contraction in WT, but it significantly enhanced Phe contraction in WT + metformin, cav-1−/−, and to a lesser extent in cav-1−/− + metformin (Fig. 3A; Table 1).
Fig. 3.
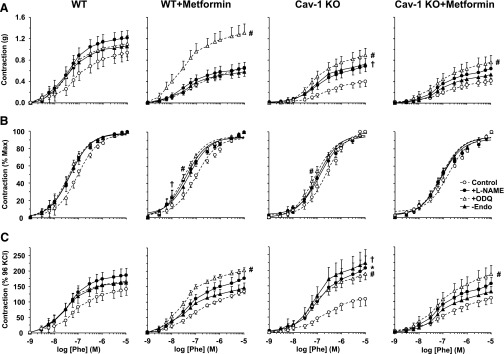
Effect of endothelium removal or blockade of NO-cGMP pathway on Phe-induced contraction (A), sensitivity to Phe (B), and Phe-induced contractile response relative to control KCl contraction (C) in aorta of WT and cav-1−/− mice with or without metformin treatment. Aortic rings from WT, WT + metformin, cav-1−/−, and cav-1 + metformin mice were either kept endothelium-intact (open circles), pretreated with the NOS inhibitor l-NAME (3 × 10−4 M) (closed circles) or the guanylate cyclase inhibitor ODQ (10−5 M) (open triangles) for 10 minutes, or endothelium-denuded (closed triangles). After measuring contraction to KCl (96 mM), the vessels were stimulated with increasing concentrations of Phe, and the contractile response was presented in grams (A), as percentage of maximum Phe contraction (B) or as percentage of KCl contraction (C). Data represent mean ± S.E.M. (n = 9–17 mice). *Measurements in l-NAME-treated arteries are significantly different (P < 0.05) from corresponding measurements in control nontreated arteries. #Measurements in ODQ-treated arteries are significantly different (P < 0.05) from corresponding measurements in control nontreated arteries. †Measurements in endothelium-denuded arteries are significantly different (P < 0.05) from corresponding measurements in intact arteries.
In all groups, when Phe contraction was presented as the percent of the maximum, the Phe concentration-response curve appeared to be shifted to the left in endothelium-denuded and l-NAME- or ODQ-treated aortas compared with control intact and nontreated aortas (Fig. 3B), suggesting a role of endothelium-dependent NO-cGMP pathway in reducing the vascular responsiveness to Phe. Further analysis of Phe ED50 showed that Phe was more potent in endothelium-denuded versus intact aortas of WT + metformin. Phe was equally potent in l-NAME-treated and nontreated vessels of WT or cav-1−/− with or without metformin treatment. ODQ significantly increased Phe potency in aortas of cav-1−/− and WT + metformin, but not WT, and cav-1 + metformin (Table 1).
Relative to KCl contraction, Phe contraction was dramatically enhanced in endothelium-denuded versus intact aortas of cav-1−/−, and this enhancement was absent in WT, WT + metformin, and cav-1−/− + metformin (Fig. 3C; Table 1). Treatment of endothelium-intact aortas with l-NAME enhanced Phe contraction in cav-1−/−, but not WT, WT + metformin, or cav-1−/− + metformin. ODQ caused minimal effects on Phe contraction in aorta of WT but significantly enhanced Phe contraction in WT + metformin, cav-1−/−, and cav-1−/− + metformin (Fig. 3C; Table 1).
ACh caused concentration-dependent aortic relaxation that was greater in cav-1−/− than in WT and cav-1−/− + metformin than in WT + metformin. ACh relaxation was not different in WT + metformin and WT but was reduced in cav-1−/− + metformin versus cav-1−/− (Fig. 4). When ACh response was measured as the percentage of maximum relaxation and ED50 was calculated, ACh was more potent in cav-1−/− than in WT and cav-1−/− + metformin than in WT + metformin. ACh was equally potent in inducing relaxation in WT + metformin and WT, but it was less potent in cav-1−/− + metformin than in cav-1−/− (Table 1).
Fig. 4.

ACh-induced relaxation in aortic rings of WT and cav-1−/− mice with and without metformin treatment. Aortic rings were precontracted with Phe (10−5 M), increasing concentrations of ACh were added, and the percent of relaxation of Phe contraction was measured. Data represent means ± S.E.M. (n = 9–17 mice). *Measurements in cav-1−/− are significantly different (P < 0.05) from corresponding measurements in WT mice. #Measurements in metformin-treated mice are significantly different (P < 0.05) from corresponding measurements in control mice without metformin treatment.
Consistent with the vasorelaxation results, Western blots revealed an increase in aortic eNOS in cav-1−/− versus WT (Fig. 5A). Metformin increased total eNOS levels in WT but reduced total eNOS in cav-1−/−. Activated and phosphorylated peNOS levels were not different in cav-1−/− and WT (Fig. 5C). Metformin treatment did not affect aortic peNOS level in WT, but it significantly decreased it in cav-1−/− (Fig. 5C). We also measured AMPK as a potential modulator of eNOS activity (Morrow et al., 2003; Osuka et al., 2009; Tanano et al., 2013) and a known target for metformin (Hauton, 2011). Western blots revealed that the amounts of total aortic AMPK (Fig. 5B) and activated pAMPK (Fig. 5D) were significantly greater in cav-1−/− than in WT. Total AMPK and pAMPK were significantly increased with metformin treatment in WT but were significantly reduced in metformin-treated cav-1−/− (Fig. 5, B and D). These observations could be explained by possible interaction between the cav-1 genotype and metformin treatment in the regulation of eNOS and AMPK protein levels or activity. Importantly, ratiometric analysis of the data suggested that peNOS/eNOS and pAMPK/AMPK ratios were not different in cav-1 and WT (Fig. 5, E and F). In addition, metformin treatment did not change the ratio of phosphorylated and total eNOS or AMPK in either genotype (Fig. 5, E and F), suggesting that the observed effects of cav-1 deficiency and chronic metformin treatment involve long-term changes in protein levels of eNOS and AMPK rather than effects on phosphorylation and activation.
Fig. 5.

Western blot analysis of total eNOS (A), total AMPK (B), peNOS (C), pAMPK (D), peNOS/total eNOS (E), and pAMPK/total AMPK (F) in the aortas of WT and cav-1−/− mice with and without metformin treatment. Data represent means ± S.E.M. (n = 6–8 mice). *Measurements in cav-1−/− are significantly different (P < 0.05) from corresponding measurements in WT. #Measurements in metformin-treated mice are significantly different (P < 0.05) from corresponding measurements in control mice without metformin treatment.
In all groups, ACh relaxation was abolished by endothelium removal, the NOS blocker l-NAME, or guanylate cyclase inhibitor ODQ (Fig. 6), supporting a role of endothelium-dependent NO-cGMP relaxation pathway.
Fig. 6.
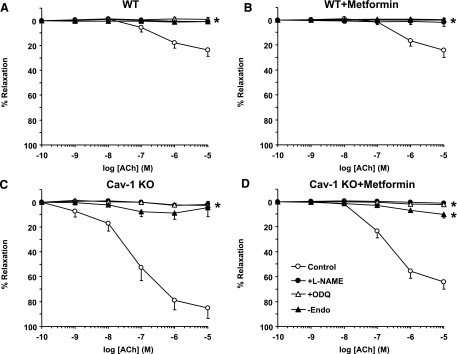
Effect of endothelium removal or blockade of the NO-cGMP pathway on ACh-induced relaxation in aortic segments of WT and cav-1−/− mice with and without metformin treatment. Aortic rings from WT (A), WT + metformin (B), cav-1−/− (C), and cav-1−/− mice treated with metformin (D) were either kept endothelium-intact (open circles), pretreated with l-NAME (3 × 10−4 M) (closed circles) or ODQ (10−5 M) (open triangles), or endothelium-denuded (closed triangles). The tissues were precontracted with Phe (10−5 M), increasing concentrations of ACh were added, and the percent of relaxation of Phe contraction was measured. Data represent means ± S.E.M. (n = 9–17 mice). *Measurements in l-NAME or ODQ-treated or endothelium-denuded arteries are significantly different from corresponding measurements in control nontreated arteries.
The exogenous NO donor SNP caused concentration-dependent relaxation of aortic segments of all groups. SNP produced similar maximal relaxation, but it was more potent in cav-1−/− than in WT (Table 1). Metformin reduced SNP-induced relaxation and reversed the enhanced SNP potency in cav-1−/− but had no significant effect in WT (Fig. 7, A and B; Table 1). Aortic sGC protein amount was greater in cav-1−/− than in WT, and metformin increased sGC in WT but did not significantly affect sGC levels in cav-1−/− (Fig. 7C).
Fig. 7.

SNP-induced vascular relaxation and sGC protein levels in WT and cav-1−/− mice. Aortic rings from WT (A) and cav-1−/− mice (B) with (closed symbols) and without metformin treatment (open symbols) were precontracted with Phe (10−5 M), increasing concentrations of SNP were added, and the percent of relaxation of Phe contraction was measured. Data represent means ± S.E.M. (n = 9–17 mice). In other experiments, aortic tissue homogenates were prepared for Western blot analysis, and sGC protein levels were assessed and normalized to β-actin levels (C). *Measurements in cav-1−/− are significantly different (P < 0.05) from corresponding measurements in WT mice. #Measurements in metformin-treated mice are significantly different (P < 0.05) from corresponding measurements in control mice without metformin treatment.
Discussion
The following are the main findings of this study: 1) cav-1−/− mice show altered vascular function and increased BP and glucose levels; 2) metformin does not affect blood glucose level or BP but affects vascular function; 3) vascular contraction to Phe and KCl is less in cav-1−/− than in WT, endothelium-denudation or inhibition of NO-cGMP enhances Phe contraction, particularly in cav-1−/−, and this enhancement is blunted with metformin; 4) endothelium-dependent ACh relaxation and eNOS and AMPK levels are enhanced in cav-1−/− versus WT, and metformin reduces ACh relaxation and both total and phosphorylated eNOS and AMPK in cav-1−/−; and 5) endothelium-independent SNP relaxation and sGC levels are enhanced in cav-1−/− versus WT, and metformin reverses the enhanced SNP relaxation.
Cav-1 is an anchoring protein and a regulator of signal transduction in VSM and endothelial cells (Feron et al., 1998; Ishizaka et al., 1998; Minshall et al., 2003; Shakirova et al., 2006; Ushio-Fukai and Alexander, 2006). We and others used cav-1−/− mice to examine the role of cav-1 in mechanotransduction, vascular remodeling, and cardiovascular function (Drab et al., 2001; Yu et al., 2006; Pojoga et al., 2010). Cav-1−/− mice show hyperproliferative and vascular abnormalities (Razani et al., 2001). Consistent with our previous reports (Pojoga et al., 2010), the present study showed that Phe contraction was reduced and ACh-induced relaxation was enhanced in cav-1−/− versus WT. The reduced Phe contraction in cav-1−/− is not due to changes in α-adrenergic receptor sensitivity as Phe ED50 was not significantly different between cav-1−/− and WT. To further examine the causes of reduced contraction in cav-1−/− mice, we measured contraction to high KCl, which is due mainly to Ca2+ influx (Murphy and Khalil, 2000). Similar to Phe, KCl-induced contraction was reduced in cav-1−/− versus WT, supporting that the reduced contraction is not specific to a particular agonist/receptor coupling mechanism in VSM. In addition, endothelium removal enhanced Phe contraction, particularly in cav-1−/−, suggesting a role of endothelium-derived vasodilators in the reduced contraction in cav-1−/− mice.
NO is a major endothelium-derived vasodilator. Under basal conditions, eNOS is bound to cav-1 in endothelial cell caveolae. An increase in endothelial cell Ca2+ by agonists such as ACh induces the release of eNOS from cav-1 to the cytosol (Feron et al., 1998; Minshall et al., 2003), where it is phosphorylated and fully activated by protein kinases that reside in caveolae, such as p38 mitogen–activated protein kinase (Anter et al., 2004) phosphatidylinositol 3-kinase-Akt (Anter et al., 2004; Uruno et al., 2005), cAMP-dependent protein kinase A, and AMPK (Tanano et al., 2013). Activated eNOS converts l-arginine to l-citrulline and increases NO release. NO diffuses into VSM, where it activates sGC (Venema et al., 2003) and increases cGMP production. The increased cGMP activates cGMP-dependent protein kinase, which promotes vascular relaxation by decreasing [Ca2+]i and inhibiting myosin light chain kinase or Ca2+ sensitization pathways of VSM contraction, such as protein kinase C (PKC) and Rho-kinase (Salamanca and Khalil, 2005). The enhanced vascular relaxation in cav-1−/− mice is likely due to increased eNOS activity and NO-cGMP because 1) Phe contraction was enhanced by the NOS inhibitor l-NAME or guanylate cyclase inhibitor ODQ; 2) ACh relaxation was blocked by endothelium removal, l-NAME, or ODQ; 3) NO donor and guanylate cyclase activator SNP was more potent in cav-1−/− than in WT; and 4) vascular levels of eNOS, AMPK, and sGC were greater in cav-1−/− than in WT. These observations suggest that cav-1 may limit the expression or activity of vascular eNOS, AMPK, and sGC and that the absence of cav-1 is associated with constitutively larger amounts of eNOS, AMPK, and sGC and upregulation of NO-cGMP.
Given the important role of vascular mechanisms in BP regulation, the reduced vascular contraction and enhanced NO-cGMP relaxation pathway in cav-1−/− are predicted to decrease BP. This is not the case, however, as BP was increased in cav-1−/− versus WT, consistent with our previous reports (Pojoga et al., 2010). The increase in BP despite increased eNOS activity in cav-1−/− mice has been partly explained by partial eNOS uncoupling, whereby increased NO production would lead to increased formation of peroxinitrites and decreased NO bioavailability (Pojoga et al., 2008). The increased BP in cav-1−/− is less likely due to increased renin-angiotensin-aldosterone system because PRA levels were not different in cav-1−/− and WT. On the other hand, hyperglycemia has been associated with hypertension and vascular dysfunction in both humans and animal models (Schulman and Zhou, 2009; Bender et al., 2013). Importantly, cav-1 is expressed in adipose cells and is an important modulator of glucose homeostasis (Cohen et al., 2003; Catalan et al., 2008). Moreover, cav-1 gene variants are associated with reduced insulin sensitivity in hypertensive, but not normotensive individuals (Pojoga et al., 2011), suggesting an interplay between cav-1, BP, and glucose homeostasis. Consistent with previous reports, the total body weight was different in cav-1−/− and WT, possibly because of altered peripheral glucose uptake (Pojoga et al., 2011). In addition, blood glucose was greater in cav-1−/− than in WT, supporting a link between cav-1 deficiency and hyperglycemia. These findings are consistent with previous reports (Cohen et al., 2003; Pojoga et al., 2011; Asterholm et al., 2012) and made it important to investigate possible relationship between hyperglycemia and BP in cav-1−/− mice.
Previous studies showed that metformin treatment (100 mg/kg) for 4 weeks decreased BP in spontaneously hypertensive rats (Bhalla et al., 1996). Although we used metformin at greater doses (400 mg/kg daily) that were reported to improve glycemic control in other models (Heishi et al., 2006; Matsumoto et al., 2008), it did not normalize glucose levels in cav-1−/− mice. This is consistent with reports that cav-1 is needed to mediate the acute effects of metformin on AMPK phosphorylation (Salani et al., 2012), an essential step in gluconeogenesis (Viollet et al., 2012). Also, metformin treatment did not change body weight or BP in cav-1−/− or WT. These results are consistent with the clinical observations that certain patients with diabetes show inadequate glycemic control with metformin monotherapy (Bailey et al., 2013), although metformin could still affect cardiovascular outcomes (Agarwal et al., 2010). These data also suggest a more complex interaction between hyperglycemia and the altered vascular function and BP and prompted us to examine methodically examine the vascular mechanisms of metformin.
Our data suggest that metformin inhibits the mechanisms of vascular contraction. Metformin reduced KCl contraction in both intact and endothelium-denuded vessels of WT mice (Table 1). Assuming that KCl contraction is due mainly to Ca2+ influx (Murphy and Khalil, 2000), the reduced KCl contraction in metformin-treated WT mice is likely due to decreased Ca2+ entry. This assumption is consistent with reports that metformin causes repolarization and decreases Ca2+ influx, [Ca2+]i and isometric force in rat tail artery (Chen et al., 1997). In comparison with KCl, α-adrenergic receptor agonists such as Phe stimulate Ca2+ entry through voltage-gated as well as ligand-gated and store-operated Ca2+ channels (Lee et al., 2001; Park et al., 2008). Phe also activates Ca2+-sensitization pathways such as PKC and Rho-kinase (Salamanca and Khalil, 2005; Dimopoulos et al., 2007). We found that in WT mice metformin caused insignificant reduction of Phe contraction in intact aortas and significantly reduced Phe contraction in endothelium-denuded vessels (Table 1), likely as a result of metformin-induced decrease in Ca2+ entry in VSM. Interestingly, relative to KCl contraction, Phe contraction was slightly greater in both intact (1.4-fold) and endothelium-denuded (1.6-fold) vessels of WT mice. Whereas Phe contraction was similar to that of KCl in endothelium-intact vessels of cav-1−/− mice (1.08-fold), endothelium removal almost doubled Phe contraction (2.23-fold) (Table 1). Again, assuming that KCl contraction is due to Ca2+ influx, the enhanced Phe contraction in endothelium-denuded vessels of cav-1−/− mice is likely due to activation of other Ca2+ channels or Ca2+ sensitization by PKC or Rho-kinase (Salamanca and Khalil, 2005). Whether these increases in Ca2+ sensitivity of VSM contraction in cav-1−/− mice are related to hyperglycemia is unclear; however, studies have shown distinct dysfunctions of mesenteric artery contraction in diabetic mice, possibly as a result of changes in Rho-kinase signaling (Nobe et al., 2012). Also, Rho-kinase and PKC promote Ca2+ sensitization in arteries from diabetic rats (Kizub et al., 2010). The effects of Ca2+ sensitization were not observed in endothelium-intact vessels of cav-1−/− mice, likely because they were obscured by increased NO-cGMP, and only with endothelium removal or blockade of NO-cGMP these enhanced contraction mechanisms could be manifested. This finding is supported by reports that NO and cGMP reduce [Ca2+]i (Cornwell and Lincoln, 1989), and cGMP-dependent protein kinase causes phosphorylation and inhibition of myosin light-chain kinase and Ca2+ sensitization pathways such as PKC and Rho-kinase (Salamanca and Khalil, 2005). The observation that metformin caused significant reduction in Phe contraction relative to KCl contraction in endothelium-denuded vessels of cav-1−/− mice (Table 1) is consistent with the possibility that metformin not only reduces Ca2+entry (Sharma and Bhalla, 1995; Bhalla et al., 1996) but could also inhibit Ca2+ sensitization pathways such as PKC (Gallo et al., 2005; Mahrouf et al., 2006) and Rho-kinase (Agard et al., 2009).
Whereas our data suggest an effect of metformin on vascular contraction mechanisms, some observations point to additional effects of metformin on vascular relaxation pathways. Metformin increased the sensitivity to Phe in intact vessels of cav-1−/− mice. Also, endothelium denudation and inhibition of NO-cGMP enhanced Phe contraction in cav-1−/− mice, and this enhancement was blunted with metformin. These findings can be explained by possible interaction of metformin with NO-cGMP, and these interactions may be augmented during increased NO-cGMP activity in cav-1−/− mice. This is supported by the observations that metformin reduced ACh and SNP induced relaxation and decreased total and phosphorylated eNOS and AMPK levels in cav-1−/− mice. Another possible interaction of metformin with the NO pathway is that during cav-1 deficiency the increased eNOS activity could lead to partial eNOS uncoupling, thus producing excess reactive oxygen species and peroxynitrites, and resulting in decreased NO bioavailability. Under these conditions, metformin could act by reducing reactive oxygen species, thus increasing NO bioavailability and reducing vascular contraction.
Other observations or limitations include the following. 1) The glucose data should be interpreted with caution as overnight fasting may represent a potential limitation given the higher metabolic demands of the mouse. However, the terminology, time course, and metabolic effects of fasting versus starvation in mice have not been clearly defined in the literature. For instance, some studies have used food deprivation in mice for periods ranging from 24 to 40 hours (Li et al., 2008; Fernandez-Rojo et al., 2013), which are longer than the overnight time period used in the present study, and still referred to this intervention as fasting. Nevertheless, all the present experiments were performed under the same conditions and using the same overnight fasting protocol for all the animal groups before performing the glucose tolerance test as previously described by other studies (Heikkinen et al., 2007; Muniyappa et al., 2008). 2) In contrast with decreasing AMPK activity in cav-1−/−, metformin appeared to increase AMPK in WT mice, and AMPK activation could affect the vascular relaxation and contraction mechanisms. Our observation that metformin increased aortic total and pAMPK in WT mice is consistent with reports that AMPK and pAMPK levels are increased in the heart of rats treated with metformin long term (Hauton, 2011) and that AMPK causes phosphorylation of eNOS (Chen et al., 1999; Tanano et al., 2013). However, the ability of metformin to enhance eNOS and AMPK appears to be compromised in cav-1−/− mice, supporting that cav-1 may be necessary for the actions of metformin on eNOS and AMPK. 3) In WT mice, metformin appeared to increase the vascular dependence on cGMP, as shown by the enhanced Phe contraction in vessels treated with the guanylate cyclase inhibitor ODQ and the increased aortic sGC levels with metformin. 4) Metformin appeared to dampen the enhanced total and phosphorylated eNOS and AMPK, but it did not alter the increased vascular sGC levels or reverse the enhanced Phe contraction by ODQ in cav-1−/− mice. Interestingly, sGC activation and cGMP production can be stimulated not only by NO but also by NO-independent mechanisms (Evgenov et al., 2006). One possibility is that in the absence of cav-1, metformin switches sGC and cGMP production from an eNOS/AMPK-dependent to an NO-independent mechanism. The observed decrease in sensitivity to SNP in cav-1−/− mice is consistent with a switch to a sGC pathway less sensitive to NO. 5) Whether the observed changes in conduit vessels also occur in resistance vessels that control BP need to be examined in future studies.
In conclusion, cav-1 deficiency is associated with increased BP, hyperglycemia, enhanced NO-cGMP–dependent relaxation, and reduced vascular contraction. Metformin does not change BP in cav-1−/− mice, likely because of its dual inhibitory effects on both vascular contraction mechanisms and vascular relaxation via NO-cGMP. Metformin does not affect blood glucose levels or body weight in cav-1−/− mice, consistent with its reported inadequate effects on glycemic control in certain patients with diabetes (Bailey et al., 2013). The data support the notion of partial independence of the vascular changes from hyperglycemia in cav-1 deficiency states. The data also suggest that hypertensive individuals with reduced insulin sensitivity, such as minor allele carriers of cav-1 gene variants (Pojoga et al., 2011), may benefit from the vascular effects of metformin, even in the absence of improvements in BP or glucose homeostasis.
Acknowledgments
The authors thank Paul Loutraris, Brigham and Women’s Hospital, for technical assistance.
Abbreviations
- Ach
acetylcholine
- AMPK
AMP-activated protein kinase
- BP
blood pressure
- cav-1
caveolin-1
- eNOS
endothelial nitric-oxide synthase
- l-NAME
Nω-nitro-l-arginine methyl ester
- NO
nitric oxide
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- Phe
phenylephrine
- PKC
protein kinase C
- PRA
plasma renin activity
- sGC
soluble guanylate cyclase
- SNP
sodium nitroprusside
- VSM
vascular smooth muscle
- WT
wild-type
Authorship Contributions
Participated in research design: Pojoga, Williams, Khalil.
Conducted experiments: Pojoga, Yao, Reslan, Khalil.
Performed data analysis: Pojoga, Opsasnick, Reslan, Khalil.
Wrote or contributed to the writing of the manuscript: Pojoga, Garza, Adler, Williams, Khalil.
Footnotes
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grants HL-104032 (to L.H.P.), HL-69208 and T32-HL007609 (to G.H.W.), and HL-65998, HL-98724, and HL-111775 (to R.A.K.)]; National Institutes of Health The Eunice Kennedy Shriver National Institute of Child Health and Human Development [Grant HD-60702] (to R.A.K.); and the American Heart Association [Grant 0735609T] (to L.H.P.).
References
- Agard C, Rolli-Derkinderen M, Dumas-de-La-Roque E, Rio M, Sagan C, Savineau JP, Loirand G, Pacaud P. (2009) Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br J Pharmacol 158:1285–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal N, Rice SP, Bolusani H, Luzio SD, Dunseath G, Ludgate M, Rees DA. (2010) Metformin reduces arterial stiffness and improves endothelial function in young women with polycystic ovary syndrome: a randomized, placebo-controlled, crossover trial. J Clin Endocrinol Metab 95:722–730 [DOI] [PubMed] [Google Scholar]
- Anter E, Thomas SR, Schulz E, Shapira OM, Vita JA, Keaney JF., Jr (2004) Activation of endothelial nitric-oxide synthase by the p38 MAPK in response to black tea polyphenols. J Biol Chem 279:46637–46643 [DOI] [PubMed] [Google Scholar]
- Asterholm IW, Mundy DI, Weng J, Anderson RG, Scherer PE. (2012) Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab 15:171–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CJ, Gross JL, Hennicken D, Iqbal N, Mansfield TA, List JF. (2013) Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med 11:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batova S, DeWever J, Godfraind T, Balligand JL, Dessy C, Feron O. (2006) The calcium channel blocker amlodipine promotes the unclamping of eNOS from caveolin in endothelial cells. Cardiovasc Res 71:478–485 [DOI] [PubMed] [Google Scholar]
- Bender SB, McGraw AP, Jaffe IZ, Sowers JR. (2013) Mineralocorticoid receptor-mediated vascular insulin resistance: an early contributor to diabetes-related vascular disease? Diabetes 62:313–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla RC, Toth KF, Tan E, Bhatty RA, Mathias E, Sharma RV. (1996) Vascular effects of metformin: possible mechanisms for its antihypertensive action in the spontaneously hypertensive rat. Am J Hypertens 9:570–576 [DOI] [PubMed] [Google Scholar]
- Brands MW, Hopkins TE. (1996) Poor glycemic control induces hypertension in diabetes mellitus. Hypertension 27:735–739 [DOI] [PubMed] [Google Scholar]
- Catalán V, Gómez-Ambrosi J, Rodríguez A, Silva C, Rotellar F, Gil MJ, Cienfuegos JA, Salvador J, Frühbeck G. (2008) Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation. Clin Endocrinol (Oxf) 68:213–219 [DOI] [PubMed] [Google Scholar]
- Chen XL, Panek K, Rembold CM. (1997) Metformin relaxes rat tail artery by repolarization and resultant decreases in Ca2+ influx and intracellular [Ca2+]. J Hypertens 15:269–274 [DOI] [PubMed] [Google Scholar]
- Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. (1999) AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett 443:285–289 [DOI] [PubMed] [Google Scholar]
- Cohen AW, Razani B, Wang XB, Combs TP, Williams TM, Scherer PE, Lisanti MP. (2003) Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol 285:C222–C235 [DOI] [PubMed] [Google Scholar]
- Cornwell TL, Lincoln TM. (1989) Regulation of intracellular Ca2+ levels in cultured vascular smooth muscle cells: reduction of Ca2+ by atriopeptin and 8-bromo-cyclic GMP is mediated by cyclic GMP-dependent protein kinase. J Biol Chem 264:1146–1155 [PubMed] [Google Scholar]
- Dimopoulos GJ, Semba S, Kitazawa K, Eto M, Kitazawa T. (2007) Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ Res 100:121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, et al. (2001) Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293:2449–2452 [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Pacher P, Schmidt PM, Haskó G, Schmidt HH, Stasch JP. (2006) NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov 5:755–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Rojo MA, Gongora M, Fitzsimmons RL, Martel N, Martin SD, Nixon SJ, Brooks AJ, Ikonomopoulou MP, Martin S, Lo HP,, et al. (2013) Caveolin-1 is necessary for hepatic oxidative lipid metabolism: evidence for crosstalk between caveolin-1 and bile acid signaling. Cell Rep 4:238–247 [DOI] [PubMed] [Google Scholar]
- Feron O, Saldana F, Michel JB, Michel T. (1998) The endothelial nitric-oxide synthase-caveolin regulatory cycle. J Biol Chem 273:3125–3128 [DOI] [PubMed] [Google Scholar]
- Gallo A, Ceolotto G, Pinton P, Iori E, Murphy E, Rutter GA, Rizzuto R, Semplicini A, Avogaro A. (2005) Metformin prevents glucose-induced protein kinase C-beta2 activation in human umbilical vein endothelial cells through an antioxidant mechanism. Diabetes 54:1123–1131 [DOI] [PubMed] [Google Scholar]
- Hall JE, Brands MW, Shek EW. (1996) Central role of the kidney and abnormal fluid volume control in hypertension. J Hum Hypertens 10:633–639 [PubMed] [Google Scholar]
- Hauton D. (2011) Does long-term metformin treatment increase cardiac lipoprotein lipase? Metabolism 60:32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen S, Argmann CA, Champy MF, and Auwerx J (2007) Evaluation of glucose homeostasis. Curr Protoc Mol Biol 29B3:1--22 [DOI] [PubMed] [Google Scholar]
- Heishi M, Ichihara J, Teramoto R, Itakura Y, Hayashi K, Ishikawa H, Gomi H, Sakai J, Kanaoka M, Taiji M, et al. (2006) Global gene expression analysis in liver of obese diabetic db/db mice treated with metformin. Diabetologia 49:1647–1655 [DOI] [PubMed] [Google Scholar]
- Ishizaka N, Griendling KK, Lassègue B, Alexander RW. (1998) Angiotensin II type 1 receptor: relationship with caveolae and caveolin after initial agonist stimulation. Hypertension 32:459–466 [DOI] [PubMed] [Google Scholar]
- Kizub IV, Pavlova OO, Johnson CD, Soloviev AI, Zholos AV. (2010) Rho kinase and protein kinase C involvement in vascular smooth muscle myofilament calcium sensitization in arteries from diabetic rats. Br J Pharmacol 159:1724–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lastra G, Dhuper S, Johnson MS, Sowers JR. (2010) Salt, aldosterone, and insulin resistance: impact on the cardiovascular system. Nat Rev Cardiol 7:577–584 [DOI] [PubMed] [Google Scholar]
- Lee CH, Poburko D, Sahota P, Sandhu J, Ruehlmann DO, van Breemen C. (2001) The mechanism of phenylephrine-mediated [Ca(2+)](i) oscillations underlying tonic contraction in the rabbit inferior vena cava. J Physiol 534:641–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Liu C, Li N, Hao T, Han T, Hill DE, Vidal M, Lin JD. (2008) Genome-wide coactivation analysis of PGC-1alpha identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metab 8:105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmeier TE, Hildebrandt DA, Warren S, May PJ, Cunningham JT. (2005) Recent insights into the interactions between the baroreflex and the kidneys in hypertension. Am J Physiol Regul Integr Comp Physiol 288:R828–R836 [DOI] [PubMed] [Google Scholar]
- Mahrouf M, Ouslimani N, Peynet J, Djelidi R, Couturier M, Therond P, Legrand A, Beaudeux JL. (2006) Metformin reduces angiotensin-mediated intracellular production of reactive oxygen species in endothelial cells through the inhibition of protein kinase C. Biochem Pharmacol 72:176–183 [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Noguchi E, Ishida K, Kobayashi T, Yamada N, Kamata K. (2008) Metformin normalizes endothelial function by suppressing vasoconstrictor prostanoids in mesenteric arteries from OLETF rats, a model of type 2 diabetes. Am J Physiol Heart Circ Physiol 295:H1165–H1176 [DOI] [PubMed] [Google Scholar]
- Minshall RD, Sessa WC, Stan RV, Anderson RG, Malik AB. (2003) Caveolin regulation of endothelial function. Am J Physiol Lung Cell Mol Physiol 285:L1179–L1183 [DOI] [PubMed] [Google Scholar]
- Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. (2003) Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem 278:31629–31639 [DOI] [PubMed] [Google Scholar]
- Muniyappa R, Lee S, Chen H, Quon MJ. (2008) Current approaches for assessing insulin sensitivity and resistance in vivo: advantages, limitations, and appropriate usage. Am J Physiol Endocrinol Metab 294:E15–E26 [DOI] [PubMed] [Google Scholar]
- Murphy JG, Khalil RA. (2000) Gender-specific reduction in contractility and [Ca(2+)](i) in vascular smooth muscle cells of female rat. Am J Physiol Cell Physiol 278:C834–C844 [DOI] [PubMed] [Google Scholar]
- Nobe K, Hashimoto T, Honda K. (2012) Two distinct dysfunctions in diabetic mouse mesenteric artery contraction are caused by changes in the Rho A-Rho kinase signaling pathway. Eur J Pharmacol 683:217–225 [DOI] [PubMed] [Google Scholar]
- Osuka K, Watanabe Y, Usuda N, Atsuzawa K, Yoshida J, Takayasu M. (2009) Modification of endothelial nitric oxide synthase through AMPK after experimental subarachnoid hemorrhage. J Neurotrauma 26:1157–1165 [DOI] [PubMed] [Google Scholar]
- Park KM, Trucillo M, Serban N, Cohen RA, Bolotina VM. (2008) Role of iPLA2 and store-operated channels in agonist-induced Ca2+ influx and constriction in cerebral, mesenteric, and carotid arteries. Am J Physiol Heart Circ Physiol 294:H1183–H1187 [DOI] [PubMed] [Google Scholar]
- Pojoga LH, Adamová Z, Kumar A, Stennett AK, Romero JR, Adler GK, Williams GH, Khalil RA. (2010) Sensitivity of NOS-dependent vascular relaxation pathway to mineralocorticoid receptor blockade in caveolin-1-deficient mice. Am J Physiol Heart Circ Physiol 298:H1776–H1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pojoga LH, Underwood PC, Goodarzi MO, Williams JS, Adler GK, Jeunemaitre X, Hopkins PN, Raby BA, Lasky-Su J, Sun B, et al. (2011) Variants of the caveolin-1 gene: a translational investigation linking insulin resistance and hypertension. J Clin Endocrinol Metab 96:E1288–E1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pojoga LH, Yao TM, Sinha S, Ross RL, Lin JC, Raffetto JD, Adler GK, Williams GH, Khalil RA. (2008) Effect of dietary sodium on vasoconstriction and eNOS-mediated vascular relaxation in caveolin-1-deficient mice. Am J Physiol Heart Circ Physiol 294:H1258–H1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, et al. (2001) Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276:38121–38138 [DOI] [PubMed] [Google Scholar]
- Salamanca DA, Khalil RA. (2005) Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem Pharmacol 70:1537–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salani B, Maffioli S, Hamoudane M, Parodi A, Ravera S, Passalacqua M, Alama A, Nhiri M, Cordera R, Maggi D. (2012) Caveolin-1 is essential for metformin inhibitory effect on IGF1 action in non-small-cell lung cancer cells. FASEB J 26:788–798 [DOI] [PubMed] [Google Scholar]
- Schulman IH, Zhou MS. (2009) Vascular insulin resistance: a potential link between cardiovascular and metabolic diseases. Curr Hypertens Rep 11:48–55 [DOI] [PubMed] [Google Scholar]
- Shakirova Y, Bonnevier J, Albinsson S, Adner M, Rippe B, Broman J, Arner A, Swärd K. (2006) Increased Rho activation and PKC-mediated smooth muscle contractility in the absence of caveolin-1. Am J Physiol Cell Physiol 291:C1326–C1335 [DOI] [PubMed] [Google Scholar]
- Sharma RV, Bhalla RC. (1995) Metformin attenuates agonist-stimulated calcium transients in vascular smooth muscle cells. Clin Exp Hypertens 17:913–929 [DOI] [PubMed] [Google Scholar]
- Tanano I, Nagaoka T, Omae T, Ishibazawa A, Kamiya T, Ono S, Yoshida A. (2013) Dilation of porcine retinal arterioles to cilostazol: roles of eNOS phosphorylation via cAMP/protein kinase A and AMP-activated protein kinase and potassium channels. Invest Ophthalmol Vis Sci 54:1443–1449 [DOI] [PubMed] [Google Scholar]
- Uruno A, Sugawara A, Kanatsuka H, Kagechika H, Saito A, Sato K, Kudo M, Takeuchi K, Ito S. (2005) Upregulation of nitric oxide production in vascular endothelial cells by all-trans retinoic acid through the phosphoinositide 3-kinase/Akt pathway. Circulation 112:727–736 [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Alexander RW. (2006) Caveolin-dependent angiotensin II type 1 receptor signaling in vascular smooth muscle. Hypertension 48:797–803 [DOI] [PubMed] [Google Scholar]
- Venema RC, Venema VJ, Ju H, Harris MB, Snead C, Jilling T, Dimitropoulou C, Maragoudakis ME, Catravas JD. (2003) Novel complexes of guanylate cyclase with heat shock protein 90 and nitric oxide synthase. Am J Physiol Heart Circ Physiol 285:H669–H678 [DOI] [PubMed] [Google Scholar]
- Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. (2012) Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 122:253–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GH. (1994) Essential hypertension as an endocrine disease. Endocrinol Metab Clin North Am 23:429–444 [PubMed] [Google Scholar]
- Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC. (2006) Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest 116:1284–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]