

Abstract
Increased numbers of macrophages are found in the lungs of smokers and those with chronic obstructive pulmonary disease. Experimental evidence shows the central role of macrophages in elaboration of inflammatory mediators such as TNF-a and the progression toward cigarette smoke-induced emphysema. We investigated the role of CX3CR1 in recruitment of mononuclear phagocytes, inflammatory cytokine responses, and tissue destruction in the lungs after cigarette smoke exposure. Using mice in which egfp is expressed at the locus of the cx3cr1 gene, we show that alveolar macrophages increased transmembrane ligand CX3CL1 expression and soluble CX3CL1 was detectable in the airspaces, but cx3cr1GFP/GFP and cx3cr1GFP/+ mice failed to show recruitment of CX3CR1+ cells into the airspaces with cigarette smoke. In contrast, cigarette smoke increased the accumulation of CX3CR1+CD11b+ mononuclear phagocytes that were spatially confined to the lung interstitium and heterogenous in their expression of CD11c, MHC class II, and autofluorescent property. Although an intact CX3CL1–CX3CR1 pathway amplified the percentage of CX3CR1+CD11b+ mononuclear phagocytes in the lungs, it was not essential for recruitment. Rather, functional CX3CR1 was required for a subset of tissue-bound mononuclear phagocytes to produce TNF-α and IL-6 in response to cigarette smoke, and the absence of functional CX3CR1 protected mice from developing tissue-destructive emphysema. Thus, CX3CR1+ “tissue resident” mononuclear phagocytes initiate an innate immune response to cigarette smoke by producing TNF-α and IL-6 and are capable of promoting emphysema.
Chronic obstructive pulmonary disease (COPD) is a leading cause of morbidity and mortality worldwide, and cigarette smoke exposure is cited as the most important risk factor (1). COPD is characterized by “an abnormal inflammatory response to noxious particles or gases” leading to variable degrees of tissue destruction, or emphysema, and airflow limitation that is influenced by the genetic susceptibility of the host (2). Physiologic gas transfer measurements suggesting emphysematous destruction may be one of the earliest measures to change even before clinically perceptible disease (3), supporting the concept of chronic cigarette smoke-induced inflammation causing repeated microinjury and eventual tissue destruction in susceptible hosts. However, the precise network of signaling events that perpetuates cigarette smoke-induced inflammation and tissue destruction is not fully understood.
The mononuclear phagocyte system plays an important role in patrolling the lung microenvironment that interfaces directly with inhaled noxious particles or gases. Although macrophages, key representatives of the mononuclear phagocyte system in the lungs, serve as a critical line of host defense against inhaled environmental particles and pathogens, experimental evidence shows those macrophages directly contribute to the pathogenesis of COPD. Macrophages orchestrate the release of mediators that promote both inflammation and tissue-destructive emphysema (4–7). Indeed, macrophages elaborate matrix metalloproteinase (MMP)-12, or macrophage elastase, which breaks down elastin and is essential for the development of cigarette smoke-induced emphysema in mice (4). Moreover, MMP-12 forms elastin fragments that are a major chemotactic factor for monocytes in cigarette smoke-induced emphysema (6) and mediates the release of bioactive TNF-α (5, 8). TNF-α, a central cytokine in the innate immune response to cigarette smoke, propagates inflammation through endothelial activation, promoting neutrophil influx and further elaboration of neutrophil-derived tissue-destructive proteases (5). The source of and process by which TNF-α production is amplified during cigarette smoke exposure is not completely understood.
We recently showed that increases in chemokine CX3CL1 gene expression is associated with recruitment of CX3CR1+ mononuclear phagocytes in the lungs during cigarette smoke-induced emphysema (9). Although cigarette smoke induced the accumulation of CX3CR1+ mononuclear phagocytes, whether an intact CX3CL1–CX3CR1 pathway was required for their recruitment was not directly tested. This is of potential relevance to COPD pathogenesis, as CX3CR1 is highly expressed by resident mononuclear phagocytes in both resting and inflamed tissues (10) and harbor the immediate potential to differentiate into macrophages (10–12), whereas inflammatory monocytes express CCR2 and the cell surface protein Ly6c (Gr1+) and are able to differentiate into inflammatory dendritic cells (10). Although considerable plasticity is involved in determining the fate of mononuclear phagocytes, the functional relevance of CX3CR1 in cigarette smoke-related lung diseases is of interest as CX3CL1 is the sole ligand for CX3CR1 and ideally suited as a specific pharmacological target. We hypothesized that cigarette smoke exposure propagates mononuclear phagocytes characterized by CX3CR1 expression that are functionally distinct in their cytokine response to promote inflammation and a tissue-destructive phenotype.
Materials and Methods
Animals
B6.129P-Cx3cr1tm1Litt/J (cx3cr1GFP/GFP) mice, in which egfp is expressed at the locus of the cx3cr1 gene, and C57BL/6 wild-type mice were purchased from The Jackson Laboratory (Bar Harbor, ME). B6.129P-cx3cr1tm1Litt/J (cx3cr1GFP/GFP) mice were crossbred with C57BL/6J wild-type mice to produce B6.129P-Cx3cr1tm1Litt/J (cx3cr1GFP/+) heterozygous mice, and a colony using a cx3cr1GFP/+ × cx3cr1GFP/GFP breeding strategy was subsequently maintained. All mice used in the experiments were age and gender matched. Experiments conducted were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh.
Cigarette smoke exposure model
For short-term cigarette smoke exposure, total body cigarette smoke exposure was performed in a stainless steel chamber using a smoking machine to produce a combination of side-stream and main-stream smoke (model TE-10; Teague Enterprises, Woodland, CA). A detailed method of our chronic environmental tobacco smoke exposure method has been previously described (9, 13, 14). The cigarette-smoking machine puffed each 4R3F University of Kentucky research cigarette for 2 s for a total of nine puffs before ejection. The smoke machine was adjusted to deliver seven cigarettes per cycle. The cigarette smoke from 100 cigarettes was delivered to the mice each day, 5 d/wk consisting of ~2.5 h in duration per day. Age-, gender-, and strain-matched mice served as controls and were exposed to air-only in the same environmental conditions. The smoking chamber atmosphere was periodically measured for total particulate matter concentrations of ~200 mg/m3. For assessment of cigarette smoke-induced emphysema, long-term cigarette smoke exposure was conducted using a smoking apparatus that delivers cigarette smoke of four unfiltered cigarettes per day (University of Kentucky, Lexington, KY), 5 d/wk for 6 mo, to mice using a nose-cone direct approach to generate a potent source of exposure that induces emphysema (4).
Isolation of bronchoalveolar lavage macrophages and peritoneal macrophages
The animals were euthanized with overdose of i.p. pentobarbital. The thorax was opened, the trachea was identified and cannulated with an 18-gauge angiocatheter, and sequential lavages of 1.2, 1.0, 1.0, and 1.0 ml bronchoalveolar lavage (BAL) fluid (0.9% saline with 0.6 mmol/l EDTAwarmed to 37°C) were instilled into the lungs. The fluid was allowed to sit for ~30 s before it was slowly removed from the lungs. The four aliquots were pooled for analysis. Cytospin slides were prepared using Shandon Cytospin 3 centrifuge (Thermo Fisher Scientific, Waltham, MA). Manual cell counts and differentials were performed and confirmed that 97–99% of the BAL cells were macrophages. Peritoneal macrophages were isolated by flushing the peritoneal cavity of mouse with 5 ml sterile PBS through a 14-gauge angiocatheter a total of 5 times per mouse.
Isolation of lung mononuclear phagocytes
After euthanization, mice underwent whole-lung lavage. The right ventricle and pulmonary artery were perfused with 5 ml cold, sterile PBS to flush blood from the lung vasculature. The lungs were removed and digested with DNAse I and collagenase A at 37°C for 45 min. Single cells were filtered through a 70-μm cell strainer. The cells were incubated with magnetic microbeads conjugated with anti-mouse CD11c Ab (Miltenyi Biotec, Bergisch Gladbach, Germany) for 15 min on ice. CD11c-expressing cells were positively selected using an LS separation column under magnetic field according to the manufacturer’s instructions. Cells were allowed to adhere onto tissue culture plates at 37°C, 5% CO2 for 2 h. The plates were gently washed twice with cell culture medium to remove unattached cells prior to use.
In vitro stimulation of cultured cells
BAL macrophages, peritoneal macrophages, RAW264.7 cells, or CD11c-selected lung mononuclear phagocytes were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA). LPS (Escherichia coli 011:B4; List Biologicals, Campbell, CA) and recombinant murine TNF-α, IFN-γ, or CX3CL1 (R&D Systems, Minneapolis, MN) was added to the cell culture at different concentrations and incubated for the indicated time. For experiments requiring flow cytometric analysis, cells were trypsinized and flushed several times to remove adherent cells from culture plates prior to counting and subsequent Ab staining. For experiments requiring RNA, cells were lysed directly in the culture dish by addition of TRIzol reagent (Invitrogen).
Flow cytometry
FITC-, PE-, PE-Cy7–, or allophycocyanin-conjugated mAbs against mouse leukocyte Ags CD11b, CD11c, I-A[b], CD80, CD86, CD40 and mAbs against murine intracellular cytokines TNF-α, IFN-γ, IL-6, and IL-10 were obtained from BD Biosciences (San Jose, CA). Anti-mouse CX3CL1 mAbs were purchased from R&D Systems. Goat anti-rat secondary Abs conjugated with fluorochromes were purchased from Jackson Immuno-Research Laboratories (West Grove, PA). Samples were stained at 4°C with optimal amounts of fluorochrome-conjugated mAbs diluted in staining buffer (PBS containing 2% FBS) for 30 min, then washed with staining buffer three times. For intracellular cytokine experiments, monensin was used as a protein transport inhibitor during cell culture and stimulation. Cell fixation and permeabilization were performed with BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit with BD GolgiStop for intracellular staining. At least 20,000 events were acquired per immunostain on a FACSCalibur flow cytometer (BD Biosciences). Flow cytometry data were analyzed with FCS Express analysis software (De Novo Software, Los Angeles, CA).
Real-time PCR
Our method of real-time PCR has been previously described (9). Adherent cells on tissue culture plates were lysed directly by addition of TRIzol reagent. After phase separation with chloroform and precipitation of RNA with isopropyl alcohol, RNA pellet was washed once with 75% ethanol and resuspended in nuclease-free water. We examined the quality of our RNA preparation by electrophoresis to exclude RNA degradation and measured concentrations by Nano-Drop. RNA samples were reverse-transcribed into cDNA using SuperScript III Reverse Transcriptase (Moloney murine leukemia virus reverse transcriptase; Invitrogen). Quantitative real-time PCR was performed according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA) by incubating cDNA samples with specified probes and primers of interest and TaqMan Universal PCR Master Mix II and measuring PCR amplification using the 7900HT Real-Time PCR System. Probes and primers for inducible NO synthase (NOS2; Mm00440502_m1), TNF (TNF-α; Mm00443258_m1), IL-6 (IL-6; Mm00446190_m1), IL-10 (IL-10; Mm01288386_m1), MMP-12 (MMP-12; Mm00500554_m1), and 18S genes were commercially available at Applied Biosystems. Gene expression was analyzed by the ΔΔ-threshold cycle (ΔΔCt) method, with 18S rRNA as the endogenous control, and average ΔCt of unstimulated wild-type controls served as the calibrator.
ELISA
Mouse CX3CL1 in BAL and lung homogenates were detected by ELISA using mouse CX3CL1/Fractalkine DuoSet kit from R&D Systems. All samples were assayed in duplicate, and standard curve was established for calculation of CX3CL1 concentration in the samples using recombinant mouse CX3CL1. For BAL samples, mouse CX3CL1 concentrations are reported as pg/ml. For lung homogenate samples, lungs were snap-frozen with liquid nitrogen and put into cytokine lysis buffer with protease inhibitor mixture and homogenized using a tissue homogenizer. The concentration of mouse CX3CL1 was normalized to the total protein concentration measured in each sample. Total protein concentration for unknown lung homogenate samples was determined using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Protein concentration for unknown samples was calculated using a standard curve generated with serial dilutions of BSA. Absorbance at 595 nm was measured with a spectrophotometer.
ELISPOT assay
Ninety-six–well polyvinylidene difluoride plates (Millipore, Billerica, MA) were coated overnight with an anti-mouse TNF-α or IL-6 Abs (BD Biosciences) according to the instructions of the manufacturer. Plates were washed two times in PBS and blocked for 2 h at 37°C with 200 μl/well DMEM containing 10% FBS. CD11c-enriched lung mononuclear phagocytes from mice were then added in a total volume of 200 μl/well of complete medium and incubated at 37°C 5% CO2 for 12 h (TNF-α) or 18 h (IL-6). Cells were removed, plates were washed, and biotinylated anti-mouse TNF-α or IL-6 detection Ab diluted in PBS with 10% FBS was added. After incubation for 2 h at room temperature, HRP-conjugated avidin diluted in PBS with 10% FBS was added for 1 h at room temperature. Plates were washed and 3,3′,5,5′-tetramethylbenzidine substrate or 3-amino-9-ethylcarbazole substrate was added (Vector Lab, Burlingame, CA). The reaction was terminated by washing with de-ionized water. After removal of the bottom, the plates were air-dried overnight at 4°C before spot counting using a CTL-ImmunoSpot reader (Cellular Technology, Shaker Heights, OH). All data are expressed as spot-forming cells per total cells and analyzed with Immunospot version 3 or 5.
Mean chord length measurements
After chronic cigarette smoke exposure, mice were sacrificed, and a tracheostomy was performed. The lungs were removed and inflated with 10% buffered formalin at a constant pressure of 25 cm H2O for 10 min via intratracheal cannula, ligated, and fixed for 24 h before embedding in paraffin. Serial midsagittal sections were obtained, stained with a modified Gill’s stain, and used to determine chord length (CL), an estimation of alveolar size, as previously described (4, 15). Ten randomly selected ×200 fields per slide were photographed and the images analyzed using Scion Image software (Scion Corporation, Frederick, MD). Airway and vascular structures were eliminated from the analysis.
Destructive index measurements
Paraffin-embedded lung tissue samples were sectioned, mounted on slides, stained with H&E, and analyzed for alveolar wall destruction using the destructive index as previously described by Saetta and colleagues (16). A minimum of 20 random images per specimen were acquired using a Zeiss Axiophot microscope with an Axiocam HRc camera and a ×20 objective. The images were imported into Adobe Photoshop, and a grid containing 20 equally spaced intersections was overlayed onto each image. The alveolar spaces or duct spaces underlying each intersection of the grid were classified as either normal (N) or destroyed (D), and the destructive index for each specimen was calculated using the following formula: destructive index (DI) = D/(D + N) × 100. An alveolar space or duct space was classified as normal when its wall was completely intact or broken in only one place. An alveolar space was classified as destroyed when the alveolar wall was broken in two or more places. A duct space was classified as destroyed when the lumen contained two or more isolated islands of lung parenchyma.
Statistics
The experimental data are expressed as mean ± SD unless otherwise stated. Statistical analysis was done using one-way ANOVA for multiple comparisons. If the data failed the test for homogeneity (Levene’s test), statistical analysis was completed using statistical method of equal variances not assumed. A p value <0.05 was considered significant. All statistics were performed using GraphPad Prism version 5.0 (GraphPad Software, La Jolla, CA) or SPSS Statistics software version 17 (SPSS, Chicago, IL).
Results
Cigarette smoke exposure increases CX3CL1 production in the lungs, and macrophages are a source of constitutive and inducible transmembrane CX3CL1 expression
We have previously shown that CX3CL1 gene expression is increased in the lungs of AKR/J mice who develop cigarette smoke-induced emphysema (9). To determine whether chronic cigarette smoke exposure increases CX3CL1 expression in C57BL/6 mice, CX3CL1 protein was measured in the lung homogenates of air-exposed and of cigarette-exposed animals. Compared with that of air-exposed controls, CX3CL1 protein increased significantly upon chronic cigarette smoke by 8 wk of exposure (10 ng/mg versus 20 ng/mg, p < 0.05) (Fig. 1A). Thus, across strains, CX3CL1 expression is upregulated with cigarette smoke exposure.
FIGURE 1.
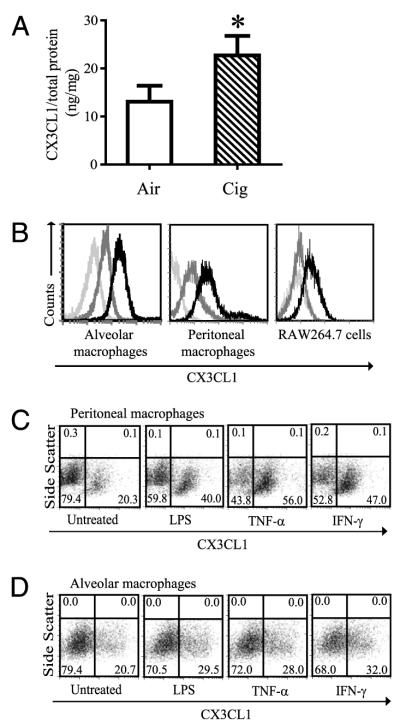
Transmembrane CX3CL1 expression on macrophages. A, Lung homogenates were obtained from C57BL/6 mice after air or cigarette smoke exposure for 8 wk and assayed for CX3CL1 expression. CX3CL1 concentration was normalized to the amount of total protein measured in each sample and is expressed as ng/mg ± SD. n = 8 mice per group. *p < 0.05. B, Mouse alveolar macrophages (left panel), peritoneal macrophages (middle panel), and mouse macrophage cell line RAW 264.7 cells (right panel) constitutively express transmembrane CX3CL1. Light-gray histogram, absence of immunostaining. Dark-gray histogram, isotype control staining. Black histogram, CX3CL1 staining. Representative images from four independent experiments are shown. C and D, Transmembrane CX3CL1 expression on peritoneal macrophages (C) and alveolar macrophages (D) is induced by TNF-α (20 ng/ml), IFN-γ (500 U/ml), and LPS (100 ng/ml) after 6 h stimulation (pooled cells from n = 9 to 12 mice).
CX3CL1 is a surface-bound molecule consisting of a chemokine domain, a mucin stalk, and a transmembrane domain (17, 18) that can mediate leukocyte adhesion (19, 20) and is expressed on inflamed endothelium (20), dendritic cells (21), epithelial cells (22), and neurons (23). Membrane-bound CX3CL1 requires cleavage by metalloproteinases to render the soluble chemokine (24, 25). We examined whether macrophages, essential for the development of emphysema (4), represent one source of CX3CL1 expression in the lungs, as previous reports have shown CX3CL1 expression by macrophages in human atherosclerotic plaques by immunohistochemistry (25). Both alveolar and peritoneal macrophages constitutively expressed transmembrane CX3CL1 (Fig. 1B). The murine macrophage cell line RAW264.7 also showed constitutive expression of transmembrane CX3CL1 (Fig. 1B). Moreover, transmembrane CX3CL1 expressed by macrophages was inducible by proinflammatory stimuli TNF-α, IFN-γ, and LPS (Fig. 1C, 1D). Thus, one source of CX3CL1 production in the lungs is macrophages that increase surface expression of this chemokine after exposure to inflammatory stimuli.
Alveolar macrophages increase transmembrane CX3CL1 with cigarette smoke exposure but fail to recruit significantly CX3CR1 mononuclear phagocytes into the airspaces
One hallmark of chronic cigarette smoke exposure is increased accumulation of mononuclear phagocytes in the lungs (26). However, the factors that mediate recruitment and compartmentalization of lung mononuclear phagocytes are not fully known. We used cx3cr1GFP/GFP mice, lacking functional CX3CR1, and cx3cr1GFP/+ mice, heterozygous for the insertion and possessing one functional CX3CR1 allele, to assess the role of the CX3CL1–CX3CR1 pathway in mononuclear phagocyte recruitment into the lungs with cigarette smoke exposure. We initially examined the airspace compartment and found that the majority of cells were macrophages that express CD11c (Fig. 2A) but lacked the surface marker CD11b (Fig. 2B), were MHC class II (MHC II) low (Fig. 2C) and autofluorescent high (Fig. 2D). The characterization of BAL macrophage surface markers are consistent with prior studies (11, 27, 28). Cigarette smoke activated transmembrane CX3CL1 in alveolar macrophages of both cx3cr1GFP/GFP and cx3cr1GFP/+ mice, with higher expression in cx3cr1GFP/+ mice than that in cx3cr1GFP/GFP mice (Fig. 2E). However, alveolar macrophages failed to express CX3CR1 either at baseline or after cigarette smoke exposure in significant numbers, with CX3CR1+ cells accounting for <1% of cells examined (Fig. 2E). Although cleaved CX3CL1 was detectable in the airspaces, the increases with cigarette smoke exposure observed were not significantly different between cx3cr1GFP/+ and cx3cr1GFP/GFP mice (Fig. 2F). Thus, cigarette smoke exposure induces transmembrane CX3CL1 expression by alveolar macrophages, but CX3CL1 activation fails to recruit significant numbers of CX3CR1+ cells into the airspaces.
FIGURE 2.
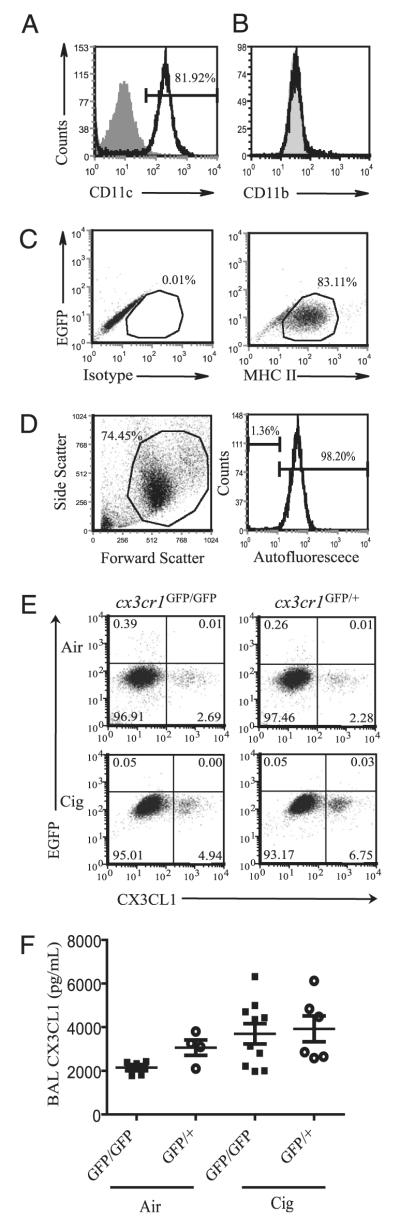
Characterization of alveolar macrophages exposed to air or cigarette smoke. A and B, Alveolar macrophages express CD11c but not CD11b. Gray-filled histogram represents isotype control staining. Black histogram represents CD11c or CD11b staining. C, The majority of alveolar macrophages lack CX3CR1 and are MHC IIlow. Isotype control staining (left panel) versus MHC II staining (right panel). The y-axis indicates EGFP signal. D, BAL macrophages are autofluorescent high. BAL cells (left panel) with histogram of gated cell population shown (right panel). Autofluorescence shown was examined in the FL-2 channel in the absence of Ab staining. Representative data shown are from n = 2 to 3 cx3cr1GFP/+ mice per group, with two independent experiments conducted (A–D). E, Alveolar macrophages isolated from cx3cr1GFP/GFP and cx3cr1GFP/+ mice exposed to ambient air or cigarette smoke for 4 wk were assayed for CX3CR1 and transmembrane CX3CL1 expression (pooled samples from n = 3 mice per group). Representative data shown are from three independent experiments. F, Cleaved CX3CL1 concentrations in the BAL fluid of cx3cr1GFP/GFP and cx3cr1GFP/+ mice, air versus cigarette smoke exposure. Each point shown represents an individual mouse sample collected after 2- to 4-wk exposure, and data are expressed as mean ± SEM. p = 0.1 (ANOVA).
Cigarette smoke exposure increases the accumulation of CX3CR1+CD11b+ mononuclear phagocytes bound to the lung interstitium
We next determined whether CX3CR1-expressing mononuclear phagocytes are present in the lung interstitial compartment and increase in number in response to cigarette smoke exposure. We defined interstitial mononuclear phagocytes as those mononuclear phagocytes isolated from collagenase-digested lung tissue after removal of alveolar macrophages by lung lavage and leukocytes from the pulmonary circulation. Because initial studies showed that cigarette smoke exposure increases the baseline autofluorescent property of lung tissue, characterizing and immunophenotyping cell populations from lung tissue digests proved challenging. Thus, we took an approach using CD11c magnetic beads to enrich for mononuclear phagocytes from lung tissue digests, as both macrophages and dendritic cells express CD11c in the lungs (29).
Testing the purity of the enrichment process, ~60% of isolated lung tissue cells were CD11c+ after positive selection (Fig. 3A). Of the CD11c-enriched population, ~40% of CX3CR1+ mononuclear phagocytes, as determined by EGFP fluorescence, expressed the β-integrin CD11c and were MHC IIhi (Fig. 3B) characteristic of surface markers expressed by dendritic cells. The majority of lung interstitial CX3CR1+ mononuclear phagocytes expressed the surface marker CD11b (Fig. 3C) that is expressed on monocytes, dendritic cells, and upregulated on macrophages during inflammation (11). High autofluorescence is attributable to macrophages rather than dendritic cells (30). Under basal conditions, CD11c-enriched CX3CR1+ lung mononuclear phagocytes contained both autofluorescent low and high populations, but the percentage of autofluorescent CX3CR1+ cells present at baseline showed a wider range across samples than other properties studied, ranging from 20 to >90% of cells examined (Fig. 3D). CX3CR1+ mononuclear phagocytes showed overall low expression of costimulatory molecules CD80, CD86, and CD40 (Fig. 3E). Thus, under homeostatic conditions, CX3CR1+ tissue-bound lung mononuclear phagocytes are a heterogenous population likely composed of dendritic cells, macrophages, and transitional mononuclear phagocytes.
FIGURE 3.
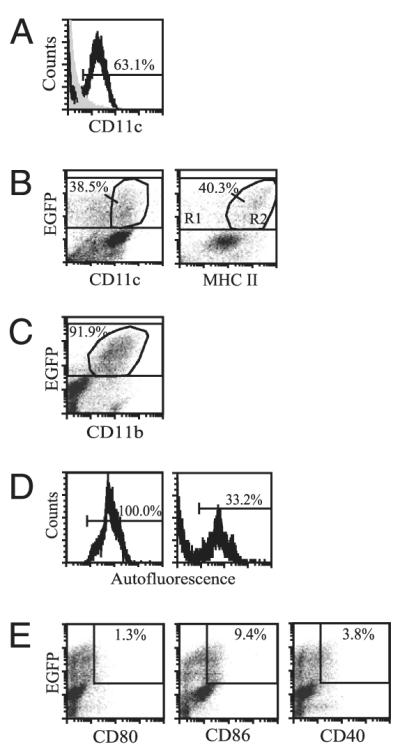
Characterization of interstitial lung mononuclear phagocytes after CD11c enrichment. A, The percentage of CD11c+ cells recovered from lung tissue digests after CD11c enrichment by magnetic bead positive selection. B, The percentage of CX3CR1+ lung mononuclear phagocytes, indicated by EGFP fluorescence, that express either CD11c (left panel) or MHC II (right panel). The percentages indicate a subset of total EGFP+ cells. C, The proportion of CX3CR1+ lung mononuclear phagocytes that express CD11b. D, The percentage of interstitial CX3CR1+ cells that are autofluorescent shows wide variability across samples, ranging from 20 to >90% (left and right panels are representative data from five independent experiments). E, The expression of costimulatory molecules CD80, CD86, and CD40 by CX3CR1+ cells is indicated. Cells were obtained from either cx3cr1GFP/GFP or cx3cr1GFP/+ mice under basal conditions as no significant differences in expression levels of surface markers were noted between cx3cr1GFP/GFP and cx3cr1GFP/+ mice. n = 2 to 3 mice from at least two independent experiments.
We examined the effect of cigarette smoke exposure on recruitment of CX3CR1+ cells given increases in CX3CL1 concentrations in the lungs. Cigarette smoke exposure doubled the numbers of CX3CR1+ cells in the lungs of both cx3cr1GFP/GFP and cx3cr1GFP/+ mice, with a higher percentage of these cells accumulating in the lung tissue of cx3cr1GFP/+ mice compared with that in cx3cr1GFP/GFP mice (25 versus 17%) (Fig. 4A). Moreover, overall percentage of CD11b+ cells recruited into the lung interstitium was higher in cx3cr1GFP/+ mice compared with that in cx3cr1GFP/GFP mice (50 versus 35%), as were the percentages of CX3CR1−CD11b+ cells (24 versus 18%). Whereas transmembrane fractalkine expression increased modestly with cigarette smoke, its expression on interstitial mononuclear phagocytes is largely independent of functional CX3CR1 (Fig. 4B). Based on these observations, CX3CR1 does not appear to be essential for recruitment of mononuclear phagocytes into the lungs after cigarette smoke exposure, but an intact CX3CL1–CX3CR1 pathway is required for amplifying a subset of mononuclear phagocytes expressing CD11b and spatially confined to the lung interstitium.
FIGURE 4.
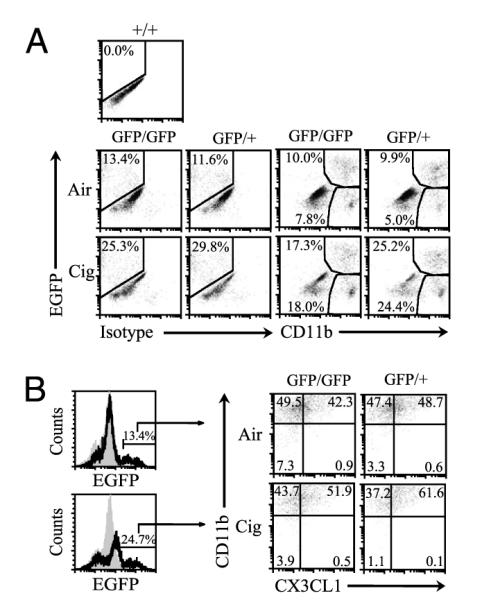
Accumulation of CX3CR1+CD11b+ mononuclear phagocytes in the lungs exposed to air or cigarette smoke. A, cx3cr1GFP/GFP and cx3cr1GFP/+ mice were exposed to ambient air or cigarette smoke for 4 wk, and CD11c-enriched interstitial cells were assayed for CD11b expression. The y-axis represents CX3CR1-expressing cells, indicated by EGFP fluorescence shift. The x-axis represents either isotype control staining or CD11b expression. EGFP fluorescence was gauged using cx3cr1+/+ mice cells set as negative control. Representative data shown from pooled cells from n = 3 mice per group, exposure time = 4 wk, two independent experiments conducted. B, The percentages of CX3CR1+ interstitial lung mononuclear phagocytes that express CD11b and transmembrane CX3CL1 after air versus cigarette smoke exposure are shown.
Lung mononuclear phagocytes require functional CX3CR1 to produce full IL-6 and TNF-α responses triggered by cigarette smoke exposure
To assess the role of cigarette smoke exposure in altering the cytokine response of CX3CR1+ lung mononuclear phagocytes, CD11c+ lung interstitial cells harvested from cx3cr1GFP/GFP and cx3cr1GFP/+ mice were assayed for CX3CR1 expression and intracellular IL-6, IL-10, TNF-α, and IFN-γ production. Lung mononuclear phagocytes of cigarette smoke-exposed cx3cr1GFP/+ mice showed IL-6 and TNF-α production, whereas cells obtained from cx3cr1GFP/GFP mice did not (Fig. 5A). No differences in IL-10 or IFN-γ production were noted above air-exposed controls in either cx3cr1GFP/+ or cx3cr1GFP/GFP mice with cigarette smoke.
FIGURE 5.
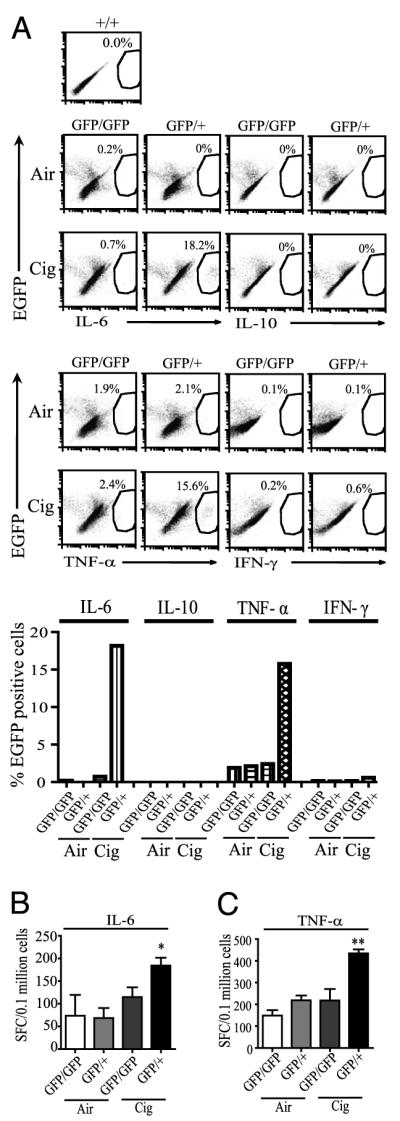
IL-6 and TNF-α responses in interstitial lung mononuclear phagocytes of cx3cr1GFP/GFP and cx3cr1GFP/+ mice exposed to air or cigarette smoke. A, CD11c-selected interstitial lung cells were isolated from mice exposed to ambient air or cigarette smoke for 4 wk, and CX3CR1-expressing cells (indicated by EGFP fluorescence shift) were assayed by intracellular cytokine immunostaining (pooled samples from n = 3 mice per group). The y-axis shows percentage of total CX3CR1 cells expressing cytokines, as indicated by EGFP fluorescence shift. EGFP fluorescence was gauged using cx3cr1+/+ mice cells set as negative control. GFP/GFP indicates cx3cr1GFP/GFP mice. GFP/+ indicates cx3cr1GFP/+ mice. B, ELISPOT data showing the number of IL-6–producing cells isolated from CD11c-enriched lung interstitial mononuclear phagocytes in cx3cr1GFP/GFP and cx3cr1GFP/+ mice exposed to air or cigarette smoke for 10 wk (pooled cells obtained from n = 5 mice in each group, performed in triplicate). *p < 0.05 (comparing cx3cr1GFP/GFP and cx3cr1GFP/+ mice; ANOVA). SFC, spot-forming cells. C, The number of TNF-α–producing cells from CD11c-enriched lung interstitial mononuclear phagocytes in cx3cr1GFP/GFP and cx3cr1GFP/+ mice exposed to cigarette smoke as determined by ELISPOT. Pooled cells were obtained from n = 2 to 3 mice in each group, 2-wk time exposure, with 6 to 12 wells per group. Data are expressed as mean ± SEM. **p < 0.01 (comparing cx3cr1GFP/GFP and cx3cr1GFP/+ mice; ANOVA).
We took advantage of the adherent properties of lung mononuclear phagocytes to confirm our findings in vitro. CD11c-enriched lung mononuclear phagocytes from air-exposed and cigarette smoke-exposed cx3cr1GFP/+ and cx3cr1GFP/GFP mice were allowed to adhere on an Elispot plate for 2 h. Nonadherent cells were washed off prior to assaying for IL-6 production. CD11c-enriched lung interstitial mononuclear phagocytes isolated from cigarette smoke-exposed cx3cr1GFP/+ mice showed higher numbers of IL-6–producing cells than that of those obtained from either air-exposed controls or cigarette smoke-exposed cx3cr1GFP/GFP mice (Fig. 5B). When exposed to cigarette smoke, higher numbers of CD11c-enriched lung mononuclear phagocytes from cx3cr1GFP/+ mice also produced TNF-α compared with that in those from cx3cr1GFP/GFP mice (Fig. 5C). Thus, functional CX3CR1 is required for full IL-6 and TNF-α responses to cigarette smoke exposure in CD11c lung mononuclear phagocytes.
CX3CL1 alone is insufficient to induce inflammatory gene expression in lung mononuclear phagocytes in vitro, but cx3cr1GFP/GFP mice show intact LPS-induced signaling
Given our findings of CX3CR1-dependent IL-6 and TNF-α production in lung mononuclear phagocytes after cigarette smoke exposure, we determined whether soluble CX3CL1 can serve as the primary signal to induce inflammatory responses in vitro because one possibility is that CX3CL1 induction in the lungs amplifies the progression of inflammatory responses during cigarette smoke exposure. Lung mononuclear phagocytes were isolated by positive selection with CD11c magnetic beads and by their ability to adhere on tissue culture plates. Cells were stimulated in vitro in the presence or absence of increasing concentrations of recombinant CX3CL1 (0, 10, 100 nM) or LPS (100 ng/ml). Under basal conditions, increasing concentrations of soluble CX3CL1 failed to induce inducible NO synthase, TNF-α, IL-6, IL-10, CXCL10, and MMP-12 gene expression in wild-type (WT) mice from baseline (Fig. 6). Thus, in the absence of cigarette smoke exposure, CX3CL1 alone is insufficient to promote inflammatory gene expression in mononuclear phagocytes. Our findings also indicate intact LPS-induced inflammatory responses in cx3cr1GFP/GFP lung mononuclear phagocytes, arguing against the possibility of a generalized hypoinflammatory response as the mechanism for reduced TNF-α and IL-6 production after cigarette smoke exposure in vivo.
FIGURE 6.

A–F, Gene expression in interstitial lung mononuclear phagocytes obtained from cx3cr1+/+ and cx3cr1GFP/GFP mice. CD11c-enriched interstitial lung mononuclear phagocytes were harvested and pooled from cx3cr1+/+ (WT; n = 12) and cx3cr1GFP/GFP (n = 9) mice under steady-state conditions. Cells were stimulated in the presence or absence of increasing concentrations of recombinant CX3CL1 (0, 10, 100 nM) or LPS (100 ng/ml). Inducible NO synthase (iNOS), TNF-α, IL-6, IL-10, CXCL10, and MMP-12 gene expression was assayed by real-time PCR using the ΔΔCt method and 18S as the endogenous control. Unstimulated WT lung mononuclear phagocytes served as the calibrator. The y-axis represents gene fold-increase above the calibrator. Each condition was assayed in triplicate.
Absence of functional CX3CR1 protects against cigarette smoke-induced emphysema
Mice develop tissue-destructive emphysema with long-term cigarette smoke exposure, and experimental models have clearly shown the role of TNF-α–mediated inflammation in the development of emphysema (5, 8, 31). We have previously noted that chronic cigarette smoke exposure causes IL-6 and TNF-α gene upregulation, accumulation of CX3CR1+ mononuclear phagocytes in the lungs, a failure to thrive syndrome, and development of emphysema in AKR/J mice (9). To assess whether functional CX3CR1 is associated with long-term consequences of chronic cigarette smoke exposure, we determined quantitatively the amounts of tissue-destructive emphysema in cx3cr1GFP/GFP mice by measuring the degree of airspace enlargement after 6-mo exposure and comparing with that of cx3cr1+/+ or WT mice. As expected, WT mice showed increases in airspace enlargement (represented by increase in mean CL) with prolonged cigarette smoke exposure. In contrast, cx3cr1GFP/GFP mice were protected from developing airspace enlargement after chronic cigarette smoke exposure (Fig. 7A, 7B). We also measured the amount of tissue destruction by methods that have been previously described by others (16) and observed that cx3cr1GFP/GFP mice were relatively protected from developing cigarette smoke-induced tissue destruction whereas the lungs of WT mice were not (Fig. 7C). Thus, the absence of functional CX3CR1 required for full TNF-α responses is protective against the development of tissue-destructive emphysema.
FIGURE 7.
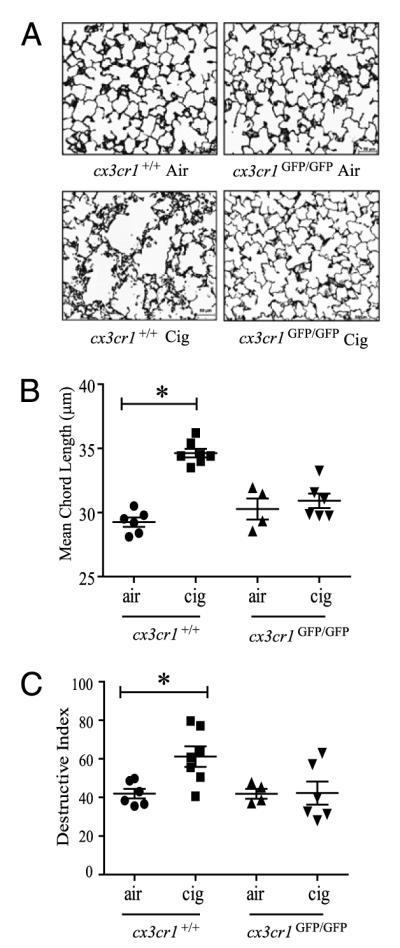
Effects of long-term cigarette smoke exposure on airspace enlargement in the lungs of cx3cr1GFP/GFP mice and cx3cr1+/+ WT control mice. A, Representative histologic sections of lungs from cx3cr1GFP/GFP and cx3cr1+/+ mice exposed to 24 wk of air versus cigarette smoke (Gill’s modified staining, original magnification ×200). B, Quantitative morphometric assessment of airspace enlargement in lung sections obtained from air-exposed and cigarette smoke-exposed cx3cr1+/+ and cx3cr1GFP/GFP mice (mean CL in μm). *p < 0.01 (ANOVA multiple comparisons). C, Destructive index of lung sections from air-exposed and cigarette smoke-exposed cx3cr1+/+ and cx3cr1GFP/GFP mice. Each point indicates individual mouse lungs studied expressed as mean ± SEM. *p < 0.05 (ANOVA).
Discussion
The main findings of our study are that cigarette smoke exposure amplifies a subset of CX3CR1+ mononuclear phagocytes spatially confined to the lung parenchyma that produces TNF-α and IL-6 and contributes to the development of tissue-destructive emphysema. Although cx3cr1GFP/GFP lung mononuclear phagocytes are fully capable of inducing inflammatory cytokine gene expression after exposure to LPS in vitro, these cells are unable to initiate robust TNF-α and IL-6 responses triggered by cigarette smoke exposure in vivo. This suggests that intact CX3CL1 signaling in vivo is important in providing a secondary signal that augments cigarette smoke-induced inflammation. Under basal conditions, increasing concentrations of recombinant CX3CL1 does not induce inflammatory gene expression in WT lung mononuclear phagocytes in vitro, suggesting that additional factors provided by cigarette smoke exposure in the lung microenvironment are required to promote inflammatory cytokine responses in a CX3CR1-dependent manner.
Although cigarette smoke induces the expression of transmembrane CX3CL1, CX3CL1 does not appear to be essential for the recruitment of mononuclear phagocytes into the airspaces. This is supported by the finding that alveolar macrophages express the transmembrane chemokine, and soluble chemokine concentrations are detectable within the airspaces, but CX3CR1-expressing cells fail to be recruited into the airspaces in significant numbers under homeostatic conditions or after chronic cigarette smoke exposure. Whereas an intact CX3CL1–CX3CR1 pathway did support a higher percentage of CX3CR1+CD11b+ mononuclear phagocytes accumulating in the lungs with cigarette smoke, the absence of functional CX3CR1 did not preclude their recruitment into the lungs, supporting the hypothesis that other factors are more prominent in mediating the initial event.
We previously reported that CX3CR1+ mononuclear phagocytes are primarily macrophages (9). In this study, CX3CR1+ interstitial lung mononuclear phagocytes expressing CD11b are not a uniform population and consist of a mixture of likely dendritic cells, macrophages, and monocytes in transition. Collectively, they are reminiscent of a previously described CX3CR1+ “resident” monocyte population with “patrolling behavior” that monitors blood vessels and tissues, initiates an early immune response by producing TNF-α, and differentiates into macrophages (10). Unlike the interstitial mononuclear phagocytes, very few, if any, resident alveolar macrophages express CX3CR1 under homeostatic conditions and after cigarette smoke exposure. Landsman et al. (11) previously showed that BAL macrophages (CD11c+ CD11b−, high autofluorescence) are CX3CR1 negative, but BAL dendritic cells (based on the expression of CD11c+CD11b+ markers, low autofluorescence) express CX3CR1. In this study, we observed minimal CX3CR1-expressing cells in the BAL fluid regardless of the presence of other surface markers.
Our prior studies also indicated that CX3CR1 localized to mononuclear cells of both the interstitium and alveolar spaces, and BAL cells showed increased CX3CR1 gene expression with cigarette smoking (9). This discrepancy with our current finding may be explained by the different approaches taken to address the question of CX3CR1 expression and localization in the lungs. Our prior study examined CX3CR1 expression using immunofluorescence staining of lung sections using a rabbit polyclonal Ab recognizing a CX3CR1 epitope. Alveolar macrophages we previously observed by immunofluorescence staining may actually represent a portion of the lung mononuclear phagocytes we observe in this study that are tightly tethered to the alveolar epithelium. The “tight tethering” exhibited by CX3CR1+ mononuclear phagocytes to the lung tissue allowed for their physical separation into the interstitial rather than alveolar compartment in this study. Moreover, the AKR/J strain used in our previous study may show differing proportions of macrophages and dendritic cells populating the airspaces than those in C57BL/6 mice under homeostatic conditions and in response to cigarette smoke.
Why cleaved CX3CL1 does not recruit CX3CR1+ mononuclear phagocytes into the airspaces despite detectable levels in the BAL fluid is unclear. CX3CR1+ mononuclear phagocytes may express a surface adhesion molecule that firmly tethers them to lung tissue preventing their migration (14). Alternatively, MMPs generated in or released into the airspaces after cigarette smoke exposure degrade chemokines into antagonistic fragments, and this may provide an additional mechanism of restraining CX3CR1+ mononuclear phagocyte movement from the interstitium. Indeed, CX3CL1 has previously been shown to be a substrate for MMP-2 (32), and ectodomain shedding of transmembrane CX3CL1 can be mediated by a disintegrin and metalloproteinase (ADAM) 10 (33), as well as by TNF-α converting enzyme (TACE; ADAM17) (24, 25). Whereas ADAM10 and ADAM17 are not known to be increased in cigarette smoke-induced emphysema, MMP-2 protein is increased in the lungs of mice after 6 mo of cigarette smoking (31), and MMP-2 activity has been previously reported to be increased in the BAL fluid of rats that develop emphysema with prolonged cigarette smoke (34). Moreover, human lungs with emphysema show higher levels of MMP-2 than those of nonemphysematous lungs (35). Thus, the lungs may provide a unique environment where the generation of antagonistic chemokine fragments by metalloproteinases under homeostatic conditions, amplified during chronic cigarette smoke exposure, contributes to the spatial confinement of CX3CR1+ mononuclear phagocytes. Indeed, we have previously shown that forced recruitment of CX3CR1+ cells into the airspaces is possible with intratracheal instillation of LPS, but this occurred in combination with high concentrations of exogenously administered CX3CL1 (9). We postulate that forced recruitment of CX3CR1+ mononuclear phagocytes occurred when concentrations of chemotactic ligand overwhelm endogenous metalloproteinase activity.
Our findings show that interstitial lung mononuclear phagocytes are one source of TNF-α and IL-6 in the lungs, and functional CX3CR1 is required for full cytokine responses to noxious stimuli such as cigarette smoke. Nonsmoking emphysematous patients exhibit increased circulating TNF-α and IL-6 concentrations compared with those of healthy never-smoker males, and these patients show a reduction in both cytokine levels after resection of inflammatory emphysematous tissue by lung volume reduction surgery (36). This finding suggests that TNF-α and IL-6 are key cytokines in the perpetuation of cigarette smoke-induced inflammation even after smoking cessation. Although direct evidence that IL-6 is involved in the pathogenesis of cigarette smoke-induced emphysema is lacking, others have previously shown that TNF-α drives the majority (~70%) of cigarette smoke-related emphysema in the mouse (31) and is the key mediator of acute smoke-induced inflammation with resulting connective tissue breakdown (8). TACE (i.e., ADAM17) or other MMPs is required for the proteolytic cleavage of the membrane-bound form of TNF-a to its active form (37). MMP-12 can also mediate TNF-α release from alveolar macrophages in response to cigarette smoke in vitro or from lungs acutely exposed to cigarette smoke (5). Thus, one mechanism by which MMP-12 can initiate and drive the progression toward emphysema is by increasing the bioactivity of TNF-α through proteolytic processing (5). It remains to be tested whether MMP-12 mediates the inflammatory phenotype of CX3CR1+ mononuclear phagocytes by increasing the bioactivity of its TNF-α release. In addition, further studies are required to test whether MMP-12 can proteolytically cleave and inactivate CX3CL1 to confine CX3CR1+ phagocytes spatially within the interstitium, as MMP-12 has been previously shown to generate ELR+ CXC chemokines in addition to CCL2, CCL7, CCL8, and CCL13 antagonists (38). Our current data suggest that other factors likely drive the initial recruitment of CX3CR1+ blood monocytes into the lungs in response to cigarette smoke but that once present within the lungs, CX3CR1+ mononuclear phagocytes bound to the interstitium promote a tissue-destructive phenotype given the phagocytes’ intimate proximity to the extracellular matrix.
Acknowledgments
We thank Mei Hulver for animal husbandry and John Cavaretta for lung morphometric analysis.
This work was supported in part by a Flight Attendants Medical Research Institute Clinical Innovator award, National Institutes of Health Grant HL086884, and a University of Pittsburgh Department of Medicine Junior Scholar award.
Abbreviations used in this article
- ADAM
a disintegrin and metalloproteinase
- BAL
bronchoalveolar lavage
- CL
chord length
- COPD
chronic obstructive pulmonary disease
- MHC II
MHC class II
- MMP
matrix metalloproteinase
- TACE
TNF-α converting enzyme
- WT
wild-type
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370:765–773. doi: 10.1016/S0140-6736(07)61380-4. [DOI] [PubMed] [Google Scholar]
- 2.Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R, van Weel C, Zielinski J, Global Initiative for Chronic Obstructive Lung Disease Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2007;176:532–555. doi: 10.1164/rccm.200703-456SO. [DOI] [PubMed] [Google Scholar]
- 3.Stockley RA, Mannino D, Barnes PJ. Burden and pathogenesis of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2009;6:524–526. doi: 10.1513/pats.200904-016DS. [DOI] [PubMed] [Google Scholar]
- 4.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 5.Churg A, Wang RD, Tai H, Wang X, Xie C, Dai J, Shapiro SD, Wright JL. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am. J. Respir. Crit. Care Med. 2003;167:1083–1089. doi: 10.1164/rccm.200212-1396OC. [DOI] [PubMed] [Google Scholar]
- 6.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. J. Clin. Invest. 2006;116:753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD. CD8+ T cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J. Immunol. 2007;178:8090–8096. doi: 10.4049/jimmunol.178.12.8090. [DOI] [PubMed] [Google Scholar]
- 8.Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am. J. Respir. Crit. Care Med. 2002;166:849–854. doi: 10.1164/rccm.200202-097OC. [DOI] [PubMed] [Google Scholar]
- 9.McComb JG, Ranganathan M, Liu XH, Pilewski JM, Ray P, Watkins SC, Choi AM, Lee JS. CX3CL1 up-regulation is associated with recruitment of CX3CR1+ mononuclear phagocytes and T lymphocytes in the lungs during cigarette smoke-induced emphysema. Am. J. Pathol. 2008;173:949–961. doi: 10.2353/ajpath.2008.071034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 11.Landsman L, Varol C, Jung S. Distinct differentiation potential of blood monocyte subsets in the lung. J. Immunol. 2007;178:2000–2007. doi: 10.4049/jimmunol.178.4.2000. [DOI] [PubMed] [Google Scholar]
- 12.Yona S, Jung S. Monocytes: subsets, origins, fates and functions. Curr. Opin. Hematol. 2010;17:53–59. doi: 10.1097/MOH.0b013e3283324f80. [DOI] [PubMed] [Google Scholar]
- 13.Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE. 2008;3:e3316. doi: 10.1371/journal.pone.0003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slebos DJ, Ryter SW, van der Toorn M, Liu F, Guo F, Baty CJ, Karlsson JM, Watkins SC, Kim HP, Wang X, et al. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am. J. Respir. Cell Mol. Biol. 2007;36:409–417. doi: 10.1165/rcmb.2006-0214OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am. J. Pathol. 2003;163:2329–2335. doi: 10.1016/S0002-9440(10)63589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saetta M, Shiner RJ, Angus GE, Kim WD, Wang NS, King M, Ghezzo H, Cosio MG. Destructive index: a measurement of lung parenchymal destruction in smokers. Am. Rev. Respir. Dis. 1985;131:764–769. doi: 10.1164/arrd.1985.131.5.764. [DOI] [PubMed] [Google Scholar]
- 17.Fong AM, Erickson HP, Zachariah JP, Poon S, Schamberg NJ, Imai T, Patel DD. Ultrastructure and function of the fractalkine mucin domain in CX(3)C chemokine domain presentation. J. Biol. Chem. 2000;275:3781–3786. doi: 10.1074/jbc.275.6.3781. [DOI] [PubMed] [Google Scholar]
- 18.Haskell CA, Cleary MD, Charo IF. Unique role of the chemokine domain of fractalkine in cell capture. Kinetics of receptor dissociation correlate with cell adhesion. J. Biol. Chem. 2000;275:34183–34189. doi: 10.1074/jbc.M005731200. [DOI] [PubMed] [Google Scholar]
- 19.Fong AM, Robinson LA, Steeber DA, Tedder TF, Yoshie O, Imai T, Patel DD. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J. Exp. Med. 1998;188:1413–1419. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 21.Papadopoulos EJ, Sassetti C, Saeki H, Yamada N, Kawamura T, Fitzhugh DJ, Saraf MA, Schall T, Blauvelt A, Rosen SD, Hwang ST. Fractalkine, a CX3C chemokine, is expressed by dendritic cells and is up-regulated upon dendritic cell maturation. Eur. J. Immunol. 1999;29:2551–2559. doi: 10.1002/(SICI)1521-4141(199908)29:08<2551::AID-IMMU2551>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 22.Lucas AD, Chadwick N, Warren BF, Jewell DP, Gordon S, Powrie F, Greaves DR. The transmembrane form of the CX3CL1 chemokine fractalkine is expressed predominantly by epithelial cells in vivo. Am. J. Pathol. 2001;158:855–866. doi: 10.1016/S0002-9440(10)64034-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsou CL, Haskell CA, Charo IF. Tumor necrosis factor-alpha-converting enzyme mediates the inducible cleavage of fractalkine. J. Biol. Chem. 2001;276:44622–44626. doi: 10.1074/jbc.M107327200. [DOI] [PubMed] [Google Scholar]
- 25.Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J. Biol. Chem. 2001;276:37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- 26.Shapiro SD. The macrophage in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;160:S29–S32. doi: 10.1164/ajrccm.160.supplement_1.9. [DOI] [PubMed] [Google Scholar]
- 27.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol. 2008;180:2562–2572. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- 28.von Garnier C, Filgueira L, Wikstrom M, Smith M, Thomas JA, Strickland DH, Holt PG, Stumbles PA. Anatomical location determines the distribution and function of dendritic cells and other APCs in the respiratory tract. J. Immunol. 2005;175:1609–1618. doi: 10.4049/jimmunol.175.3.1609. [DOI] [PubMed] [Google Scholar]
- 29.Landsman L, Jung S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J. Immunol. 2007;179:3488–3494. doi: 10.4049/jimmunol.179.6.3488. [DOI] [PubMed] [Google Scholar]
- 30.Arora M, Poe SL, Oriss TB, Krishnamoorthy N, Yarlagadda M, Wenzel SE, Billiar TR, Ray A, Ray P. TLR4/MyD88-induced CD11b(+)Gr-1(int)F4/80(+)non-migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol. 2010;3:578–593. doi: 10.1038/mi.2010.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am. J. Respir. Crit. Care Med. 2004;170:492–498. doi: 10.1164/rccm.200404-511OC. [DOI] [PubMed] [Google Scholar]
- 32.Dean RA, Overall CM. Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol. Cell. Proteomics. 2007;6:611–623. doi: 10.1074/mcp.M600341-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102:1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 34.March TH, Wilder JA, Esparza DC, Cossey PY, Blair LF, Herrera LK, McDonald JD, Campen MJ, Mauderly JL, Seagrave J. Modulators of cigarette smoke-induced pulmonary emphysema in A/J mice. Toxicol. Sci. 2006;92:545–559. doi: 10.1093/toxsci/kfl016. [DOI] [PubMed] [Google Scholar]
- 35.Ohnishi K, Takagi M, Kurokawa Y, Satomi S, Konttinen YT. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab. Invest. 1998;78:1077–1087. [PubMed] [Google Scholar]
- 36.Mineo D, Ambrogi V, Cufari ME, Gambardella S, Pignotti L, Pompeo E, Mineo TC. Variations of inflammatory mediators and alpha1-antitrypsin levels after lung volume reduction surgery for emphysema. Am. J. Respir. Crit. Care Med. 2010;181:806–814. doi: 10.1164/rccm.200910-1476OC. [DOI] [PubMed] [Google Scholar]
- 37.Gearing AJ, Beckett P, Christodoulou M, Churchill M, Clements J, Davidson AH, Drummond AH, Galloway WA, Gilbert R, Gordon JL, et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature. 1994;370:555–557. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- 38.Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, Overall CM. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood. 2008;112:3455–3464. doi: 10.1182/blood-2007-12-129080. [DOI] [PubMed] [Google Scholar]