

Summary
Viral infections are often detrimental to host survival and reproduction. Consequently, hosts have evolved a variety of mechanisms to defend themselves against viruses. A component of this arsenal is a set of proteins, termed restriction factors, which exhibit direct antiviral activity. Among these are several classes of proteins (APOBEC3, TRIM5, Tetherin and SAMHD1) that inhibit the replication of human and simian immunodeficiency viruses. Here, we outline the features, mechanisms, and evolution of these defense mechanisms. We also speculate on how restriction factors arose, how they might interact with the conventional innate and adaptive immune systems and how an understanding of these intrinsic cellular defenses might be usefully exploited.
Introduction
There are many examples in nature wherein antagonistic co-evolution results in rapid adaptation of genes, physiology and behavior. Classic examples include those involving relationships between parasites and hosts, or predators and prey. Predators possess characteristics such as strong jaws and sharp teeth; speed, camouflage and stealth for ambush; keen eyesight for hunting. In their turn, prey species adapt to sense, evade, and defend themselves against predators by acquiring, for example, keen odor detection, tremendous speeds, and specialized defense structures such as body shells, spines or horns. The evolution of these traits is driven by the reciprocal selective pressures that each group applies on the other. In a similar way, the invasion of hosts by viruses represents another example of an antagonistic evolutionary struggle. Because viral infections are often detrimental to host survival and reproduction, hosts have evolved a variety of mechanisms to sense, evade, and defend themselves against a variety of viral threats. A component of this arsenal is a set of proteins with direct antiviral activity. These can be thought of as comprising an autonomously functioning, `intrinsic' immune system (Bieniasz, 2004), or as a specialized component of the conventional innate immune system. These antiviral proteins, often termed `restriction factors', inhibit the replication of viruses, which then adapt, to evade and defend themselves against this form of host immunity. Thus, antagonistic conflict begets defense and counter defense measures, iteratively shaping viral and host functions and genomes.
Human and simian Immunodeficiency Viruses (HIVs and SIVs), have come to represent a model system in virology that has been instrumental in expanding our understanding of how viruses and hosts interact. In this review, we focus our attention on restriction factors that are known to inhibit the replication of this group of viruses, highlighting the features, mechanisms, and evolution of these defense systems. We also speculate on how these particular restriction factors arose, how they might interact with the conventional immune systems and influence the course of disease, and how an understanding of intrinsic cellular defenses might be usefully exploited.
General features of restriction factors
Restriction factors often possess certain properties that differentiate them from most other gene products (Malim and Bieniasz, 2012). Specifically, they (i) are dominantly and autonomously acting proteins that exhibit antiviral activity in simple cell-culture based assays, (ii) are often constitutively expressed in some cell types, but are sometimes upregulated by interferons, (iii) employ unique and unanticipated mechanisms to inhibit specific processes in viral replication, (iv) have unusually diverse amino acid sequences as a consequence of antagonistic co-evolution with viruses, and (v) are often (but not always) antagonized by viral accessory proteins. There are four currently known classes of restriction factors that target HIV-1 and other primate lentiviruses (Figure 1): the APOBEC3 proteins (Sheehy et al., 2002), TRIM5 proteins (Stremlau et al., 2004), Tetherin (Neil et al., 2008; Van Damme et al., 2008) and SAMHD1 (Hrecka et al., 2011; Lahouassa et al., 2012). Five classes of primate lentivirus proteins: Vif (Sheehy et al., 2002), Vpu (Neil et al., 2008; Van Damme et al., 2008), Vpx (Hrecka et al., 2011; Lahouassa et al., 2012), Nef (Jia et al., 2009; Zhang et al., 2009) and Env (Gupta et al., 2009b; Le Tortorec and Neil, 2009) have each evolved the ability to antagonize a specific antiviral protein (Figure 1).
Figure 1. Overview of restriction factors that target HIV and SIV and their viral antagonists.
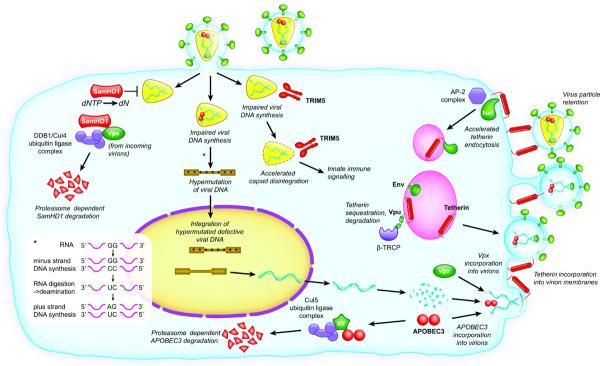
The key mechanisms by which restriction factors directly act upon the retroviral replication cycle, and their counteraction by viral accessory proteins are depicted. The process of APOBEC3-mediated hypermutation is indicated in the inset panel.
Restriction factors are generally autonomous inhibitors of viral replication
In general, antiretroviral restriction factors that have been identified thus far act as simple self-sufficient entities, rather than being components of complex pathways. Moreover, they act in a cell-autonomous fashion, i.e. their activity is evident in simple cell-culture based viral replication or infectivity assays. Thus, their existence was indicated by early studies that defined cell lines to be “restrictive” or “permissive”, depending on whether wild type or mutant viral strains could efficiently replicate therein (Malim and Bieniasz, 2012). Often, cell fusion experiments between restrictive and permissive cell lines demonstrated that heterokaryons exhibited the restrictive phenotype, suggesting a dominant mode of action for putative restriction factors (Malim and Bieniasz, 2012). In one case, otherwise permissive cells exhibited a restrictive phenotype after treatment with interferon (Neil et al., 2007). Sometimes, viruses lacking a particular accessory gene failed to complete a particular steps in the viral life cycle, specifically in restrictive cell lines, and it was posited that the accessory gene was required to antagonize an unknown inhibitor (Malim and Bieniasz, 2012). These concepts underlined the design of experiments that established the identification of several restriction factors. In each case, expression of a single gene could convert the phenotype of a cell line from permissive to restrictive (Sheehy et al., 2002; Stremlau et al., 2004; Neil et al., 2008; Van Damme et al., 2008; Lahouassa et al., 2012).
Restriction factors are constitutively expressed or induced by interferons
Adaptive immunity differs from innate (or intrinsic) immunity in various respects. One key difference is the rapidity with which restriction factors can exert their effects. T- and B-lymphocyte receptor diversity is generated via somatic recombination, meaning that only tiny numbers of cells with antiviral potential are present at the onset of a new infection. Effector cells are clonally expanded upon exposure to the pathogen, requiring several days to weeks to accumulate sufficient numbers to mount an effective adaptive immune response. In contrast, fully active forms of restriction factors are encoded in the germ line and are sometimes constitutively expressed. Indeed, prototype human antiretroviral restriction factors, i.e. APOBEC3G and TRIM5, were found to be constitutively expressed in cell types targeted by primate lentiviruses, leading to the notion of `intrinsic' immunity (Bieniasz, 2004), whereby cells, and perhaps whole organisms, are resistant to infection in the apparent absence of any signaling event. While it was subsequently shown that each of the known primate lentivirus restriction factors can be increased by interferon in some cell types (Neil and Bieniasz, 2009), IFN-induction is not required for profound antiretroviral activities to be evident in primate cells. Thus, although the distinction between intrinsic and innate immunity may not be as clear as originally envisaged (Bieniasz, 2004), there is a clear, perhaps biologically critical, distinction between intrinsic, pre-existing, immunity and interferon-dependent innate immunity.
Restriction factors have diverse mechanisms of action
In the case of adaptive immune responses and antiretroviral drugs that target viral proteins, resistance is most often acquired through one or a few amino acid substitutions that prevent drug or antibody binding, or prevent epitope recognition by, or presentation to, cytotoxic T-cells. Like antiretroviral drugs, restriction factors target specific steps in the HIV-1 life cycle, but unlike antiretroviral drugs, do so in a way that makes it difficult for HIV-1 to evolve resistance to the inhibitor through simple evasive mutations. Rather, the mechanisms employed by restriction factors often impose a requirement for the virus to gain new functions to counter the inhibitor. The mechanisms employed by restriction factors are diverse, elegant and unanticipated (Figure 1).
Hypermutation
The APOlipoprotein B Editing Catalytic subunit-like 3 family of proteins includes seven members in humans (APOBEC3A, B, C, DE, F, G, and H), each of which is characterized by the presence of cytidine deaminase (CDA) domains (Harris and Liddament, 2004). APOBEC3G (384 amino acids, 46.4 kDa, Figure 2A) is the prototype antiretroviral cytidine deaminase (Sheehy et al., 2002), but several members of this family are capable of inhibiting retroviral infection. In most cases, antiretroviral activity requires that the APOBEC3 protein is expressed in virus infected cells and incorporated into progeny virions (Sheehy et al., 2002). In the case of APOBEC3G, the viral nucleocapsid (NC) and associated viral or cellular RNAs that are incorporated into virions drive APOBEC3G packaging (Malim and Bieniasz, 2012). Upon virion entry into a new target cell, APOBEC3G acts during reverse transcription, primarily during the synthesis of the negative sense DNA strand (Yu et al., 2004). Specifically, cysteine residues in the conserved active site ((C/H)-X-E-X23~28-P-C-X-2~4-C) coordinate a Zn2+ ion allowing the catalytic glutamate to deaminate the C4 position of 2'-deoxycytidine producing 2'-deoxyuridine. Several structures of the CDA domain of APOBEC3G have been reported but there are discrepancies in the conformation of a terminal β-strand, β2, as well as the nature of the DNA binding site (Autore et al., 2010) (Figure 2B). APOBEC3G preferentially acts on the third cytosine of the sequence 5'-CCCA-3' (Yu et al., 2004) but deviations from this consensus are tolerated and a remarkably high fraction (up to 10% of nascent deoxycytidines) can be deaminated. This causes major disruption of the coding potential of the viral genome, generally rendering it replication defective.
Figure 2. Structure and antiretroviral activity of Restriction Factors.

A. Architecture of APOBEC3G. B. Ribbon representation of the C-terminal CDA domain of APOBEC3G (PDB entry 3IR2) consisting of a five-stranded β-sheet core surrounded by six α-helices. The β2-sheet (shown in green) is distorted to various degrees in all reported NMR (PDB entries 2JYW, 2KBO, and 2KEM) and X-ray structures (PDB entries 3IQS and 3IR2), due to the differential hydration of residues in each structure. The three flexible loops near the CDA catalytic site (shown in red) contribute to substrate binding. The residues coordinating the zinc atom (black sphere), either directly or via a water molecule (blue sphere), are shown as a stick representation. C. Architecture of TRIM5α. D. Left panel: NMR structure (PDB entry 2ECV; residues 1 to 78) of the RING domain of human TRIM5α. The putative E2 enzyme-binding domain is shown in brown. The residues coordinating the zinc atom (black sphere) are shown as a stick representation. Right panel: NMR structure (PDB entry 2YRG; residues 86 to 131) of the B-box domain of human TRIM5α. A hydrophobic cluster of residues (shown in pink) and Arg 119 (shown in green) in particular are critical for higher-order oligomerization. E. A proposed model of TRIM5α activity suggests that TRIM5α forms a complementary three-dimensional lattice around the incoming capsid. The RING domain (green circles), coiled-coil and BBOX domains (black lines) and the SPRY domain (pink rectangles) are indicated. F. Architecture of Tetherin. G. A model for the possible configurations adopted by tetherin dimers (PDB entry 2XG7) during virion tethering. Tetherin dimers might trap virions by the incorporation of one pair of anchors into the viral envelope (left and center panels). Alternatively, tethering might be achieved through the multimerization of tetherin molecules that are distributed between virion envelope and cell membrane (right panel). N and C represent the termini of tetherin. H. Architecture of SAMHD1. I. Ribbon representation of the HD domain of SAMHD1 (PDB entry 3U1N) with an expanded view of the active site. The residues coordinating the zinc atom (grey sphere), water molecule (blue sphere) and the phosphate ion are shown as a stick representation. For panels A, C, F, and H; domains and motifs critical for function are highlighted in color and numbers indicate the amino acid positions. Stars indicate catalytic site residues.
Aside from APOBEC3G, several other APOBEC3 proteins, including APOBEC3A, APOBEC3B, APOBEC3D, APOBEC3F and APOBEC3H, can significantly restrict primate lentivirus infection in in vitro assays (Bishop et al., 2004). While most work via broadly similar mechanisms to those employed by APOBEC3G, the other APOBEC3 proteins preferentially catalyze the deamination of cytidines in a different sequence context to APOBEC3G, and can have different domain organization with respect to determinants of virion incorporation and catalytic activity (e.g. APOBEC3H, has only one CDA domain). Notably, in addition to their hypermutation activity, APOBEC3G and APOBEC3F can inhibit the accumulation of reverse transcripts even when mutated to a catalytically inactive form (Newman et al., 2005), but the precise mechanism underpinning this activity is yet to be elucidated. In the case of APOBEC3A, a quite different role in the restriction of primate lentiviruses has been proposed, whereby APOBEC3A expressed in myeloid target cells acts on incoming viruses. The principal effect of APOBEC3A appears to be the reduction in the amount of viral DNA. Furthermore, SIVMAC Vpx reportedly counteracts APOBEC3A by inducing its degradation (Berger et al., 2011).
Viral capsid disruption
TRIM5α (497 amino acids, 57.3 kDa, Figure 2C) is a member of the TRIpartite Motif family of proteins that share a common architecture, but have distinct functions. TRIM5α targets the incoming viral capsid prior to reverse transcription (Stremlau et al., 2004), and a consequence of restriction is capsid disruption and/or degradation (Stremlau et al., 2006). TRIM proteins are characterized by an N-terminal domain comprised of RING, B-Box and coiled-coil domains. The RING domain of TRIM5α binds two zinc atoms tetrahedrally and possesses E3 ubiquitin ligase activity (Figure 2D, left panel). Whilst RING domain mutants have residual restriction activity, the RING domain greatly increases antiretroviral potency (Stremlau et al., 2004). B-Boxes are also zinc-binding domains that are unique to the TRIM family and appear to mediate the formation of higher-order multimers of TRIM5α (Diaz-Griffero et al., 2009). Notably, certain solvent-exposed B-box residues are critical for higher order multimerization (Figure 2D, right panel), and are also critical for antiviral activity. Absent the B-box domain, TRIM5 forms dimers, driven by the coiled-coil domain (Diaz-Griffero et al., 2009). Again, this intermolecular TRIM5 interaction is required for antiviral activity.
The C-terminal domain of TRIM5α is a SPRY domain, portions of which are highly variable among primate species. This domain contains the determinants that dictate the spectrum of retroviruses that are restricted by a particular TRIM5 variant and is almost certainly responsible for directly binding to incoming retroviral capsids. Indeed, in owl monkeys and macaques, LINE-mediated retrotransposition events have inserted cyclophilin A (CypA) cDNAs into the respective TRIM5 loci (Nisole et al., 2004; Sayah et al., 2004; Stoye and Yap, 2008). These insertions result in the expression of fusion proteins known as TRIMCyp, in which the C-terminal SPRY domain is replaced by a CypA domain. Several primate lentiviral capsids bind CypA, and those that do are generally restricted by TRIMCyp proteins. Notably, owl monkey and macaque TRIMCyp proteins arose and have evolved independently, with remodeling of the capsid binding site on CypA to alter restriction specificity (Price et al., 2009). It is striking that very similar, intuitively rare, retrotransposition events leading to the genesis of TRIMCyp proteins have occurred, and been fixed, at least twice in primate genomes within the past few million years.
Although it is reasonably clear that direct binding of TRIM5 protein to incoming capsids is required for restriction, subsequent events and the mechanistic basis of TRIM5α-mediated inhibition remain somewhat enigmatic (Sastri and Campbell, 2011). Restriction occurs within minutes of the entry of the viral capsid into the cytoplasm (Perez-Caballero et al., 2005), reverse transcription is blocked, and the biochemical behavior of the capsid protein on sucrose gradients is altered, consistent with the notion that TRIM5 induces the premature disruption of the capsid structure (Stremlau et al., 2006). However, both inhibition of reverse transcription, and capsid disruption can be blocked by the application of proteasome inhibitors, without affecting the antiviral activity of TRIM5α (Wu et al., 2006). This finding suggests that the reported biochemical effects are not central to the mechanism by which TRIM5 proteins inhibit infection. Intriguingly, a recombinant chimeric TRIM21-TRIM5α protein self-assembles in vitro into a hexagonal lattice that is complementary to that formed by the HIV-1 capsid protein (Figure 2E) (Ganser-Pornillos et al., 2011) and it is possible that this activity, in itself, underlies its inhibitory activity. However, more complex indirect models, involving TRIM5-dependent signaling (see below), and mobilization of as yet unidentified antiviral effectors have also been suggested to underlie the antiretroviral activity of TRIM5 (Pertel et al., 2011) (see discussion by Iwasaki in this issue).
Tethering nascent virions
Tetherin (180 amino acids, 19.7 kDa, Figure 2F; also known as CD317, BST-2 or HM1.24) is a type II transmembrane protein that also encodes a C-terminal glycosylphosphatidylinositol (GPI) anchor. Tetherin traps nascent but mature virions at the cell surface (Neil et al., 2008; Van Damme et al., 2008), from where they may be endocytosed. Unlike the other restriction factors, tetherin exhibits activity against enveloped viruses from several different viral families (Evans et al., 2010). Structurally, tetherin is comprised of a short cytoplasmic N-terminal domain and an extracellular ~170Å α-helical domain that is flanked by the two membrane anchors. Disulfide bonds between extracellular cysteines stabilize tetherin dimers in a parallel coiled-coil conformation but the N-terminal portion of the coiled coil appears quite flexible (Weissenhorn et al., 2012). Tetherin is mainly located in the trans-Golgi network (TGN) and at the cell surface, and appears to continuously shuttle between these locations.
An engineered protein, constructed from protein domains that have similar configuration, but almost no sequence homology to domains found in tetherin, restricts virion release in a similar manner to wild type tetherin (Perez-Caballero et al., 2009). This finding suggests that tetherin: (i) acts alone and does not require cellular co-factors, and, (ii) does not recognize a specific viral protein component. In fact, tetherin is concentrated at sites of virion budding and is incorporated into the lipid envelope of virions (Perez-Caballero et al., 2009; Fitzpatrick et al., 2010; Hammonds et al., 2010). Thus, tetherin likely traps virions simply by the partitioning of linked membrane anchors between virion and cell membranes. The specific orientation that the tetherin dimer adopts during virion tethering is not entirely clear, but both the GPI and transmembrane anchors are capable of driving tetherin incorporation into virions. Moreover, virions become tethered not only to the cell membrane, but also to each other, a scenario that is only compatible with the notion that both membrane anchors can be incorporated into virion membranes. There are a few configurations that could be adopted by tetherin during virion tethering (Figure 2G), and which, if any, of these predominates is yet to be fully resolved.
While tetherin retains viruses on infected cells and thereby inhibits transmission to distal cells, it may enhance direct cell-to-cell transmission by concentrating viral particles in the vicinity of adjacent, uninfected cells. Cell-to-cell transmission is putatively mediated by the formation of virological synapses, i.e. concentrations of viral particles at sites of direct contact between cells. Tetherin mediated enhancement of this mode of spread can be observed in vitro (Jolly et al., 2010). However, an in vivo model for retroviral infection has demonstrated that tetherin reduces viral burden and inhibits pathogenesis, suggesting that the net effect of tetherin in vivo is to inhibit retroviral replication (Liberatore and Bieniasz, 2011).
Depletion of deoxynucleotide triphosphates
Whereas the aforementioned restriction factors directly interact with viral components (such as genome, capsid, and lipid envelope), the Sterile Alpha Motif- and HD-domain containing protein 1 (SAMHD1) (626 amino acids, 72.2 kDa, Figure 2H) exerts an indirect antiretroviral effect (Hrecka et al., 2011; Laguette et al., 2011). Specifically, it reduces intracellular nucleotide concentrations, and thus inhibits reverse transcription (Lahouassa et al., 2012). SAMHD1 is composed of N-terminal SAM domain and a C-terminal HD domain. SAM domains typically mediate protein-protein interactions, occasionally forming higher order polymers, and can also possess specific RNA binding activity. HD domains contain conserved histidine and aspartate residues and are found in metalloenzymes bearing phosphohydrolytic activity. Indeed, SAMHD1 possesses an unusual phosphohydrolytic activity that underlies its ability to inhibit primate lentivirus infection. Specifically, it is a dGTP-activated triphosphohydrolase that hydrolyzes deoxynucleoside triphosphates (dNTPs) to deoxynucleosides and inorganic triphosphate (Goldstone et al., 2011; Powell et al., 2011). While all four dNTPs are substrates of SAMHD1, dGTP promotes its dimerization and acts as both activator and substrate for the enzyme. The SAMHD1 catalytic core (Figure 2I) fortuitously crystallizes with a phosphate ion from the crystallization buffer, marking the putative active site (Goldstone et al., 2011).
Although lentiviral reverse transcriptases (RTs) have evolved to replicate under lower concentrations of dNTPs than gammaretrovirus RTs, HIV-1 reverse transcribes with slower kinetics in myeloid cells than in activated T cells (Diamond et al., 2004). This finding is attributable to expression of SAMHD1 in myeloid cells, which deplete dNTPs to concentrations below the Michaelis constant (KM) of HIV-1 RT. Consistent with this notion, a HIV-1 bearing a mutant RT with lower affinity for dNTPs is particularly sensitive to SAMHD1-mediated restriction, and replicates in activated T cells, but not in myeloid cells (Diamond et al., 2004; Lahouassa et al., 2012). Moreover, SAMHD1 restriction is relieved by artificially elevating dNTPs to a concentration at which reverse transcription can efficiently occur.
Antagonism of restriction factors by viral accessory genes
Presumably because APOBEC3, tetherin and SAMHD1 target steps of the viral replication cycle that are virtually unalterable, and are hence especially difficult to escape via evasive mutations, these restriction factors have driven primate lentiviruses to acquire specialized countermeasures. In general, these countermeasures involve binding of the restriction factor by a viral protein, followed by removal of the restriction factor from the cell, or movement of the restriction factor to a subcellular location where it is ineffective. During antagonism, viral proteins often exploit the normal cellular degradation or transport machinery. For example, Vif proteins bind to a number of APOBEC3 proteins, and also recruit a cellular ubiquitin ligase complex composed of cullin5, elongins B and C, and Rbx2 (Yu et al., 2003). This results in polyubiquitination and subsequent degradation of APOBEC3 proteins, and effectively denudes cells of the antiviral protein. A similar mechanism is used by SIV Vpr and Vpx proteins to remove SAMHD1 from cells, although in this case, Vpr and Vpx are brought into a newly infected cell as a component of the incoming virion. Vpx appears to have originated from a duplication of the Vpr gene, and is only present in SIVSMM and SIVRCM lineages. At some point in the distant past, prior to gene duplication, some Vpr genes may have acquired the ability to degrade SAMHD1 (Lim et al., 2012). Thus, among modern primate lentiviruses, only some Vpr proteins, but all known Vpx proteins, have the ability to induce degradation of SAMHD1. They do so by recruiting a cullin4A-based ubiquitin ligase complex through a DDB1–DCAF1 adaptor (Le Rouzic et al., 2007; Schrofelbauer et al., 2007). Simultaneous Vpr or Vpx binding to SAMHD1 leads to SAMHD1 ubiquitination and degradation by proteasomes (Hrecka et al., 2011; Laguette et al., 2011).
Remarkably, no less than three different primate lentiviral proteins have acquired the ability to antagonize tetherin. HIV-1 Vpu is a small transmembrane protein that interacts, likely directly, with tetherin via their transmembrane domains (Iwabu et al., 2009; McNatt et al., 2009; Dube et al., 2010). Thereafter, multiple effects appear to contribute to Vpu's antagonist activity, including increased tetherin endocytosis, entrapment of de novo synthesized, and/or recycling tetherin to the Golgi and tetherin degradation. (Douglas et al., 2009; Goffinet et al., 2009; Iwabu et al., 2009; Dube et al., 2010; Skasko et al., 2011). Vpu recruits a β-TrCP-based ubiquitin ligase complex (Margottin et al., 1998) and induces ubiquitination of the tetherin cytoplasmic tail (Tokarev et al., 2011). This modification and engagement of the ESCRT pathway (Janvier et al., 2011; Kueck and Neil, 2012) likely underlies at least some of the aforementioned antagonistic effects of Vpu on tetherin.
Many simian immunodeficiency viruses do not encode a Vpu protein and in these instances, other viral proteins have assumed the role of tetherin antagonist. Nef is associated with the cytoplasmic face of cell membranes and regulates the levels of a variety of cellular surface proteins including CD4 (Kirchhoff et al., 2008). Some SIV Nef proteins have acquired the ability to antagonize tetherin (Jia et al., 2009; Zhang et al., 2009) using the same basic mechanism that they employ to dowenregulate CD4, namely recruitment of the AP-2 clathrin adaptor complex and accelerated tetherin endocytosis (Zhang et al., 2011).
HIV-2 also lacks Vpu and has adapted to use its Env protein to counteract tetherin. HIV-2 Env appears to interact with tetherin via their respective ectodomains, promoting tetherin internalization and/or sequestration (Le Tortorec and Neil, 2009). Interestingly, pathogenic revertants of the normally nonpathogenic SIVMAC (ΔNef) arise in rhesus macaques, in which SIVMAC Env has also adapted to antagonize tetherin (Serra-Moreno et al., 2011). Curiously, unlike HIV-2 Env, it appears that that SIVMAC Env cytoplasmic tail interacts with tetherin. These diverse ways in which primate lentiviruses have adapted to antagonize tetherin (i.e. employing three different proteins and four apparently distinct mechanisms) underscore their enormous plasticity and ability to innovate in response to selective pressures and, in turn, emphasize the role of tetherin in shaping the evolution of primate lentiviruses.
Restriction factors have evolved under positive selection
Infectious diseases can serve as potent agents of natural selection, and it is likely that antagonistic coevolution of interactions between viruses and their hosts represent a major driver of evolutionary change in both. Restriction factors variants that confer an advantage are selected by detrimental viral infections and can become widespread or even fixed in the host population. The resulting reduction in viral fitness in the newly adapted host provides the impetus for the selection of viral variants that have acquired mutations or new functions that relieve restriction and restore fitness. Iterative cycles of this genetic conflict constitute a molecular “arms race” and can result in the rapid evolution of restriction factors and their viral targets or antagonists (Figure 3A).
Figure 3. Evolution of restriction factor and accessory gene function.

A. Nef proteins of SIVs antagonize tetherin by interacting with the tetherin cytoplasmic tail. The diagram is a schematic representation of the genetic conflict between them. Colored figures indicate tetherin sequences in the cytoplasmic tail that are recognized by Nef and are hence rapidly evolving under positive selection. B. Cumulative frequency distribution of dN/dS ratios for 12,404 Human-Chimpanzee orthologous gene pairs. Adapted from previously computed data (Chimpanzee Sequencing and Analysis Consortium, 2005). Positive selection (dN/dS>1) and purifying selection (dN/dS<1) are indicated by purple and orange arrows respectively. The dN/dS value for each restriction factor is indicated by the dotted lines. The percentage of orthologous gene pairs with lower dN/dS ratios is indicated by the solid lines C. Evolution of Vpu and Nef function as primate lentiviruses were transmitted between species. See text for details.
A molecular arms race between protein-coding genes often means that mutations that change the amino acid sequence (non-synonymous mutations) are fixed more frequently than those that do not (synonymous mutations). Selection of beneficial alleles is termed diversifying or positive selection and contrasts with purifying or negative selection in that the latter is driven by the need to preserve protein function through the elimination of deleterious mutations. If multiple cycles of positive selection drive the evolution of a protein-coding gene, then non-synonymous mutations (dN) are observed more frequently than synonymous mutations (dS) in alignments of genes, portions of genes, or individual codons (dN/dS>1). As intuitively expected, the majority of human genes have evolved under purifying selection (dN/dS <1) but a subset have evolved under positive selection (Figure 3B) (Chimpanzee Sequencing and Analysis Consortium, 2005; Meyerson and Sawyer, 2011). Genes that exhibit signatures of positive selection include those involved in sensory perception, likely driven by temporal or migration-induced changes in the need to sense such things as environment, food, or predators. Predictably, positively selected genes also include those involved in immune responses and pathogen defense (Kosiol et al., 2008), including restriction factors. Notably, APOBEC3G and TRIM5α have among the highest dN/dS ratios of all human genes (Sawyer et al., 2004; Sawyer et al., 2005; Song et al., 2005) (Figure 3B).
Positive selection typically acts on domains or codons that participate in the interaction between proteins and their targets. For restriction factors, positive selection can be driven by the advantage conferred by increased binding to a viral target or decreased binding to a viral antagonist. For example, portions of TRIM5α that bear the signatures of positive selection are found only in the SPRY domain, which directly binds to the incoming retroviral capsid (Sawyer et al., 2005; Song et al., 2005). In the case of tetherin, the overall dN/dS ratio is quite low (Figure 3B), but portions of its coding sequences in the cytoplasmic tail and transmembrane domain that are targeted by viral antagonists, have evolved under positive selection (Gupta et al., 2009a; McNatt et al., 2009). Similarly, SAMHD1 sequences that are determinants for Vpx sensitivity also exhibit positive selection (Laguette et al., 2012; Lim et al., 2012).
Notably, the extent to which positive selection is observed in restriction factors could, in principle, be masked by biochemical constraints. For example, the transmembrane domain of tetherin is targeted by HIV-1 Vpu, which could drive positive selection, yet the need to maintain a hydrophobic helical character likely drives purifying selection of the same sequence. Moreover, the virus-driven appearance of positive selection requires a specific circumstance – namely recurrent challenges to a population of host variants by different viral species or strains. Thus, the mere presence or absence of signatures of positive selection in a given gene cannot, by itself, be construed as diagnostic of the presence or absence of a virus-host interaction. Nevertheless, each of the four classes of HIV and SIV restriction factors exhibit signatures of positive selection over at least a portion of their sequence, in at least some mammalian lineages.
Intrinsic cellular defenses are determinants of viral host range
Positive selection causes high interspecies protein sequence variability in restriction factors. Consequently, viral adaption to antagonize or evade a particular restriction factor variant in one host species can come at the cost of susceptibility to restriction factor variants in another potential host. Thus, antagonistic co-evolution of virus and a particular host can reduce the probability that an individual viral species can evade or antagonize the array of defense mechanisms that confront it when the opportunity to colonize a new host species arises.
Blocks to cross species transmission are imposed by APOBEC3G and TRIM5 proteins that appear particularly powerful, perhaps because (i) these two restriction factors are especially potent inhibitors, and (ii) they are constitutively expressed in the natural target cells of primate lentiviruses. HIV-1 and SIV Vif proteins are universally capable of antagonizing APOBEC3G proteins in their natural hosts, but are often impotent when confronted with APOBEC3G proteins from other primates (Mariani et al., 2003; Malim and Bieniasz, 2012). Indeed, the inability of many SIV Vif proteins to induce degradation of human APOBEC3G might explain why many SIVs have not been found in humans. Notably, SIVCPZ Vif and SIVSMM Vif are both active against human APOBEC3G (Gaddis et al., 2004), and both of these lineages have successfully colonized humans (as HIV-1 and HIV-2, respectively).
Although human TRIM5α is largely ineffective as an inhibitor of primate lentiviruses (Kratovac et al., 2008), TRIM5α variants found in monkeys can be very potent inhibitors, and those found in some species are capable of restricting an impressively diverse array of retroviruses (Bieniasz, 2004). Nevertheless, the fact that many monkey species harbor primate lentiviruses indicates that blocks imposed by TRIM5α can be overcome via adaptation of the capsid protein. Tetherin and SAMHD1 may also impede cross-species transmission of primate lentiviruses. Vpu and Nef proteins are generally only able to antagonize tetherin from a restricted range of species. For example, most HIV-1 Vpu proteins antagonize human tetherin but are inactive against monkey tetherin proteins (Gupta et al., 2009a; McNatt et al., 2009). Moreover, whereas SIV Nef proteins are often active against tetherin variants found in their natural host species, none are active against human tetherin, due to the deletion of five amino acids in the human tetherin cytoplasmic tail (Jia et al., 2009; Zhang et al., 2009). In fact, a rather interesting series of adaptations appears to have occurred as primate lentiviruses were transmitted from species to species en route to colonizing humans, as HIV-1 (Figure 3C) (Sauter et al., 2009). Specifically, SIVCPZ, the immediate ancestor of HIV-1, arose (in chimpanzees) through recombination between two SIV lineages found in monkeys upon which chimpanzee prey, one of which employs Vpu as a tetherin antagonist while the other employs Nef. Adaptation of the recombinant SIVCPZ in chimpanzees resulted in the selection of Nef as the tetherin antagonist and the loss of tetherin antagonist function by Vpu. However, upon transmission to humans, SIVCPZ encountered a tetherin in which the Nef target site was deleted and, presumably as a consequence, Vpu adapted to regain a lost function and antagonize human tetherin (Sauter et al., 2009). Provocatively, analysis of the Vpu proteins from HIV-1 groups M, N, and O reveals that only the Vpu protein of HIV-1 M strains have fully evolved to efficiently antagonize human tetherin, as well as retain its function of CD4 down regulation (Sauter et al., 2009). It is possible that this factor might have contributed to the relative success of this viral lineage in spreading through human populations. Species-specific SAMHD1 degrading activities are also evident among Vpr and Vpx proteins (Laguette et al., 2012; Lim et al., 2012) and some Vpx and Vpr appear to have specialized to deal with SAMHD1 variants that are present in the natural host species. However, SAMHD1 may not be an especially powerful barrier to cross species transmission, because HIV-1 apparently lacks the ability to antagonize human SAMHD1, yet has spread globally in human populations. Rather, the ability or otherwise of a particular primate lentivirus to antagonize SAMHD1 may determine the extent to which myeloid cell types are infected within a given host species.
Perspectives and questions about restriction factors
What are the origins of antiviral factors?
While the aforementioned restriction factors share some common properties, their mechanisms of action and evolutionary origins are utterly different from each other. How did such a diverse array of antiviral proteins arise? In principle, restriction factors could arise de novo as completely new genes, or through redirection of existing genes that have antiviral potential. In the case of tetherin, it is difficult (albeit not impossible) to imagine a precursor gene with a cellular function – there are no known cellular proteins that have related sequence, structure or function, and tetherin appears to be present only in mammals. Moreover, tetherin is not expressed in the vast majority of cells unless they are treated with interferon, and mice that lack a tetherin gene have no obvious deficiencies (Liberatore and Bieniasz, 2011). Thus, the absence of homologous genes and dispensability for viability are consistent with the notion that tetherin arose de novo, purely as an antiviral gene.
Alternatively, restriction factors may arise through relatively minor adaptations of existing cellular activities that have pre-existing potential to inhibit viral replication. The APOBEC3 family is likely derived from duplicated copies of cytidine deaminases (AID and APOBEC1) that have specific roles in editing cellular DNA and RNA. Thus, in this case, a normal cellular function was simply redirected to hypermutate viral genomes. In a similar manner, one might imagine that enzymatic regulation of cellular dNTP levels by SAMHD1 might have served some important regulatory function that was subsequently exploited by cells to inhibit the replication of retroviruses (and perhaps other DNA viruses). TRIM5 likely represents an intermediate example, whereby genes with some pre-existing, but mechanistically unrelated, function were mutated to a form with antiretroviral activity. Consistent with this idea, most of the dozens of TRIM proteins do not possess intrinsic antiretroviral activity, but two examples, TRIM1 and TRIM34, can exhibit weak antiretroviral activity when overexpressed (Yap et al., 2004; Zhang et al., 2006). Moreover, a variety of TRIM genes, that otherwise lack antiretroviral function, can be endowed with anti-HIV-1 activity if their C-terminal SPRY domains are replaced with cyclophilin (Yap et al., 2006; Zhang et al., 2006). These data suggest that the architecture of TRIM proteins, perhaps a propensity to assemble hexameric lattices (Ganser-Pornillos et al., 2011), lends itself to the acquisition of antiretroviral activity.
Are there more restriction factors to be discovered?
A number of lines of evidence hint at the existence of undiscovered antiretroviral factors that target primate lentiviruses. First, certain cell types and cell lines are resistant to lentiviral infection, depending on their state of differentiation and/or interferon induction (Goujon and Malim, 2009), and it is entirely plausible, even likely, that this resistance is due to the expression of unknown restriction factors. Second, it is well known that Vpr has the ability to induce the degradation of proteins to which it binds (Schrofelbauer et al., 2005). While the role of some SIV Vpr proteins appears to be the removal of SAMHD1 from cells (Lim et al., 2012), many other Vpr proteins lack the ability to induce SAMHD1 degradation, suggesting that another target for Vpr induced degradation exists. Vpr is dispensible for HIV-1 replication in cell culture and SIVMAC replication in vivo (Gibbs et al., 1995), but humans and chimpanzees infected with HIV-1 strains encoding defective Vpr proteins develop revertant mutants (Goh et al., 1998). Thus, Vpr may degrade a restriction factor that is only encountered in vivo, and has only a modest effect on the overall level of viral replication. Finally, the poorly understood ability of Nef to increase the intrinsic infectiousness of primate lentivirus particles (Chowers et al., 1994; Miller et al., 1994) could well be due to antagonism of a restriction factor, particularly given the known propensity of Nef to down regulate various cell-surface proteins. Large scale RNAi and cDNA expression screens (Brass et al., 2009; Schoggins et al., 2011) are beginning to uncover the range of proteins that inhibit the replication of a variety of viruses and it seems likely that the identification of additional proteins with antiretroviral activity will be forthcoming.
How did viral infections in the distant past shape the array of modern restriction factors?
Although restriction factors are most frequently studied because of their ability to inhibit modern, clinically important retroviruses, their existence in modern genomes is the result of selection pressures imposed by viruses in the distant past. Most retroviral lineages are long extinct, but some ancient retroviruses are `fossilized' in modern genomes, through their ability to infect germline cells and become inherited in a Mendelian manner. Although the viral fossil record likely represents only a small fraction of the retroviruses that have ever existed, the increasing availability of genome sequences, and convenience of gene synthesis has made it possible to reconstitute ancient retroviral proteins, or even complete retroviruses in functional form (Dewannieux et al., 2006; Lee and Bieniasz, 2007). These advances have enabled attempts to reconstruct evolutionary history and illuminate how restriction factors evolved into their modern forms. Although the incompleteness of the retroviral fossil record urges circumspection in the interpretation of such studies, it has been possible to suggest scenarios by which ancient hosts and viruses interacted.
For example, reconstitution of ancient lentiviral capsid proteins from endogenous sequences in lagomorphs and prosimians indicates that the ability of retroviral capsids to bind CypA was acquired millions of years ago (Goldstone et al., 2010). Indeed, it is plausible that CypA binding was widespread, perhaps the norm among ancient lentiviruses. Given the apparent absence of CypA binding in any other retroviral lineage, ancient CypA-binding lentiviruses might have been responsible for fixation of TRIMCyp encoding genes in owl monkeys and macaques, even though lentiviruses are not known to be present in these two modern species.
Other examples of ancient host-virus interactions involve APOBEC3 proteins where hypermutation can be readily observed in fossilized proviruses. Specifically, APOBEC3G was clearly responsible for the hypermutation and inactivation of two HERV-K proviruses in the human genome (Lee et al., 2008). Furthermore there are many gammaretrovirus genomes in chimpanzee, macaque and murine genomes that were inactivated by APOBEC3G and other APOBEC3 proteins (Jern et al., 2007; Perez-Caballero et al., 2008).
Although these examples illustrate how it has been possible to infer interactions between hosts and viruses, it has not yet been possible to unequivocally demonstrate that specific past retroviral infections were responsible for the acquisition, or evolution of any particular restriction factor. Nor has it been possible to demonstrate that any particular restriction factor was responsible for extinction of any retrovirus. The sparse nature of the viral fossil record may make such definitive findings impossible to achieve. However, this is an active area of research, and it is even possible that reconstitution and study of ancient retroviruses might uncover novel restriction factors to which modern retroviruses have become resistant.
How might restriction factors interact with other immune functions?
Because several restriction factors interact with and alter the fate of virions and virion components, they have the potential to modify innate and adaptive immune responses. Additionally, they are, in principle, positioned to recognize pathogen-specific molecular patterns and perhaps directly participate in triggering innate immune signaling pathways. Alternatively, the action of restriction factors could alter the spectrum of cells that become infected (e.g specific protection of myeloid cells by SAMHD1), and in so doing alter the way that viral components are presented to the innate and adaptive immune systems, as discussed in an accompanying review (Iwasaki, 2012, this issue).
Moreover, in addition to directly inhibiting viral infection, TRIM5α appears to trigger signaling pathways. Indeed, the UBC13-UEV1A E2 ubiquitin-conjugating enzyme interacts with human TRIM5α to generate the formation of K63-linked ubiquitin chains that are unattached to substrates (Pertel et al., 2011). These ubiquitin chains can activate the AP-1 and NF-κB signaling pathways, potentially leading to cytokine production and modulation of innate and adaptive immune responses as discussed in an accompanying review (Iwasaki, 2012, this issue). Whether other restriction factors, such as tetherin, that directly interact with virions, can initiate signaling cascades, is under investigation.
Tetherin might affect immune responses because of the substantial effect it has on the fate of virions, concentrating them on the surface of infected cells, from where a significant fraction can be endocytosed (Neil et al., 2008; Van Damme et al., 2008). Depending on which cell type is infected, this phenomenon could well affect how virion proteins are presented to the immune system on MHC-II molecules. Alternatively, internalization could increase the exposure of virion components to endosomal Toll-like receptors (TLRs), e.g. TLR7, which is known to respond to endocytosed HIV-1 particles (Beignon et al., 2005). Conversely, by inhibiting infection of myeloid cells, dendritic cells in particular, SAMHD1-mediated restriction might reduce the exposure of HIV-1 to TLRs as well as yet undiscovered cytoplasmic sensors in these cells (Manel et al., 2010) as discussed in an accompanying review (Iwasaki, 2012, this issue). Again, this might affect how innate and adaptive immune systems are mobilized, with the potential for both positive and negative effects on host and virus.
Does variation in restriction factors contribute to AIDS susceptibility in humans?
There is apparently wide variation among humans in the sensitivity to, and the outcomes of, viral infection. While at least some of the variation in HIV/AIDS susceptibility in humans can be attributed to CCR5 and MHC polymorphisms, variation in these genes does not entirely account for variation in AIDS susceptibility, and it is also possible that genetic variability in restriction factor loci might also contribute. Sub-populations of humans encode inactivating or destabilizing lesions in genes encoding TRIM5α (Torimiro et al., 2009), APOBEC3B (Kidd et al., 2007), and APOBEC3H (OhAinle et al., 2008; Harari et al., 2009). There is also variation among APOBEC3H haplotypes in terms of their sensitivity to antagonism by Vif. Additionally, more subtle polymorphic mutations are found in TRIM5α (H43Y) that decrease activity in in vitro assays (Sawyer et al., 2006), as well as in APOBEC3G (H186R) (An et al., 2004).
Unfortunately, attempts to link the restriction factor polymorphisms with altered HIV/AIDS susceptibility in humans have yielded contradictory results. This is true for an APOBEC3B deletion (An et al., 2009; Itaya et al., 2010), the TRIM5α (H43Y) mutation(van Manen et al., 2008; Liu et al., 2011) and the APOBEC3G (H186R) mutation (Bizinoto et al.; An et al., 2004; Do et al., 2005). In each case, studies showing polymorphism-associated alteration in HIV-1 infection rate or disease progression have been contradicted by studies showing no effect, or even opposing effects. It is unclear whether the lack of concordance in these studies is due to methodological problems, or genuine race-based modifying effects. Conversely, in rhesus macaques, wherein the variation in the sequence and activity of restriction factors appears more extensive, there is clear evidence that TRIM5α polymorphisms can affect the course of SIV infection. Specifically, a group of TRIM5 alleles (designated TRIM5TFP), and in some cases TRIMCyp, appear to suppress SIV replication in vivo while others (designated TRIM5Q) are essentially inactive (Kirmaier et al., 2010; Lim et al., 2010). In some cases, TRIM5 variants select changes in SIV capsid sequences conferring resistance to TRIM5 restriction in vivo (Kirmaier et al., 2010).
Can knowledge of restriction factors aid the development of better animal models of AIDS?
The formidable barriers to zoonotic transmission imposed by restriction factors have very likely protected humans from potentially lethal viral pandemics that original in nonhuman animals. Reciprocally, HIV-1 cannot replicate in most nonhuman animals, including nonhuman primates (NHP). Because NHP models of HIV-1 infection could serve as better platforms to evaluate drug and vaccine candidates prior to human clinical trials an appreciation of how variation in restriction factors limits cross species transmission, has guided the construction of simian-tropic HIV-1 (stHIV) strains that encode SIVMAC capsid and Vif sequences (to evade and antagonize macaque TRIM5α and APOBEC3 proteins). These stHIV-1 strains replicate well in macaque cells and led to the conclusion that these two restriction factors constitute major barriers to cross-species primate lentivirus transmission (Hatziioannou et al., 2006). Fortuitously, pigtailed macaques encode a TRIMCyp protein that cannot restrict HIV-1 (Virgen et al., 2008). Consequently, stHIV-1 stains in which the only alteration is to substitute SIVMAC Vif sequences can successfully mount a spreading infection in pigtailed macaques (Hatziioannou et al., 2009). Although stHIV-1 infected pigtailed macaques do not yet develop disease, there is clear potential for the development of improved models of human AIDS based on engineered resistance to restriction factors.
Can an understanding of restriction factors be exploited therapeutically?
A possible consequence of the increased understanding of restriction factors is the prospect that they might be mobilized, or otherwise exploited, in the context of new therapies. For example, inhibiting the antagonistic effect of accessory proteins might usefully mobilize the activity of antiviral restriction factors. Although such work is clearly in its infancy, small molecule inhibitors of Vif-mediated APOBEC3G degradation have been identified that inhibit HIV-1 replication in vitro (Nathans et al., 2008; Cen et al., 2010). In principle, conceptually similar approaches could be used to inhibit Vpu-tetherin interactions and mobilize the latter's antiviral activity. An alternative approach is to mimic or potentiate the effects of restriction factors. For instance, a small molecule inhibitor of HIV-1 (PF74) that seems to promote premature viral uncoating mimics the action of TRIM5α (Shi et al., 2011). Finally, artificially engineered restriction factors to which HIV-1 is sensitive, could be considered for use in gene therapy approaches. As a proof of principle, engineered TRIMCyp or tetherin proteins assembled from human components (Neagu et al., 2009; Perez-Caballero et al., 2009) have been demonstrated to have activity against HIV-1 in vitro.
In conclusion, the identification of restriction factors that inhibit primate lentiviruses and the elucidation of the means by which they act, evolve, and affect the biology of hosts and viruses has been a rich scientific endeavor. Although much remains to be learned, there is clear, albeit yet to be realized, potential for restriction factors to be exploited for practical benefit.
ACKNOWLEDGEMENTS
We apologize to those investigators whose work was not cited due to space constraints. We are grateful to V. Chandrasekaran and W. Sundquist for providing the high resolution image of HIV-1 capsid. Work in our laboratory on restriction factors is supported by the NIH (grants R01AI050111 and R37AI0664003). P.D.B. is an Investigator of the Howard Hughes Medical Institute.
REFERENCES
- An P, Bleiber G, Duggal P, Nelson G, May M, Mangeat B, Alobwede I, Trono D, Vlahov D, Donfield S, et al. APOBEC3G genetic variants and their influence on the progression to AIDS. J Virol. 2004;78:11070–11076. doi: 10.1128/JVI.78.20.11070-11076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An P, Johnson R, Phair J, Kirk GD, Yu XF, Donfield S, Buchbinder S, Goedert JJ, Winkler CA. APOBEC3B deletion and risk of HIV-1 acquisition. J Infect Dis. 2009;200:1054–1058. doi: 10.1086/605644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autore F, Bergeron JR, Malim MH, Fraternali F, Huthoff H. Rationalisation of the differences between APOBEC3G structures from crystallography and NMR studies by molecular dynamics simulations. PLoS One. 2010;5:e11515. doi: 10.1371/journal.pone.0011515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, Larsson M, Gorelick RJ, Lifson JD, Bhardwaj N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115:3265–3275. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger G, Durand S, Fargier G, Nguyen XN, Cordeil S, Bouaziz S, Muriaux D, Darlix JL, Cimarelli A. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog. 2011;7:e1002221. doi: 10.1371/journal.ppat.1002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz PD. Intrinsic immunity: a front-line defense against viral attack. Nat Immunol. 2004;5:1109–1115. doi: 10.1038/ni1125. [DOI] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Bizinoto MC, Leal E, Diaz RS, Janini LM. Loci polymorphisms of the APOBEC3G gene in HIV type 1-infected Brazilians. AIDS Res Hum Retroviruses. 27:137–141. doi: 10.1089/aid.2010.0146. [DOI] [PubMed] [Google Scholar]
- Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen S, Peng ZG, Li XY, Li ZR, Ma J, Wang YM, Fan B, You XF, Wang YP, Liu F, et al. Small molecular compounds inhibit HIV-1 replication through specifically stabilizing APOBEC3G. J Biol Chem. 2010;285:16546–16552. doi: 10.1074/jbc.M109.085308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowers MY, Spina CA, Kwoh TJ, Fitch NJ, Richman DD, Guatelli JC. Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J Virol. 1994;68:2906–2914. doi: 10.1128/jvi.68.5.2906-2914.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Dewannieux M, Harper F, Richaud A, Letzelter C, Ribet D, Pierron G, Heidmann T. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 2006;16:1548–1556. doi: 10.1101/gr.5565706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, Kim B. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem. 2004;279:51545–51553. doi: 10.1074/jbc.M408573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Griffero F, Qin XR, Hayashi F, Kigawa T, Finzi A, Sarnak Z, Lienlaf M, Yokoyama S, Sodroski J. A B-box 2 surface patch important for TRIM5alpha self-association, capsid binding avidity, and retrovirus restriction. J Virol. 2009;83:10737–10751. doi: 10.1128/JVI.01307-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H, Vasilescu A, Diop G, Hirtzig T, Heath SC, Coulonges C, Rappaport J, Therwath A, Lathrop M, Matsuda F, Zagury JF. Exhaustive genotyping of the CEM15 (APOBEC3G) gene and absence of association with AIDS progression in a French cohort. J Infect Dis. 2005;191:159–163. doi: 10.1086/426826. [DOI] [PubMed] [Google Scholar]
- Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Fruh K, Moses AV. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J Virol. 2009;83:7931–7947. doi: 10.1128/JVI.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube M, Roy BB, Guiot-Guillain P, Binette J, Mercier J, Chiasson A, Cohen EA. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010;6:e1000856. doi: 10.1371/journal.ppat.1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DT, Serra-Moreno R, Singh RK, Guatelli JC. BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol. 2010;18:388–396. doi: 10.1016/j.tim.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick K, Skasko M, Deerinck TJ, Crum J, Ellisman MH, Guatelli J. Direct restriction of virus release and incorporation of the interferon-induced protein BST-2 into HIV-1 particles. PLoS Pathog. 2010;6:e1000701. doi: 10.1371/journal.ppat.1000701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaddis NC, Sheehy AM, Ahmad KM, Swanson CM, Bishop KN, Beer BE, Marx PA, Gao F, Bibollet-Ruche F, Hahn BH, Malim MH. Further investigation of simian immunodeficiency virus Vif function in human cells. J Virol. 2004;78:12041–12046. doi: 10.1128/JVI.78.21.12041-12046.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, Yeager M. Hexagonal assembly of a restricting TRIM5alpha protein. Proc Natl Acad Sci U S A. 2011;108:534–539. doi: 10.1073/pnas.1013426108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JS, Lackner AA, Lang SM, Simon MA, Sehgal PK, Daniel MD, Desrosiers RC. Progression to AIDS in the absence of a gene for vpr or vpx. J Virol. 1995;69:2378–2383. doi: 10.1128/jvi.69.4.2378-2383.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffinet C, Allespach I, Homann S, Tervo HM, Habermann A, Rupp D, Oberbremer L, Kern C, Tibroni N, Welsch S, et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe. 2009;5:285–297. doi: 10.1016/j.chom.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Goh WC, Rogel ME, Kinsey CM, Michael SF, Fultz PN, Nowak MA, Hahn BH, Emerman M. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: a mechanism for selection of Vpr in vivo. Nat Med. 1998;4:65–71. doi: 10.1038/nm0198-065. [DOI] [PubMed] [Google Scholar]
- Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- Goldstone DC, Yap MW, Robertson LE, Haire LF, Taylor WR, Katzourakis A, Stoye JP, Taylor IA. Structural and functional analysis of prehistoric lentiviruses uncovers an ancient molecular interface. Cell Host Microbe. 2010;8:248–259. doi: 10.1016/j.chom.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Goujon C, Malim MH. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J Virol. 2009;84:9254–9266. doi: 10.1128/JVI.00854-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Hue S, Schaller T, Verschoor E, Pillay D, Towers GJ. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 2009a;5:e1000443. doi: 10.1371/journal.ppat.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Mlcochova P, Pelchen-Matthews A, Petit SJ, Mattiuzzo G, Pillay D, Takeuchi Y, Marsh M, Towers GJ. Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST-2/CD317 by intracellular sequestration. Proc Natl Acad Sci U S A. 2009b;106:20889–20894. doi: 10.1073/pnas.0907075106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammonds J, Wang JJ, Yi H, Spearman P. Immunoelectron microscopic evidence for Tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog. 2010;6:e1000749. doi: 10.1371/journal.ppat.1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari A, Ooms M, Mulder LC, Simon V. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J Virol. 2009;83:295–303. doi: 10.1128/JVI.01665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- Hatziioannou T, Ambrose Z, Chung NP, Piatak M, Jr., Yuan F, Trubey CM, Coalter V, Kiser R, Schneider D, Smedley J, et al. A macaque model of HIV-1 infection. Proc Natl Acad Sci U S A. 2009;106:4425–4429. doi: 10.1073/pnas.0812587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatziioannou T, Princiotta M, Piatak M, Jr., Yuan F, Zhang F, Lifson JD, Bieniasz PD. Generation of simian-tropic HIV-1 by restriction factor evasion. Science. 2006;314:95. doi: 10.1126/science.1130994. [DOI] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itaya S, Nakajima T, Kaur G, Terunuma H, Ohtani H, Mehra N, Kimura A. No evidence of an association between the APOBEC3B deletion polymorphism and susceptibility to HIV infection and AIDS in Japanese and Indian populations. J Infect Dis. 2010;202:815–816. doi: 10.1086/655227. author reply 816–817. [DOI] [PubMed] [Google Scholar]
- Iwabu Y, Fujita H, Kinomoto M, Kaneko K, Ishizaka Y, Tanaka Y, Sata T, Tokunaga K. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J Biol Chem. 2009;284:35060–35072. doi: 10.1074/jbc.M109.058305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janvier K, Pelchen-Matthews A, Renaud JB, Caillet M, Marsh M, Berlioz-Torrent C. The ESCRT-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 2011;7:e1001265. doi: 10.1371/journal.ppat.1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jern P, Stoye JP, Coffin JM. Role of APOBEC3 in genetic diversity among endogenous murine leukemia viruses. PLoS Genet. 2007;3:2014–2022. doi: 10.1371/journal.pgen.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, Johnson WE, Westmoreland S, Evans DT. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009;5:e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly C, Booth NJ, Neil SJ. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J Virol. 2010;84:12185–12199. doi: 10.1128/JVI.01447-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 2007;3:e63. doi: 10.1371/journal.pgen.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff F, Schindler M, Specht A, Arhel N, Munch J. Role of Nef in primate lentiviral immunopathogenesis. Cell Mol Life Sci. 2008;65:2621–2636. doi: 10.1007/s00018-008-8094-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O'Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, et al. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosiol C, Vinar T, da Fonseca RR, Hubisz MJ, Bustamante CD, Nielsen R, Siepel A. Patterns of positive selection in six Mammalian genomes. PLoS Genet. 2008;4:e1000144. doi: 10.1371/journal.pgen.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratovac Z, Virgen CA, Bibollet-Ruche F, Hahn BH, Bieniasz PD, Hatziioannou T. Primate lentivirus capsid sensitivity to TRIM5 proteins. J Virol. 2008;82:6772–6777. doi: 10.1128/JVI.00410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueck T, Neil SJ. A cytoplasmic tail determinant in HIV-1 Vpu mediates targeting of tetherin for endosomal degradation and counteracts interferon-induced restriction. PLoS Pathog. 2012;8:e1002609. doi: 10.1371/journal.ppat.1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette N, Rahm N, Sobhian B, Chable-Bessia C, Munch J, Snoeck J, Sauter D, Switzer WM, Heneine W, Kirchhoff F, et al. Evolutionary and functional analyses of the interaction between the myeloid restriction factor SAMHD1 and the lentiviral Vpx protein. Cell Host Microbe. 2012;11:205–217. doi: 10.1016/j.chom.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. 2012;13:223–228. doi: 10.1038/ni.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rouzic E, Belaidouni N, Estrabaud E, Morel M, Rain JC, Transy C, Margottin-Goguet F. HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle. 2007;6:182–188. doi: 10.4161/cc.6.2.3732. [DOI] [PubMed] [Google Scholar]
- Le Tortorec A, Neil SJ. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J Virol. 2009;83:11966–11978. doi: 10.1128/JVI.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YN, Bieniasz PD. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 2007;3:e10. doi: 10.1371/journal.ppat.0030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YN, Malim MH, Bieniasz PD. Hypermutation of an ancient human retrovirus by APOBEC3G. J Virol. 2008;82:8762–8770. doi: 10.1128/JVI.00751-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberatore RA, Bieniasz PD. Tetherin is a key effector of the antiretroviral activity of type I interferon in vitro and in vivo. Proc Natl Acad Sci U S A. 2011;108:18097–18101. doi: 10.1073/pnas.1113694108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ES, Fregoso OI, McCoy CO, Matsen FA, Malik HS, Emerman M. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe. 2012;11:194–204. doi: 10.1016/j.chom.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SY, Rogers T, Chan T, Whitney JB, Kim J, Sodroski J, Letvin NL. TRIM5alpha Modulates Immunodeficiency Virus Control in Rhesus Monkeys. PLoS Pathog. 2010;6:e1000738. doi: 10.1371/journal.ppat.1000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu FL, Qiu YQ, Li H, Kuang YQ, Tang X, Cao G, Tang NL, Zheng YT. An HIV-1 resistance polymorphism in TRIM5alpha gene among Chinese intravenous drug users. J Acquir Immune Defic Syndr. 2011;56:306–311. doi: 10.1097/QAI.0b013e318205a59b. [DOI] [PubMed] [Google Scholar]
- Malim MH, Bieniasz PD. Cold Spring Harbor Perspectives in Medicine. 2012. HIV Restriction Factors and Mechanisms of Evasion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, Littman DR. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature. 2010;467:214–217. doi: 10.1038/nature09337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998;1:565–574. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- McNatt MW, Zang T, Hatziioannou T, Bartlett M, Fofana IB, Johnson WE, Neil SJ, Bieniasz PD. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009;5:e1000300. doi: 10.1371/journal.ppat.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MD, Warmerdam MT, Gaston I, Greene WC, Feinberg MB. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J Exp Med. 1994;179:101–113. doi: 10.1084/jem.179.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathans R, Cao H, Sharova N, Ali A, Sharkey M, Stranska R, Stevenson M, Rana TM. Small-molecule inhibition of HIV-1 Vif. Nat Biotechnol. 2008;26:1187–1192. doi: 10.1038/nbt.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neagu MR, Ziegler P, Pertel T, Strambio-De-Castillia C, Grutter C, Martinetti G, Mazzucchelli L, Grutter M, Manz MG, Luban J. Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J Clin Invest. 2009;119:3035–3047. doi: 10.1172/JCI39354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil S, Bieniasz P. Human immunodeficiency virus, restriction factors, and interferon. J Interferon Cytokine Res. 2009;29:569–580. doi: 10.1089/jir.2009.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil SJ, Sandrin V, Sundquist WI, Bieniasz PD. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe. 2007;2:193–203. doi: 10.1016/j.chom.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15:166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- Nisole S, Lynch C, Stoye JP, Yap MW. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci U S A. 2004;101:13324–13328. doi: 10.1073/pnas.0404640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe. 2008;4:249–259. doi: 10.1016/j.chom.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Caballero D, Hatziioannou T, Zhang F, Cowan S, Bieniasz PD. Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J Virol. 2005;79:15567–15572. doi: 10.1128/JVI.79.24.15567-15572.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Caballero D, Soll SJ, Bieniasz PD. Evidence for restriction of ancient primate gammaretroviruses by APOBEC3 but not TRIM5alpha proteins. PLoS Pathog. 2008;4:e1000181. doi: 10.1371/journal.ppat.1000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009;139:499–511. doi: 10.1016/j.cell.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 2011;472:361–365. doi: 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell RD, Holland PJ, Hollis T, Perrino FW. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J Biol Chem. 2011;286:43596–43600. doi: 10.1074/jbc.C111.317628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AJ, Marzetta F, Lammers M, Ylinen LM, Schaller T, Wilson SJ, Towers GJ, James LC. Active site remodeling switches HIV specificity of antiretroviral TRIMCyp. Nat Struct Mol Biol. 2009;16:1036–1042. doi: 10.1038/nsmb.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastri J, Campbell EM. Recent insights into the mechanism and consequences of TRIM5alpha retroviral restriction. AIDS Res Hum Retroviruses. 2011;27:231–238. doi: 10.1089/aid.2010.0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter D, Schindler M, Specht A, Landford WN, Munch J, Kim KA, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, et al. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe. 2009;6:409–421. doi: 10.1016/j.chom.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Emerman M, Malik HS. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004;2:E275. doi: 10.1371/journal.pbio.0020275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Wu LI, Akey JM, Emerman M, Malik HS. High-frequency persistence of an impaired allele of the retroviral defense gene TRIM5alpha in humans. Curr Biol. 2006;16:95–100. doi: 10.1016/j.cub.2005.11.045. [DOI] [PubMed] [Google Scholar]
- Sawyer SL, Wu LI, Emerman M, Malik HS. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A. 2005;102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayah DM, Sokolskaja E, Berthoux L, Luban J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004;430:569–573. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrofelbauer B, Hakata Y, Landau NR. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc Natl Acad Sci U S A. 2007;104:4130–4135. doi: 10.1073/pnas.0610167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrofelbauer B, Yu Q, Zeitlin SG, Landau NR. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol. 2005;79:10978–10987. doi: 10.1128/JVI.79.17.10978-10987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Moreno R, Jia B, Breed M, Alvarez X, Evans DT. Compensatory changes in the cytoplasmic tail of gp41 confer resistance to tetherin/BST-2 in a pathogenic nef-deleted SIV. Cell Host Microbe. 2011;9:46–57. doi: 10.1016/j.chom.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhou J, Shah VB, Aiken C, Whitby K. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J Virol. 2011;85:542–549. doi: 10.1128/JVI.01406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skasko M, Tokarev A, Chen CC, Fischer WB, Pillai SK, Guatelli J. BST-2 is rapidly down-regulated from the cell surface by the HIV-1 protein Vpu: evidence for a post-ER mechanism of Vpu-action. Virology. 2011;411:65–77. doi: 10.1016/j.virol.2010.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Gold B, O'Huigin C, Javanbakht H, Li X, Stremlau M, Winkler C, Dean M, Sodroski J. The B30.2(SPRY) domain of the retroviral restriction factor TRIM5alpha exhibits lineage-specific length and sequence variation in primates. J Virol. 2005;79:6111–6121. doi: 10.1128/JVI.79.10.6111-6121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoye JP, Yap MW. Chance favors a prepared genome. Proc Natl Acad Sci U S A. 2008;105:3177–3178. doi: 10.1073/pnas.0800667105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc Natl Acad Sci U S A. 2006;103:5514–5519. doi: 10.1073/pnas.0509996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarev AA, Munguia J, Guatelli JC. Serine-threonine ubiquitination mediates downregulation of BST-2/tetherin and relief of restricted virion release by HIV-1 Vpu. J Virol. 2011;85:51–63. doi: 10.1128/JVI.01795-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torimiro JN, Javanbakht H, Diaz-Griffero F, Kim J, Carr JK, Carrington M, Sawitzke J, Burke DS, Wolfe ND, Dean M, Sodroski J. A rare null allele potentially encoding a dominant-negative TRIM5alpha protein in Baka pygmies. Virology. 2009;391:140–147. doi: 10.1016/j.virol.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Manen D, Rits MA, Beugeling C, van Dort K, Schuitemaker H, Kootstra NA. The effect of Trim5 polymorphisms on the clinical course of HIV-1 infection. PLoS Pathog. 2008;4:e18. doi: 10.1371/journal.ppat.0040018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgen CA, Kratovac Z, Bieniasz PD, Hatziioannou T. Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc Natl Acad Sci U S A. 2008;105:3563–3568. doi: 10.1073/pnas.0709258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W, Miguet N, Aschman N, Renesto P, Usami Y, Gottlinger HG. Structural basis of tetherin function. Curr HIV Res. 2012;10:298–306. doi: 10.2174/157016212800792487. [DOI] [PubMed] [Google Scholar]
- Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc Natl Acad Sci U S A. 2006;103:7465–7470. doi: 10.1073/pnas.0510483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap MW, Dodding MP, Stoye JP. Trim-cyclophilin A fusion proteins can restrict human immunodeficiency virus type 1 infection at two distinct phases in the viral life cycle. J Virol. 2006;80:4061–4067. doi: 10.1128/JVI.80.8.4061-4067.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap MW, Nisole S, Lynch C, Stoye JP. Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc Natl Acad Sci U S A. 2004;101:10786–10791. doi: 10.1073/pnas.0402876101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol. 2004;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Zhang F, Hatziioannou T, Perez-Caballero D, Derse D, Bieniasz PD. Antiretroviral potential of human tripartite motif-5 and related proteins. Virology. 2006;353:396–409. doi: 10.1016/j.virol.2006.05.035. [DOI] [PubMed] [Google Scholar]
- Zhang F, Landford WN, Ng M, McNatt MW, Bieniasz PD, Hatziioannou T. SIV Nef proteins recruit the AP-2 complex to antagonize Tetherin and facilitate virion release. PLoS Pathog. 2011;7:e1002039. doi: 10.1371/journal.ppat.1002039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. 2009;6:54–67. doi: 10.1016/j.chom.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]