

Abstract
Adhesion of Plasmodium falciparum infected erythrocytes (IE) to human endothelial receptors during malaria infections is mediated by expression of PfEMP1 protein variants encoded by the var genes.
The haploid P. falciparum genome harbors approximately 60 different var genes of which only one has been believed to be transcribed per cell at a time during the blood stage of the infection. How such mutually exclusive regulation of var gene transcription is achieved is unclear, as is the identification of individual var genes or sub-groups of var genes associated with different receptors and the consequence of differential binding on the clinical outcome of P. falciparum infections. Recently, the mutually exclusive transcription paradigm has been called into doubt by transcription assays based on individual P. falciparum transcript identification in single infected erythrocytic cells using RNA fluorescent in situ hybridization (FISH) analysis of var gene transcription by the parasite in individual nuclei of P. falciparum IE1.
Here, we present a detailed protocol for carrying out the RNA-FISH methodology for analysis of var gene transcription in single-nuclei of P. falciparum infected human erythrocytes. The method is based on the use of digoxigenin- and biotin- labeled antisense RNA probes using the TSA Plus Fluorescence Palette System2 (Perkin Elmer), microscopic analyses and freshly selected P. falciparum IE. The in situ hybridization method can be used to monitor transcription and regulation of a variety of genes expressed during the different stages of the P. falciparum life cycle and is adaptable to other malaria parasite species and other organisms and cell types.
Keywords: Genetics, Issue 68, Infectious Diseases, Immunology, Molecular Biology, nuclei, transcription, var genes, PfEMP1, infected erythrocytes (IE), Plasmodium falciparum, fluorescent in situ hybridization (FISH)
Protocol
1. Generation of Freshly Selected Infected Erythrocytes
For this assay, the best results are obtained when using freshly selected cultures for surface expression of PfEMP1 protein. In this particular experiment the 3D7 P. falciparum lineage was selected using specific antibodies as previously described1.
Day 1
Harvest 200 μl of packed blood cells from a P. falciparum culture containing 2-5 % late stage IE by centrifugation at 800 x g for 8 min at room temperature.
Re-suspend the blood pellet in 2 ml of 37 °C warm 0.75 % gelatin solution in a 14 ml sterile tube, and leave for 15-20 min at 37 °C to allow the uninfected and ring-stage IE to sediment.
Harvest the late-stage IE, i.e. the upper phase, into a new 14 ml tube and wash twice with 10 ml of 37 °C warm RBC-wash media.
Dilute 50 μl of specific anti-PfEMP1 antibodies in 2 ml of 37 °C warm RBC-wash media and sterile filter.
Mix the washed late-stage IE with the sterilized diluted antisera in a 14 ml sterile tube and incubate for 30 min at 37 °C with gentle agitation.
Spin down at 800 x g for 8 min at room temperature, and wash twice with 10 ml of 37 °C warm RBC-wash media.
Wash 50 μl of Dynabead Protein A suspension twice with RBC-wash media using a DynaMaq-15 magnet.
Resuspend the Dynabeads in 300 μl RBC-wash media and add them to the washed late-stage IE. Incubate for 30 min at 37 °C with gentle agitation.
Transfer the tube to the DynaMaq-15 magnet. Leave for 1-2 min and remove all the liquid.
Resuspend the Dynabeads in 4-5 ml of RBC-wash media. Transfer the tube back onto the magnet and leave it for 1-2 min before removing all the liquid. Repeat the washing step once.
Transfer the washed beads to a 25 cm2 sterile culture flask containing 200 μl freshly packed red blood cells. Add 5-6 ml of Albumax media into the culture. Store overnight at 5% CO2 at 37 °C.
Day 2
Remove the Dynabeads from the culture when the selected IE has reinvaded the fresh red blood cells by transferring all the culture to a 14 ml sterile tube. Put the tube onto the DynaMaq-15 magnet and leave for 1-2 min. Then, pour all the liquid back to a culture flask.
Culture for as few cycles as possible until a parasitemia of 5-10 % and parasites are at the ringstage.
IE should be analyzed by FACS to make sure that the correct surface phenotype, i.e. protein expression of either PFD1235w and/orPF11_0008 has been obtained1.
2. Preparation of Probes
The antisense RNA probes were generated from the most variable regions of the PFD1235w and the PF11_0008 var genes from P. falciparum 3D7 genomic DNA. DNA was amplified by PCR and cloned into the pSPT18 or 19 vector for transcription, respectively according to the manufacturer's description (Roche). The probes (580 base pairs (bp) and 590 bp in length) were labeled with Digoxigenin (DIG) or Biotin using a DIG RNA labeling Kit or a Biotin RNA labeling Mix, respectively. We confirmed the specificity of the probes both by Northern blot analyses and by single-label FISH analysis1.
3. Thin Smear and Fixation of Parasites Prior to In Situ Hybridization
Day 1
All steps are performed in an RNase-free environment and all reagents are RNase-free or pre-treated with diethyl pyrocarbonate (DEPC) or RNase Zap (Invitrogen). The slides and cover slips should be cleaned with alcohol to remove residual manufacturing grease. It is important to perform the protocol without any pause in between steps in order to minimize the risk of nuclease contamination.
Make a thin blood film by the standard spreading method3. Using the 100x oil objective lens of the microscope, count the IE and calculate the percentage of IE in the culture. Check that the parasite stages are in approximate synchrony, in the ring and early trophozoite stages of the IE cycle. These are the pre-replication, uni-nucleate stages, expressing var gene mRNA and suitable for single cell FISH transcription assays of this type.
Transfer 50 μl of the parasite culture, at a parasitemia of 5-10 %, to a 1.5 ml RNase-free Eppendorf tube. Centrifuge for 1 min at 2,000 rpm at room temperature.
Remove the media and add 50 μl PBS pH 7.2 in DEPC-treated water. Agitate the tube gently. Repeat the centrifugal sedimentation step twice to wash the cells free from media and cell debris.
Make four good quality thin smears. For each smear, make one smear onto one 11 mm diameter well of a 4 well slide, i.e. four glass slides in total, in a RNase-free hood (a critical step). Wave slides briefly into the air to dry. Make three extra slides for negative controls–one slide for single probe staining for any other irrelevant gene by using a specific probe, another slide for RNase treatment and a third slide containing IE not expressing the gene of interest. Leave the slides to dry on the bench for 10-20 min.
Fix the thin films by adding 60 μl of fixation solution containing 4 % paraformaldehyde and 5 % glacial acetic acid into the wells. Leave for 10 min on the bench at room temperature.
Wash the slides in 2x SSPE buffer for 5 min by gentle agitation at room temperature. Briefly remove excess liquid by touching the wells containing the cells gently with a paper tissue.
Cover the slides with 0.01 % pepsin in 0.01 M HCl for 2 min at 37 °C (an extremely critical step). Wash the slides in 2x SSPE for 5 min at room temperature.
Dilute the RNase solution to a final concentration of 10 μg/ml. Cover the negative control smears with 60 μl of the RNase solution. Incubate for 30 min at 37 °C and then wash the slides in 2x SSPE for 5 min at room temperature.
Proceed immediately to the hybridization step.
4. Hybridization of RNA Probes to Fixed Slides
Day 1
Prepare the hybridization chamber, such as a ThermoStar 100 HC4 or an empty pipette tip box put in a clean oven, by spraying with RNase Zap and wiping it clean with a paper tissue. Fill the channels of the hybridization chamber or the bottom of the box with DEPC-treated water to make sure that the slides will not dry out during hybridization. Close the chamber and preheat to 48 °C.
Dilute the probes into the hybridization solution to a final concentration of 12 ng/μl. Denature the probe solution at 65 °C for 5 min in a heating block. Immediately chill on ice.
Briefly air dry the slides after the 2x SSPE wash. Carefully remove excess liquid around the wells with a paper tissue.
Open the hybridization chamber and place the slides in the chamber. Apply one or both probes in a total volume of 60 μl per well. Carefully cover the liquid drop with an RNase-free cover slip using sterile forceps.
Close the chamber and hybridize the slides at 48 °C for at least 16 hr over night.
5. Antibody Conjugation and Fluorescence Amplification
Day 2
The following steps are based on the TSA Plus Fluorescence Palette System from Perkin Elmer. All the steps are done at room temperature.
Wash the slides 3 times in TNT buffer for 5 min with gentle agitation.
Block the wells in 150 μl of TNB buffer for 30 min.
Dilute the HRP-conjugated α-DIG antibody in TNB buffer at a ratio of 1:100 and the HRP-conjugated α-biotin antibody in TNB buffer, at a ratio of 1:500. Remove any excess TNB buffer from the slides with a paper tissue. When detecting two transcripts simultaneously, both antibodies can go at once. Apply a total amount of 100 μl of the antibody-TNB solution per well and carefully cover with a cover slip. Incubate the slides for 2 hr.
Remove the cover slips carefully. Wash the slides in TNT buffer 3 times for 5 min.
Dilute the FITC-fluorophore in the tyramide amplification reagent at a ratio of 1:500 and apply 100 μl of the solution to each well. Cover the wells with cover slips and incubate for 12 min.
Remove the cover slips carefully and wash the slides 3 times for 5 min in TNT buffer by gentle agitation.
Dilute the Cyan3-fluorophore in the tyramide amplification reagent at a ratio of 1:500. Add 100 μl per well of this solution. Cover the wells with cover slips and incubate for 12 min.
Remove the cover slips carefully and then rinse the slides quickly by immersion in TNT buffer.
Wash the slides 3 times with TNT buffer for 5 min.
Immerse the slides quickly in DEPC-treated water.
Leave the slides to air dry. Mount the slides with anti-fade reagent containing DAPI. Seal the corners of the cover slips with nail polish.
The slides are now ready for microscopy. Keep the slides covered with aluminum foil and store at 4 °C if the experiment is to be completed the next day. A complete sealing of the sides of the slides can be carried out 24 hr after applying the anti-fade reagent. This will allow the slides to be imaged for up to one week after the protocol has been completed.
6. Visualization of Hybridized Probes
Turn on the microscope. A standard immunofluorescence microscope is sufficient for this kind of experiment, but if you have access to a confocal microscope you can also use this for 3D images or to obtain videos of your stained cells. Allow microscope to warm up for 15-30 min. Switch on the computer and the software.
Check the quality of the staining on the immunofluorescence microscope. Choose a spot containing a few parasites.
Adjust the gain of each laser manually. Adjust the pixel dwell to 4-6 m-1s-1 and the steps to 512 x 512 pixels.
Take a test picture using Frame Lambda. Readjust the laser gain.
Take a minimum of 20 good pictures of fluorescent single cells. At least 2-5 pictures of a field containing 5-20 parasites are needed in order to obtain a fair overview of the percentage of positive parasites.
Representative Results
Figure 1 illustrates a flowchart of the major steps and the time duration of the RNA FISH methodology.
A series of representative images of well and poorly preserved stained mRNA FISH experiments using single P. falciparum IE are shown in Figure 2. This intracellular protozoan has a small (1-1.5 μm diameter) nucleus which is stained blue with DAPI. Twenty four hours after erythrocyte invasion in P. falciparum the process of schizogony, i.e. nuclear division, begins. Optimal FISH detection of var gene transcription occurs at around 10-22 hr into this 48 hr intra-erythrocytic lytic cycle. Probes generally hybridize to mRNA species which appear adjacent to the main nuclear body, in what is probably nuclear envelope-associated endoplasmic reticulum. The good quality images in row A show FITC- (green) and Cyan3- (red) labeled probes corresponding to the PFD1235w and PF11_0008 var genes, respectively, in the double selected P. falciparum sub-line, 3D7 PFD1235w/PF11_0008. Co-localization of the genes is apparent but not absolute. Row B shows poor hybridization of both probes to parasites which have either degraded or largely ceased transcribing var mRNA. In row C, a good FISH hybridization is obtained but the cellular preservation is poor, as shown by badly spread and aggregating parasites.
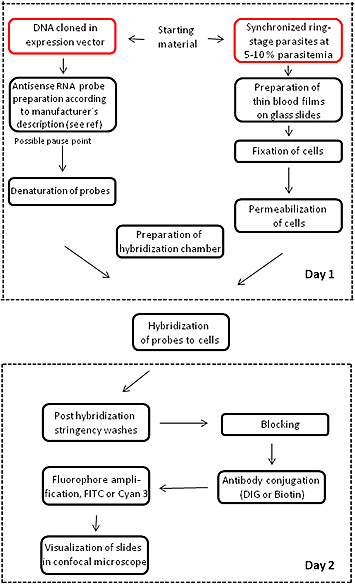 Figure 1. RNA FISH experiment flowchart. The flowchart illustrates the main points and timing of the FISH procedure.
Figure 1. RNA FISH experiment flowchart. The flowchart illustrates the main points and timing of the FISH procedure.
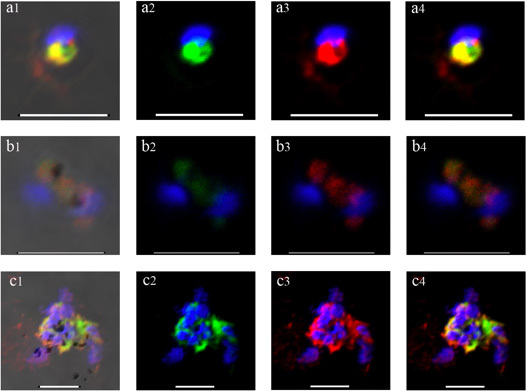 Figure 2. Confocal images of RNA-FISH in double selected P. falciparum indicating simultanous mRNA transcription of two different var genes labeled with either FITC (green) or Cyan3 (red) signal. DAPI staining (blue) indicates the parasite nuclear DNA. The vertical row a1, b1 and c1 show the transmission images of the parasites. Scale bar 5 μm.
Figure 2. Confocal images of RNA-FISH in double selected P. falciparum indicating simultanous mRNA transcription of two different var genes labeled with either FITC (green) or Cyan3 (red) signal. DAPI staining (blue) indicates the parasite nuclear DNA. The vertical row a1, b1 and c1 show the transmission images of the parasites. Scale bar 5 μm.
Discussion
RNA FISH analysis, in contrast to methods such as Northern blotting and RT-PCR, allows discrimination of specific mRNA transcripts at the single cell level. This makes it possible to discriminate between transcriptionally active and inactive cells, in this example, P. falciparum parasitic protozoa inside human red blood cells. Such whole-cell observations are often necessary and may unravel important and novel transcriptional patterns1.
Although other RNA FISH methods have been described4-10, there has been a need for development of a new and refined protocol for the study of simultaneous var mRNA transcription in P. falciparum. By using asRNA probes as detection tools, the special-temporal patterns of mRNA activity can be easily localized in the cells. In addition, asRNA probes can also be used in other experimental systems which would support their specificity1. Other critical parameters that are important when developing a new protocol includes fixation and permeabilzation of the cells. These steps were empirically defined by testing various chemical reagents as suggested by other authors5. We concluded that by combining the slow acting cross linker paraformaldehyde together with the coagulant fixative acetic acid we could obtain a good preservation of the tissue morphology. In addition, we found out that pre-blocking the cells before adding the probes was not necessary, similarly to other described RNA FISH protocols5. Furthermore, in the present protocol we use gentle washes, a mild protease (pepsin) at low amounts and a short incubation period to allow for the probe and the antibodies to diffuse into the nucleus and hybridize with the mRNA attached on the ER, minimizing the damage to surrounding membranes (Figure 2a1-2a4).
In addition, the RNA FISH and the high resolution images generated by the camera attached to the confocal microscope reveals whether a particular cell is positive for staining or not and also gives valuable information about the spatial relationships between the stained antisense RNA and the DAPI stained nucleus. As resolved in the images in Figure 2a4, the mRNA appears to be attached next to the nucleus, probably in the endoplasmic reticulum, and not on top of it, as expected in a DNA-FISH image11-12.
Studies of single cells require observations of numerous parasites and at different time points to get a statistically significant result. Thus, RNA FISH analysis is a rather time consuming technique that demands patience and considerable trial-and-error experimentation to calibrate the technique. As RNA FISH is a technique of many parameters a trouble-shooting guide to the main steps of this protocol are given in Table 1.
| Problem | Possible cause | Recommendation |
| Weak signal or no signal |
|
|
| Poor cellular preservation |
|
|
| High Background |
|
|
| The RNase treated control is positive |
|
|
| False positive detection of negative control parasites |
|
|
Table 1. Troubleshooting guidance.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors would like to thank Michael Alifrangis and Ulla Abildtrup for genotyping of parasites and Christina Holm for excellent technical assistance. This work was funded by Howard Hughes Medical Institute (grant 55005511), The Lundbeck Foundation (grant R9-A840) and by the Niels Bohr Foundation.
References
- Joergensen L, Bengtsson DC, Bengtsson A, Ronander E, Berger SS, Turner L, Dalgaard MB, Cham GK, Victor ME, Lavstsen T, Theander TG, Arnot DE, Jensen AT. Surface co-expression of two different PfEMP1 antigens on single Plasmodium falciparum-infected erythrocytes facilitates binding to ICAM1 and PECAM1. PLoS Pathog. 2010;2:1001083. doi: 10.1371/journal.ppat.1001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinaud R, Mello CV, Velho TA, Wynne RD, Tremere LA. Detection of two mRNA species at single-cell resolution by double-fluorescence in situ hybridization. Nat. Protoc. 2008;3:1370–1379. doi: 10.1038/nprot.2008.115. [DOI] [PubMed] [Google Scholar]
- Cheeseborough M. District Laboratory Practice in Tropical Countries. Cambridge: Cambridge University Press; 2006. [Google Scholar]
- Brolin KJ, Ribacke U, Nilsson S, Ankarklev J, Moll K, Wahlgren M, Chen Q. Simultaneous transcription of duplicated var2csa gene copies in individual Plasmodium falciparum parasites. Genome Biol. 2009;10:R117. doi: 10.1186/gb-2009-10-10-r117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epp C, Li F, Howitt CA, Chookajorn T, Deitsch KW. Chromatin associated sense and antisense noncoding RNAs are transcribed from the var gene family of virulence genes of the malaria parasite Plasmodium falciparum. RNA. 2009;15:116–127. doi: 10.1261/rna.1080109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas-Junior LH, Hernandez-Rivas R, Ralph SA, Montiel-Condado D, Ruvalcaba-Salazar OK, Rojas-Meza AP. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell. 2005;121:25–36. doi: 10.1016/j.cell.2005.01.037. [DOI] [PubMed] [Google Scholar]
- Obernosterer G, Martinez J, Alenius M. Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nature Protoc. 2007;2:1508–1514. doi: 10.1038/nprot.2007.153. [DOI] [PubMed] [Google Scholar]
- Li F, Sonbuchner L, Kyes SA, Epp C, Deitsch KW. Nuclear non-coding RNA are transcribed from the centromeres of Plasmodium falciparum and are associated with centromeric chromatin. J. Biol. Chem. 2008;283:5692–5698. doi: 10.1074/jbc.M707344200. [DOI] [PubMed] [Google Scholar]
- Thompson J, Sinden RE. In situ detection of Pbs21 mRNA during sexual development of Plasmodium berghei. Mol. Biochem. Parasitol. 1994;68:189–196. doi: 10.1016/0166-6851(94)90164-3. [DOI] [PubMed] [Google Scholar]
- Thompson J. In situ detection of RNA in blood- and mosquito-stage malaria parasites. Methods Mole Med. 2002;72:225–235. doi: 10.1385/1-59259-271-6:225. [DOI] [PubMed] [Google Scholar]
- Arnot DE, Ronander E, Bengtsson DC. The progression of the intra-erythrocytic cell cycle of Plasmodium falciparum and the role of the centriolar plaques in asynchronous mitotic division during schizogony. Int. J. Parasitol. 2011;41:71–80. doi: 10.1016/j.ijpara.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, Fraser P. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 2004;36:1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]