

Abstract
HIV-1 replication can be inhibited by type-I interferon (IFN), and the expression of a number of gene products with anti HIV-1 activity is induced by type-I IFN1,2. However, none of the known antiretroviral proteins can account for the ability of type-I IFN to inhibit early, preintegration, phases of the HIV-1 replication cycle in human cells3,4. By comparing gene expression profiles in cell lines that differ in their ability to support the inhibitory action of IFNα on early steps of the HIV-1 replication cycle, we identified Myxovirus resistance-2 (Mx2) as an interferon-induced inhibitor of HIV-1 infection. Expression of Mx2 reduced permissiveness to a variety of lentiviruses, while depletion of Mx2 using RNA interference reduced the anti-HIV-1 potency of IFNα. HIV-1 reverse transcription proceeded normally in Mx2-expressing cells, but 2-LTR circular forms of HIV-1 DNA were less abundant, suggesting that Mx2 inhibits HIV-1 nuclear import, or destabilizes nuclear HIV-1 DNA. Consistent with this notion, mutations in the HIV-1 capsid protein that are known, or suspected to alter the nuclear import pathways used by HIV-1 conferred resistance to Mx2, while preventing cell division increased Mx2 potency. Overall, these findings indicate that Mx2 is an effector of the anti-HIV-1 activity of type-I IFN, and suggest that Mx2 inhibits HIV-1 infection by inhibiting capsid-dependent nuclear import of subviral complexes.
We and others have previously identified proteins with antiretroviral activity based on their differential expression in cells that are permissive or non-permissive with respect to particular steps in the HIV-1 life cycle5,6. We noticed that monocytoid cell lines varied in their ability to support the anti-HIV-1 activity of type-I IFN. Specifically, IFNα treatment of THP-1 cells caused an ~40-fold reduction in infection by an HIV-1 based GFP-reporter vector, while treatment of K562 and U937 cells had little effect (Fig. 1a). When these cell lines were differentiated into a macrophage-like state by treatment with phorbol 12-myristate 13-acetate (PMA), the inhibitory effect of IFNα was accentuated in THP-1 cells, accentuated to a lesser extent in U937 cells, but remained nearly absent in K562 cells (Fig. 1a).
Figure 1. Differential effects of IFNα on HIV-1 infection of monocytoid cell lines correlates with Mx2 expression.

a, Undifferentiated, or PMA-differentiated THP-1, K562 and U937 cells with or without IFNα treatment (1000 U/ml) were challenged with a GFP-expressing HIV-1 vector (CSGW). b, RNA extracted from cells treated identically to those shown in a was analyzed on microarrays. The array signal is plotted in arbitrary units, and the data points representing Mx2 are highlighted. c, Western blot analysis of Mx2 and tubulin expression in monocytoid cell lines treated for 24h with the indicated doses of IFNα. Numbers below each lane indicated fold increase in Mx2 protein levels relative to untreated cells.
To identify candidate effectors of the antiviral action of IFNα, we used microarrays to measure messenger RNA levels in the aforementioned cell lines. Twenty-two genes whose induction, or non-induction, by IFNα correlated to varying degrees with the ability or inability of IFNα to inhibit HIV-1-GFP vector infection in the monocytoid cell lines were selected for further study (Fig. 1b Extended Data Fig. 1, 2). Among these candidates, Mx2, a gene that was not previously thought to exhibit antiviral activity7, was of particular interest as we recently identified it as a ‘hit’ in an overexpression screen in a T-cell line during which Mx2 modestly inhibited infection by HIV-18. Western blot analyses confirmed that Mx2 expression was strongly induced by IFNα in THP-1 cells but not K562 cells, and a basal level of Mx2 expression was slightly increased by IFNα treatment in U937 cells (Fig. 1c). Mx2 was expressed at a basal level in primary CD4+ T-cells and macrophages, and was induced to varying degrees by IFNα, depending on the individual donor, and how cells were activated (Extended Data Fig. 3).
Extended Data Figure 1. Candidate anti-HIV-1 genes from the microarray analysis.

Messenger RNA levels, determined using Illumina BeadChips, and given in arbitrary units, for genes whose differential induction in undifferentiated and phorbol 12-myristate 13-acetate (PMA) treated THP-1, K562 and U937 cells correlated best with the anti-HIV-1 effect of IFNα.
Extended Data Figure 2. Additional candidate anti-HIV-1 genes from the microarray analysis.

Messenger RNA levels, determined using Illumina BeadChips, and given in arbitrary units, for genes whose differential induction in undifferentiated and phorbol 12-myristate 13-acetate (PMA) treated THP-1, K562 and U937 cells correlated to some degree with the anti-HIV-1 effect of IFNα.
Extended Data Figure 3. Induction of Mx2 by IFNα in primary CD4+ T-cells and macrophages.
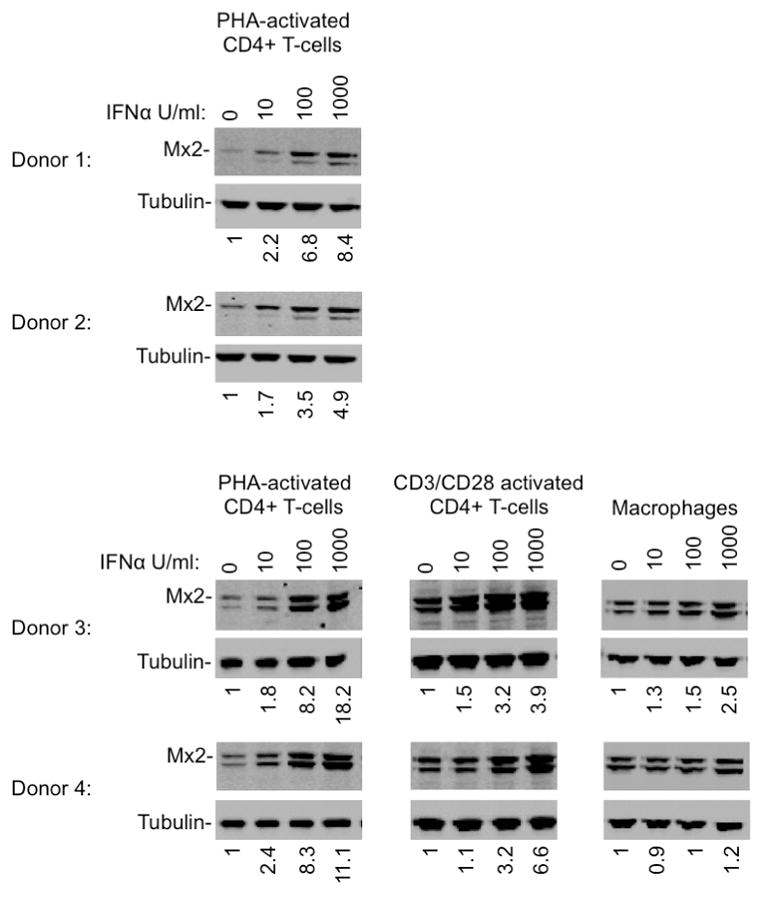
Western blot analysis of Mx2 and tubulin expression in purified CD4+ T-cells, activated with PHA or antiCD3/CD28, and macrophages treated for 24h with the indicated doses of IFNα. Numbers below each lane indicate fluorescence intensity associated with the Mx2 band. The second more rapidly migrating Mx2 species that was detected inconsistently is of unknown provenance, and may represent a proteolytic breakdown product, or may arise through the use of an alternative start codon at amino acid 25, generating an Mx2 protein that lacks the nuclear localization signal.
Expression of the 22 candidate and control genes in K562 cells revealed that only Mx2 and a control antiviral gene, rhesus macaque (rh) TRIM5α9 inhibited HIV-1 infection. (Fig. 2a). A rhesus macaque variant of Mx2 also inhibited HIV-1 infection to a similar degree as human Mx2, while Mx1 was inactive against HIV-1 (Fig. 2a), even though it inhibits a variety of other viruses7. Although Mx2 clearly inhibited HIV-1 infection (Fig 2a – d), the fact that U937 cells (Fig. 1a), primary macrophages and αCD3/CD28-stimulated CD4+ T-cells are readily infected by HIV-1, despite expressing appreciable levels of Mx2 (Fig 1c, Extended Data Fig. 3), indicates that the block imposed by Mx2 is not absolute, or that Mx2 potency is perhaps influenced by the cellular environment or cofactors.
Figure 2. Inhibition of lentivirus infection by WT and mutant Mx2, but not other differentially interferon-induced genes.

a, Infection of K562 cells, previously transduced with an HIV-1 vector (SCRPSY) expressing negative (luciferase) or positive (rhTRIM5α) control genes, or candidate antiviral genes, with GFP-expressing HIV-1 vector (CSGW) at the indicated multiplicity of infection (MOI). b, Western blot analysis of Mx2 and tubulin expression in K562 cell clones transduced with an HIV-1 vector (CSIB) expressing WT and mutant Mx2 proteins. c, Infection of the same K562 cells as in b with an HIV-1-GFP reporter virus. d, e, Infection of HOS cells, previously transduced with an Mx2 expressing or empty HIV-1 vector (SCRPSY), with various GFP-reporter viruses. Titers are mean+SD, n=3 technical replicates, representative of four experiments.
Mx1 and Mx2 are members of a family of dynamin-like GTPases7, but only Mx2 is localized to the nucleus by virtue of a basic nuclear localization signal (NLS) contained within its N-terminal 25 amino acids10,11. Notably, the N-terminal 25 amino acids that encode the Mx2 NLS were strictly required for antiviral activity (Fig. 2b, c). Conversely, mutations K131A and T151A that inhibit GTP binding and hydrolysis, respectively11 did not block the anti-HIV-1 activity of Mx2 (Fig. 2b, c). This result is in contrast to findings with Mx1, whose antiviral activity is GTPase dependent7, but should be interpreted cautiously given the reported ability of these Mx2 mutants to induce a generalized perturbation of nucleocytoplasmic transport11. In addition to its activity against HIV-1 and HIV-1 (Fig. 2d), Mx2 expression in HOS cells inhibited infection by GFP-reporter viruses based on a variety of primate lentiviruses, including HIV-2, SIVmac, SIVagmTan and SIVagmSab, with some variation in Mx2 antiviral potency (Fig. 2e). The nonprimate lentiviruses, equine infectious anemia virus, and feline immunodeficiency virus were less potently inhibited, while a gammaretrovirus, murine leukemia virus, was only marginally sensitive to Mx2.
The experiments described above all represented single-cycle infection assays, using vesicular stomatitis virus glycoprotein (VSV-G) pseudotyped reporter viruses. However, expression of Mx2 in GHOST-R5 cells also inhibited infection by two full-length primary HIV-1 strains, suggesting that Mx2 inhibition was independent of the route of entry, and not counteracted by HIV-1 accessory genes (Fig. 3a). Moreover, Mx2 expression in GHOST-X4 cells inhibited spreading infection by full-length replication-competent HIV-1NL4-3 (Fig. 3b), reducing the number of infected cells by ~20-fold during the exponential phase of viral growth. Reduction of Mx2 expression in THP-1 cells (Fig. 3c, d) or in HOS cells (Extended Data Fig. 4a, b) using shRNAs reduced, but did not eliminate the antiviral effect of IFNα. Thus, Mx2 is required for the full potency of IFNα, but is not solely responsible for the inhibitory action of IFNα on the early steps of the HIV-1 replication cycle.
Figure 3. Mx2 inhibits replication competent HIV-1 and is required for the full antiviral activity of IFNα.

a, Infection of empty vector (CSIB) or Mx2-expressing, GHOST-R5 cells with full-length primary HIV-1 strains. Titers are mean+SD, n=3 technical replicates, representative of two experiments. b, Growth of replication competent HIV-1NL4-3 in empty vector (CSIB) or Mx2-expressing GHOST-X4 cells (containing an HIV-2 LTR-GFP indicator gene). c, Western analysis of Mx2 and tubulin expression in IFNα-treated THP-1 cells expressing control or Mx2-targeted shRNAs. Numbers below each lane indicate fluorescence intensity associated with the Mx2 band. d, HIV-1 GFP reporter virus infection of shRNA-expressing THP-1 cells from c, with or without IFNα treatment. Titers are mean+SD, n=3 technical replicates, p values calculated using unpaired T-test, representative of three experiments.
Extended Data Figure 4. Mx2 is required for the full antiviral activity of IFNα in HOS cells.
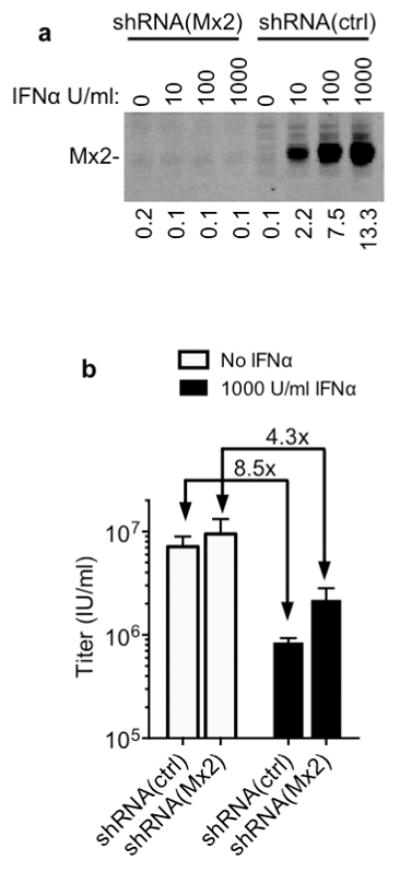
a, Western blot analysis of Mx2 expression HOS cells transduced with vectors expressing control or Mx2-targeted shRNAs, and treated with IFNα. Numbers below each lane indicate fluorescence intensity associated with the Mx2 band. d, Infectious titer of an HIV-1 GFP reporter virus determined using the shRNA containing HOS cells from a, with or without IFNα treatment. Titers are mean+SD, n=3 technical replicates, representative of three experiments.
Consistent with this conclusion, IFNα treatment reduced the accumulation of HIV-1 reverse transcripts in HOS cells (Fig. 4a), as has previously been reported for other cell types12. Conversely, Mx2 expression did not inhibit reverse transcript accumulation in either HOS or K562 cells (Fig. 4a, Extended Data Fig. 5). However, Mx2 did reduce the generation of 2-LTR circles (Fig. 4a, Extended Data Fig. 5), which are thought to form only after retroviral DNA has accessed the nucleus of infected cells. Mx2 may, therefore, inhibit the entry of HIV-1 into the nucleus, or perhaps cause destabilization of viral DNA in the nucleus. Consistent with previous reports10,11, we found that that N- or C-terminally HA-tagged forms of Mx2 were particularly concentrated at nuclear pores marked by the nucleoporin Nup98 (Extended Data Fig. 6). The Mx2 (K131A) mutant is primarily cytoplasmic but nevertheless inhibits nucleocytoplasmic transport11 and also retains antiviral activity (Fig. 2c). Therefore, alteration of the fate of incoming HIV-1 DNA with respect to the nucleus may underlie the antiviral activity of Mx2, even though stable physical association with nuclear pores may not be required for antiviral function.
Figure 4. Mx2 activity reduces levels of nuclear HIV-1 DNA, is capsid dependent and is more potent in non-dividing cells.

a, Quantitative PCR analysis of reverse transcript (left) and 2-LTR circle (right) abundance in inhibitor treated or Mx2-expressing HOS cells. b, WT or CA-mutant HIV-1 GFP-reporter virus infection of vector or Mx2-expressing HOS cells. Titers are mean+SD, n=3 technical replicates, representative of four experiments. c, Infectivity of WT and N57S CA mutant HIV-1 GFP-reporter viruses in untreated and IFNα-treated THP-1 cells. Titers are mean+SD, n=3 technical replicates, representative of four experiments, fold inhibition is the ratio of the mean titers on untreated and IFNα treated cells. d, HIV-1 GFP reporter virus infection of dividing and non-dividing (aphidicolin treated) vector or Mx2-expressing HOS cell clones.
Extended Data Figure 5. Mx2 activity reduces levels of nuclear HIV-1 DNA in K562 cells.
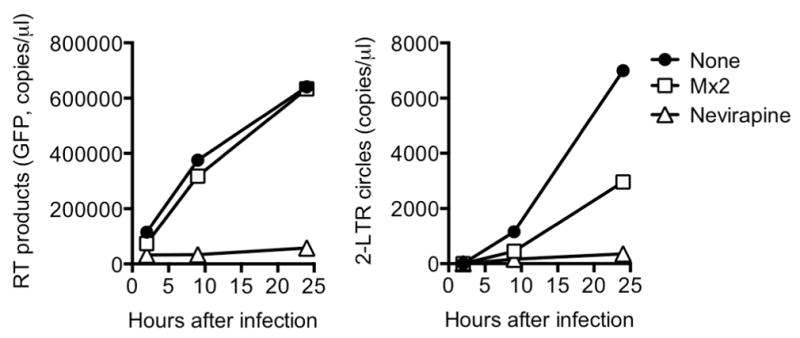
Quantitative PCR analysis of reverse transcript (left) and 2-LTR circle (right) abundance in empty vector untreated (None) nevirapine-treated or Mx2-expressing K562 cells.
Extended Data Figure 6. Localization of Mx2 at nuclear pores.

a, Deconvolution microscopic images (single optical sections) of immunofluorescently stained Nup98 (red), HA-tagged Mx2 (green, expressed using CSIB vectors), and DAPI stained DNA (blue) in HOS cells. The upper set of panels is an optical section approximately through the center of the vertical dimension of the nucleus, while the center and lower panels are an optical section approximately coincident with the dorsal surface of the nucleus. The lower panels are an expanded view of a portion of the center panels. Scale bars represents 10μm (upper panels), 5μm (center panels) or 1μm (lower panels). b, Pearson’s coefficient for colocalization of Mx1 or Mx2 and Nup98. Each data point represents an individual cell and the horizontal bar is the mean (n=6 for Mx1, n=10 for Mx2).
The HIV-1 capsid protein (CA) is a key determinant required for infection of non-dividing cells and nuclear entry of subviral complexes13–15. Indeed, HIV-1 CA mutations have been shown to change the requirement for specific nucleoporins (e.g. Nup358/RanBP2, Nup85, Nup153, Nup155) during HIV-1 infection, and to alter the distribution of sites at which HIV-1 DNA integrates into host chromosomes16–18. Therefore, we tested whether a number of CA mutations that are known or suspected to affect the pathway used by HIV-1 DNA into the nucleus also affected sensitivity to inhibition by Mx2 (Fig. 4b). Of these, a mutation (N57S) that confers cell-cycle dependence on HIV-1 infection19, and presumably restricts HIV-1 nuclear entry to the mitotic phase of the cell cycle, conferred resistance to Mx2 (Fig. 4b). Another mutation (G89V), that abolishes CypA binding by HIV-1 CA, and the requirement for Nup358 during HIV-1 infection17, also conferred apparently complete Mx2 resistance (Fig. 4b). Other CA mutations, N74D that abolishes CA interaction with cleavage and polyadenylation specificity factor-6 (CPSF6)16, reduced but did not eliminate sensitivity to Mx2, while mutations that confer cyclophilin A-sensitivity (cyclosporin A dependence) during early replication steps20 slightly reduced Mx2 sensitivity (Fig. 4b). These data demonstrate that the viral capsid governs the sensitivity of HIV-1 to Mx2. Additionally, they show that the antiviral activity of Mx2 is specific, and unlikely to be the result of some generalized perturbation of cell physiology. Notably, the Mx2-resistant CA mutant, N57S, exhibited a modest degree of resistance to IFNα, relative to WT HIV-1, in THP-1 cells and HOS cells (Fig. 4c and Extended Data Fig. 7), supporting the notion that Mx2 is one, but not the only, effector of the antiviral activity of IFNα during the early steps of HIV-1 replication cycle.
Extended Data Figure 7. The N57S capsid mutation reduces HIV-1 sensitivity to IFNα in HOS cells.
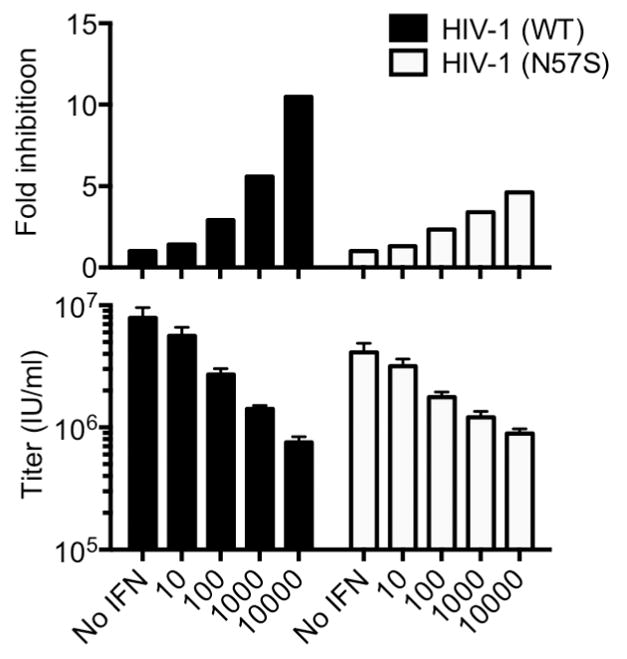
Infectivity of WT and N57S CA mutant HIV-1 GFP-reporter viruses in untreated and IFNα-treated HOS cells. Titers are mean+SD n=3, technical replicates, representative of three experiments, fold inhibition is the ratio of the titers on untreated and IFNα treated cells.
Because the cell-cycle dependent HIV-1 CA-mutant (N57S) was not inhibited by Mx2 (Fig. 4b), we reasoned that arresting the cell cycle and thereby restricting HIV-1 infection to non-mitotic cells might potentiate the antiviral activity of Mx2. Growth arrest of HOS or K562 cells with aphidicolin blocked infection by a control cell-cycle dependent retrovirus (MLV) irrespective of Mx2 expression, while HIV-1 was almost unaffected, as expected (Fig. 4d and Extended Data Fig. 8a, b). However, the inhibitory activity of Mx2 was increased in non-dividing cells (Fig. 4d and Extended Data Fig. 8), in which it inhibited a single-cycle of replication by ~30-fold. Put another way, Mx2 both inhibited and conferred a degree of cell-cycle dependence on WT HIV-1 infection.
Extended Data Figure 8. Effect of Mx2 on HIV-1 and MLV infection in dividing and non-dividing cells.

a, MLV-GFP reporter virus infection of dividing and non-dividing (aphidicolin treated) vector or Mx2 expressing HOS cell clones. b, HIV-1 GFP reporter virus infection of dividing and non-dividing (aphidicolin treated) vector or Mx2 expressing K562 cell clones. c, MLV-GFP reporter virus infection of dividing and non-dividing (aphidicolin treated) vector or Mx2 expressing K562 cell clones.
Type-I IFN inhibits HIV-1 replication at multiple points in the life cycle, both before and after the point at which Mx2 appears to act2,6,12. Thus Mx2 is one of multiple effectors that contribute to the overall anti-HIV-1 activity of type-I IFN. A few potential mechanisms might underlie the anti-HIV-1 activity of Mx2. First, Mx2 might directly target the incoming viral capsid, in a manner akin to the TRIM5α and Fv1 antiretroviral proteins2,9,21, or mutant cytoplasmic forms of CPSF616. Like Mx2, one consequence of the action of these capsid-targeting proteins is inhibition of the import of viral DNA into the nucleus, and in some cases their potency is enhanced in non-dividing cells16,22. A second possibility is that Mx2 inhibits particular nuclear import pathways, without regard to the precise nature of the import cargo, as mutant forms of Mx2 have been shown to inhibit the nuclear accumulation of model cargos unrelated to HIV-111. A third possibility is that Mx2 acts after nuclear entry to destabilize viral DNA and/or inhibit integration. In these scenarios, CA mutations (G89V, N57S) could confer resistance by inhibiting interaction with Mx2, by modulating the timing or extent of capsid uncoating, or by directing HIV-1 to alternative nuclear entry pathways. We note that the Mx2-resistant G89V and N57S mutants exhibit reduced infectiousness in human cells, raising the possibility that the mutations abolish the use of pathways or processes during infection that are inhibited by Mx2. Finally, it is formally possible that Mx2 acts indirectly, for example by affecting the nuclear-cytoplasmic distribution of other cellular proteins that can interact with the viral capsid. However, the poor correlation in the degree of Mx2 (Fig. 4) and CPSF623 resistance/sensitivity exhibited by HIV-1 CA mutants argues that redistribution of CPSF6 is unlikely to underlie the antiviral action of Mx2. While further work will be required to precisely define the molecular mechanisms involved, our findings demonstrate that Mx2 is an effector in the anti-HIV-1 activity of type-I IFN and underscore the remarkable diversity of proteins that cells can mobilize as antiretroviral defenses.
Methods
Microarray analyses
Total RNA was extracted, using the RNeasy Plus Mini kit (Qiagen), from THP-1, K562 and U937 cells that were either undifferentiated, or differentiated using phorbol-12-myristate-13-acetate, and untreated, or treated with 1,000 U/ml IFNα for 24 h before harvest. cRNA was prepared and probed using Human HT12 Expression Beadchip (Illumina), containing ~48,000 transcript probes, according to the manufacturer’s instructions.
Plasmid Construction
The HIV-1 based expression vector SCRPSY expresses a red fluorescent protein (TagRFP) and puromycin resistance from a TagRFP-FMDV2A-Puro cassette in the unspliced viral transcript (in place of Gag coding sequence). SCRPSY also expresses spliced transcripts that encode HIV-1 Tat and Rev proteins, as well as genes inserted in Gateway compatible sequences in place of Nef encoding sequences. The HIV-1 based expression vectors CSIB, HA-CSIB and CSIB-HA are derived from CSGW24 by replacing GFP coding sequences with a multi-cloning site (with or without sequences encoding a HA tag) followed by an IRES sequence and a blasticidin resistance cassette.
Candidate antiviral genes, including Mx2, were also transferred into the SCRPSY vector from a library of interferon-stimulated genes8 in Gateway-compatible entry vectors using the clonase reaction (Life Technologies). Alternatively, open reading frames were amplified from cDNA prepared from THP-1 cells following treatment with 1000U/ml IFNα for 24h. The GTP hydrolysis defective Mx2 mutant (T151A), the GTP binding defective Mx2 mutant (K131A), Mx2 lacking the N-terminal nuclear localization signal (delN25)10,11 were generated using PCR–based site directed mutagenesis. For insertion into CSIB based vectors, Mx1, WT and mutant Mx2 genes were amplified by PCR and inserted following SfiI restriction digestion.
Cells
The adherent cell lines 293T and HOS were maintained in DMEM. The suspension cell lines U937 and K562, were maintained in RPMI. THP-1 cells were maintained in RPMI supplemented with 0.05mM beta-mercaptoethanol These cell lines were obtained from the ATCC. Single-cell clones of GHOST cells expressing CXCR4 or CCR5 obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH from Dr. Vineet N. KewalRamani and Dr. Dan R. Littman were derived by limiting dilution and maintained in DMEM supplemented with 2.5μg/ml puromycin, 50μg/ml hygromycin, and 500μg/ml G418. All growth media were supplemented with 10% fetal calf serum and gentamicin.
Derivatives of K562 and GHOST cells expressing WT and mutant Mx2 proteins were generated by transduction with CSIB-based vectors followed by selection in 5μg/ml blasticidin. HOS cells expressing WT or mutant Mx2 proteins were prepared by transduction with SCRPSY-based vectors followed by selection in 2.5μg/ml puromycin. HOS cells expressing WT or mutant HA-tagged Mx1 or Mx2 proteins were prepared by transduction with HA-CSIB based vectors followed by selection in 5μg/ml blasticidin. In general, the Mx2-expressing cells were used as populations of blasticidin or puromycin resistant cells, but some experiments employed single-cell clones of Mx2-expressing HOS and K562 cells, which were derived from puromycin or blasticidin-resistant populations by limiting dilution. For experiments in which the panels of candidate antiviral genes were screened (Fig. 2a), K562 cells were transduced with SCRPSY-based vectors, but were not selected in puromycin; infection was measured in the tagRFP-positive population. To generate the Mx2-expressing or candidate gene cell lines, SCRPSY or CSIB vector stocks for transduction were generated by co-transfection of 293T cells with a vesicular stomatitis virus G protein (VSV-G) expression plasmid, an HIV-1NL4-3 GagPol expression plasmid, and an Mx2-expressing vector using polyethyleneimine (PolySciences).
Primary CD4+ T-cells were isolated from human blood by Ficoll-paque gradient centrifugation and negative selection (RosetteSep Human CD4+ T Cells Enrichment Cocktail, StemCell Technologies). Cells were activated with phytohemagglutinin (PHA-P, Sigma, 5μg/ml) or anti-CD3/CD28 beads (Dynabeads Human T-Activator CD3/CD28, Gibco) for 48h, cultured in the presence of IL-2 (50U/ml, Peprotech) and treated with, or without, IFNα for 24h.
Primary macrophages were differentiated from fresh human peripheral blood mononuclear cells. Cells were plated in serum free medium for 3h at 37°C, the supernatant with non-adherent cells was discarded and adherent monocytes were cultured in RPMI with 10%FCS, 1% L-glutamine and GM-CSF (100ng/ml, Peprotech) for 6 days and treated with, or without IFNα for 24h.
Viruses
All viruses were generated by transfection in 293T cells using polyethylenimine (PolySciences). For the GFP-reporter proviral plasmids; HIV-1NL4-3 dEnv-GFP (WT, G89V, A92E and G94D CA mutants)25, NHGCapNM (WT and N57S CA mutant)19, pLai3ΔenvGFP (WT and N74D CA mutant)14, SIVmac dEnv-GFP, SIVagmTan dEnv-GFP, SIVagmSab dEnv-GFP, HIV-2ROD dEnv-GFP 26, 10 μg of proviral plasmid was co-transfected with 1 μg of VSV-G expression plasmid. For HIV-1 (in Fig. 1, 2a), MLV, EIAV and FIV, three-plasmid vector systems were also used to generate GFP reporter viruses, whereby 5 μg of GagPol, 5 μg of packageable genome and 1 μg of VSV-G expression plasmids 27,28 were co-transfected. Plasmids containing full-length replication-competent ‘transmitted founder’ HIV-1 proviruses (pTRJO.c/2851 and pREJO.c/2864)29 were obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH from Dr. John Kappes and Dr. Christina Ochsenbauer. At 48 hours after transfection, viral supernatants were harvested and their infectivity was determined using MT4 or GHOST target cells.
Infection assays
Single-cycle infectivity in HOS, K562, THP-1 and U937 cell lines was measured in cells seeded in 96 well plates at 5 × 103 cells/well and inoculated with serial-dilutions of VSV-G pseudotyped GFP reporter viruses in the presence of 5μg/ml polybrene. For replication competent primary HIV-1 strains, GHOST-R5 cells, which contain an HIV-2 LTR-GFP reporter construct, were used. Two days post-infection, cells were trypsinized (adherent cells only), and fixed in 2% paraformaldehyde. In some experiments, cells were pre-treated with IFNα (Sigma-Aldrich) for 24 hours, re-plated, followed by inoculation with GFP reporter viruses.
For experiments in which infection of dividing and non-dividing cells was compared, single cell clones of K562 and HOS cells transduced with Mx2-expressing or empty vector (CSIB in the case of K562, SCRPSY in the case of HOS) were seeded at 3 × 104 cells/well in 48 well plates. Cells were treated with aphidicolin (1μg/ml, Sigma-Aldrich) or an equivalent volume of DMSO alone for 24 hours prior to infection with HIV-1 or MLV GFP-reporter viruses.
For spreading replication assays, empty vector or Mx2-expressing HIV-1 (CSIB) transduced GHOST(X4) cell lines (which contain an HIV-2 LTR-GFP indicator gene) were seeded at 1 × 105 cells in a 6 well plate. Thereafter they were inoculated with HIV-1NL4-3 at an MOI of 0.01. Cells were split at a 1:4 dilution, and the percentage of infected (GFP-expressing) cells measured, every two days until the empty vector cultures died due to HIV-1-induced cytopathic effects. In all infection assays, infected cells (%GFP positive of viable cells) were enumerated by FACS analysis using a CyFlow cytometer (Partec) coupled to a Hypercyte Autosampler (Intellicyt). For cells transduced with the SCRPSY vector, the percentage of infected cells was determined as the percentage of RFP/GFP double positive cells in the total RFP-positive population.
RNA interference
Lentiviral Mx2-specific shRNA (Target sequence: 5′-TCG CTA TTC CTG GCT GCT TCA AGA GCA GA-3′), or scrambled negative control shRNA, pGFP-C-shLenti expression plasmids (OriGene) were digested with Xba1 and Bsu361 to excise the CMV promotor and GFP. Virus stocks for transducing the modified shRNA-expression vectors were generated by co-transfection of 293T cells with VSV-G and HIV-1NL4-3 GagPol expression plasmids. THP-1 and HOS cells were transduced and selected in 1μg/ml and 2.5μg/ml puromycin, respectively. Puromycin-selected cells were treated with IFNα for 24 hours prior to assessment of Mx2 expression by western blotting and inoculation with GFP reporter viruses.
Measurement of HIV-1 DNA species in infected cells
For analysis of HIV-1 reverse transcription products and 2-LTR circles in infected cells, 1×105 HOS or K562 cells were seeded in 24-well culture plates and infected with a VSV-G pseudotyped HIV-1 reporter virus (CCGW), which is derived from CSGW but encodes a GFP reporter under the control of CMV promoter. The virus inoculum was pretreated with 20 U/ml RNase-free DNase I (Roche) for 1 hour at 37 °C in the presence of 6 mM MgCl2. In some experiments, cells were pretreated with IFN-α for 24 hours, or were treated with nevirapine starting at the time of infection. Cells were collected at 2, 12, and 24 hours post-infection, washed with 1x phosphate-buffered saline and total DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen). The resulting DNA samples were used as template for quantitative real-time PCR (qPCR) using FastStart Universal SYBR Green Master Mix (Roche) and ABI 7500 Fast PCR system. The primer pairs used in this study are as follows: for GFP (late reverse transcription products): Forward: 5′ AAGTTCATCTGCACCACCGGCAA Reverse: 5′ TGCACGCCGTAGGTCAGG; for 2-LTR circles: Forward: 5′ GACTCTGGTAACTAGAGATCCCTC Reverse: 5′ TTGGGAGTGAATTAGCCCTTCCA
Western blotting
Cell suspensions were normalized for cell number, lysed in SDS sample buffer, separated by electrophoresis on NuPage 4–12% Bis-Tris Gels (Novex) and blotted onto nitrocellulose membranes (GE Healthcare). Membranes were incubated with rabbit anti-Mx2 (Novus Biologicals) and mouse anti-tubulin (Sigma) antibodies, followed by incubation with goat anti-rabbit IRDye® 800CW and goat anti-mouse IRDye® 680RD, respectively (LI-COR Biosciences). A LI-COR Odyssey scanner was used to detect and quantify fluorescent signals.
Microscopy
HOS cells expressing N-terminally HA-tagged Mx2 and Mx1 were seeded onto 24 well gelatin coated glass bottomed dishes (MatTek) and immunostained using a combination of anti-HA (Covance) and anti-Nup98 (Cell Signaling Technology) antibodies followed by goat-anti-mouse Alexa488 and goat-anti-rabbit Alexa594 secondary antibodies (Molecular Probes). Cells were visualized by deconvolution microscopy as described previously30. Image generation and co-localization analysis and were completed with the SoftWorx software suite (Applied Precision). Pearson’s Coefficient values were derived for Mx2 or Mx1 (as a control) and Nup98 by analysis of optical sections coincident with the dorsal nuclear surface, for 6–10 individual cells.
Acknowledgments
We thank members of The Rockefeller University Genomics Resource Center for assistance with the microarray experiments and members of the Bieniasz laboratory for discussion and advice. This work was supported by grants from the NIH; R37AI64003 (to P.D.B.), R01AI078788 (to T.H.) R01AI100720 (to M.Y.) AI091707 to C.M.R., AI057158 (to I. Lipkin, Northeast Biodefense Center, subcontract to C.M.R.), DK095031 to J.W.S., the Greenberg Medical Research Institute and the Starr Foundation (C.M.R.) and by the Howard Hughes Medical Institute.
Footnotes
Author contributions
M.K., S.Y., J.B., S.B.K., T.Z. and S.J.W., designed and executed the experiments and analyzed the data. J.W.S., C.M.R. provided an interferon stimulated gene library and advice. M.Y. provided reagents and advice. T.H. provided reagents and advice and supervised the work. P.D.B. conceived the study, supervised the work and wrote the paper, with additional input from all authors.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Ho DD, et al. Recombinant human interferon alfa-A suppresses HTLV-III replication in vitro. Lancet. 1985;1:602–604. doi: 10.1016/s0140-6736(85)92144-0. [DOI] [PubMed] [Google Scholar]
- 2.Neil S, Bieniasz P, et al. Human immunodeficiency virus, restriction factors, and interferon. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2009;29:569–580. doi: 10.1089/jir.2009.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bitzegeio J, Sampias M, Bieniasz PD, Hatziioannou T, et al. Adaptation to the interferon-induced antiviral state by human and simian immunodeficiency viruses. Journal of virology. 2013;87:3549–3560. doi: 10.1128/JVI.03219-12. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 4.Goujon C, et al. Evidence for IFNalpha-induced, SAMHD1-independent inhibitors of early HIV-1 infection. Retrovirology. 2013;10:23. doi: 10.1186/1742-4690-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheehy AM, Gaddis NC, Choi JD, Malim MH, et al. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 6.Neil SJ, Zang T, Bieniasz PD, et al. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 7.Haller O, Staeheli P, Kochs G, et al. Interferon-induced Mx proteins in antiviral host defense. Biochimie. 2007;89:812–818. doi: 10.1016/j.biochi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 8.Schoggins JW, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stremlau M, et al. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 10.Melen K, et al. Human MxB protein, an interferon-alpha-inducible GTPase, contains a nuclear targeting signal and is localized in the heterochromatin region beneath the nuclear envelope. The Journal of biological chemistry. 1996;271:23478–23486. doi: 10.1074/jbc.271.38.23478. [DOI] [PubMed] [Google Scholar]
- 11.King MC, Raposo G, Lemmon MA, et al. Inhibition of nuclear import and cell-cycle progression by mutated forms of the dynamin-like GTPase MxB. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8957–8962. doi: 10.1073/pnas.0403167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goujon C, Malim MH, et al. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. Journal of virology. 2010;84:9254–9266. doi: 10.1128/JVI.00854-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamashita M, Perez O, Hope TJ, Emerman M, et al. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS pathogens. 2007;3:1502–1510. doi: 10.1371/journal.ppat.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamashita M, Emerman M, et al. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. Journal of virology. 2004;78:5670–5678. doi: 10.1128/JVI.78.11.5670-5678.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dismuke DJ, Aiken C, et al. Evidence for a functional link between uncoating of the human immunodeficiency virus type 1 core and nuclear import of the viral preintegration complex. Journal of virology. 2006;80:3712–3720. doi: 10.1128/JVI.80.8.3712-3720.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee K, et al. Flexible use of nuclear import pathways by HIV-1. Cell host & microbe. 2010;7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaller T, et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS pathogens. 2011;7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koh Y, et al. Differential effects of human immunodeficiency virus type 1 capsid and cellular factors nucleoporin 153 and LEDGF/p75 on the efficiency and specificity of viral DNA integration. Journal of virology. 2013;87:648–658. doi: 10.1128/JVI.01148-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rihn SJ, et al. Extreme genetic fragility of the HIV-1 capsid. PLoS pathogens. 2013;9:e1003461. doi: 10.1371/journal.ppat.1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sokolskaja E, Sayah DM, Luban J, et al. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. Journal of virology. 2004;78:12800–12808. doi: 10.1128/JVI.78.23.12800-12808.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoye JP. Fv1, the mouse retrovirus resistance gene. Rev Sci Tech. 1998;17:269–277. doi: 10.20506/rst.17.1.1080. [DOI] [PubMed] [Google Scholar]
- 22.Yamashita M, Emerman M, et al. Cellular restriction targeting viral capsids perturbs human immunodeficiency virus type 1 infection of nondividing cells. Journal of virology. 2009;83:9835–9843. doi: 10.1128/JVI.01084-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Iaco A, et al. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology. 2013;10:20. doi: 10.1186/1742-4690-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bainbridge JW, et al. In vivo gene transfer to the mouse eye using an HIV-based lentiviral vector; efficient long-term transduction of corneal endothelium and retinal pigment epithelium. Gene therapy. 2001;8:1665–1668. doi: 10.1038/sj.gt.3301574. [DOI] [PubMed] [Google Scholar]
- 25.Hatziioannou T, Cowan S, Von Schwedler UK, Sundquist WI, Bieniasz PD, et al. Species-specific tropism determinants in the human immunodeficiency virus type 1 capsid. Journal of virology. 2004;78:6005–6012. doi: 10.1128/JVI.78.11.6005-6012.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatziioannou T, Cowan S, Goff SP, Bieniasz PD, Towers GJ, et al. Restriction of multiple divergent retroviruses by Lv1 and Ref1. The EMBO journal. 2003;22:385–394. doi: 10.1093/emboj/cdg042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitrophanous K, et al. Stable gene transfer to the nervous system using a non-primate lentiviral vector. Gene therapy. 1999;6:1808–1818. doi: 10.1038/sj.gt.3301023. [DOI] [PubMed] [Google Scholar]
- 28.Kemler I, Barraza R, Poeschla EM. Mapping the encapsidation determinants of feline immunodeficiency virus. Journal of virology. 2002;76:11889–11903. doi: 10.1128/JVI.76.23.11889-11903.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ochsenbauer C, et al. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. Journal of virology. 2012;86:2715–2728. doi: 10.1128/JVI.06157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jouvenet N, et al. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS biology. 2006;4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]