

Abstract
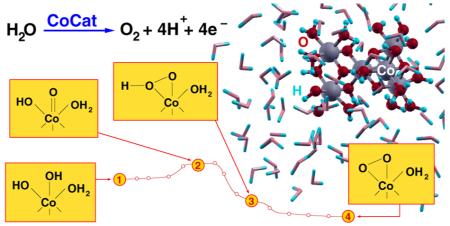
The in-depth understanding of the molecular mechanisms regulating the water oxidation catalysis is of key relevance for the rationalization and the design of efficient oxygen evolution catalysts based on earth-abundant transition metals. Performing ab initio DFT+U molecular dynamics calculations of cluster models in explicit water solution, we provide insight into the pathways for oxygen evolution of a cobalt-based catalyst (CoCat). The fast motion of protons at the CoCat/water interface and the occurrence of cubane-like Co-oxo units at the catalyst boundaries are the keys to unlock the fast formation of O–O bonds. Along the resulting pathways, we identified the formation of Co(IV)-oxyl species as the driving ingredient for the activation of the catalytic mechanism, followed by their geminal coupling with O atoms coordinated by the same Co. Concurrent nucleophilic attack of water molecules coming directly from the water solution is discouraged by high activation barriers. The achieved results suggest also interesting similarities between the CoCat and the Mn4Ca-oxo oxygen evolving complex of photosystem II.
INTRODUCTION
Photosynthetic processes represent the most important source of energy produced by biological systems, which have been designed by evolution to capture sunlight very efficiently and convert it into chemical energy, i.e., organic molecules.1,2 A crucial role in this task is played by small clusters containing transition metals (TM), the earth abundant Mn and Fe, embedded into a complex biochemical environment. In detail, an oxygen evolving complex (OEC), contained into the photosystem (II) (PS(II)), promotes the oxidation of water molecules to dioxygen, protons and electrons, the former released in the air, and the latter ones stored into chemical fuel (e.g., oligo- and polysaccharides) by further biochemical systems. Inspired by such natural processes, the goal of artificial photosynthesis is to develop simplified but still efficient routes to generate chemical fuels (e.g., H2) directly from sunlight by means of “artificial leafs”,3,4 i.e., technologically relevant and low-cost photoelectrolytic cells performing photosynthetic tasks.
Several heterogeneous and homogeneous TM-based oxygen evolving catalysts,5 containing different metal–oxygen cores (Mn,6–8 Ni,9,10 Ru,7,11,12 Co13–16), have been recently proposed. An inorganic cobalt-based catalyst film (CoCat) has attracted much interest because it is efficient at neutral pH, is very stable (self-repairing) under working conditions, operates close to the Nernstian potentials for the H2O/O2 half-cell reaction, and is self-assembled from low-cost materials.14,17 A fine comparison between X-ray absorption spectroscopy (XAS) measurements18–20 and theoretical calculations21,22 revealed that the CoCat structure is likely formed by several Co(III)O6 octahedra assembled in incomplete and complete cubane units, building blocks of the amorphous extended system (an example is shown in Figure 1). The formation of active catalyst films is not affected by the presence of other species, e.g., Cl−, PO43−, K+, Li+ ions, depending upon the composition of the starting solution.23 Moreover, theoretical results suggested that terminal O atoms, whose occurrence at the CoCat boundaries is expected for all of the catalyst models proposed so far,11,18–20,24 are likely to form low-barrier H bonds, thus enhancing the proton mobility at the CoCat/water interface.21 Finally, a first mechanistic study,24 based on electrokinetic experiments and isotopic labeling of O atoms, indicated the extrusion of O atoms from the catalyst and a predominant Co(III) or higher oxidation state during the O2 evolution process. An EPR fingerprint of Co(IV) species has been indeed estimated to arise from 3% of the cobalt centers in the catalyst film subjected to prolonged electrolysis.25 The above electrochemical measurements suggested also that even if the oxygen evolution reaction ideally requires the global transfer of four protons and four electrons, in agreement with the stoichiometric balance
| (1) |
it is actually characterized by two main events occurring at the CoCat/water interface, namely, a one-electron, one-proton equilibrium step, identified as
| (2) |
followed by a single, unspecified chemical step.24 However, it has to be noted that the extended, nonstoichiometric nature of the CoCat can lead to oxidation state assignments which may be poorly representative of electronic structure, since the charge may be delocalized between the Co and O atoms comprising the active site.24
Figure 1.

Upper panel: Equilibrium geometry of the H saturated Co6O23H28 c1 cluster. The “2” label indicates one of the protonated di-μ2-O sites, also enclosed into a yellow circle. The “3” label indicates one of the nonprotonated di-μ3-O sites, also enclosed into a green circle. The “T” label indicates one of the terminal O atoms, also enclosed into a blue circle. A dashed orange line divides a complete cubane unit (right-hand side) from an incomplete one (left-hand side). Lower panel: Total (black curve) and Projected on O 2p (red curve) and Co 3d (blue curve) atomic orbitals DOS of the Co6O23H28 c1 cluster. A 0.02 Ry (0.27 eV) Gaussian broadening of Kohn–Sham eigenvalues has been applied to simulate the DOS of the amorphous CoCat. A zero energy value has been assigned to the valence band maximum.
Founding on such preliminary investigations of the structural motifs, dynamical and mechanistic properties characterizing the catalyst film, we present here the results of a careful study of the reaction mechanism for oxygen evolution promoted by the CoCat, performed by means of ab initio simulations. These first bricks used to pave the pathways of the complex water oxidation and oxygen evolution processes promoted by metal-oxo cores are of crucial importance in the understanding of the potentialities of the CoCat, compared to other promising materials. Among several results and indications, discussed in detail in the following sections, we would focus on the following main achievements of the present study: (i) The fast H+ mobility at the CoCat/water interface is responsible for an optimal distribution of terminal Co(III)–OH groups which favors the localization at these sites of injected holes (left back by the removal of electrons from the catalyst due to the applied external bias). Such a localization is preferred in the case of complete cubane units. (ii) The oxygen evolution process starts with the release of a proton from one of such terminal Co–OH sites, possibly favored by proton-acceptor species in solution, which leads to the formation of a Co(IV)═O• oxyl radical, in agreement with the above equilibrium step.24 (iii) The coupling of Co═O radicals with geminal (i.e., bonded to the same Co atom) Co–OH or Co-μO–Co groups to form hydroperoxo and peroxo intermediates represents the irreversible chemical step of the process, a nucleophilic attack of an external water molecule to the Co═O species being discouraged by a high-energy barrier.
THEORETICAL METHODS
The reaction pathways for oxygen evolution promoted by CoCat have been investigated by using ab initio molecular dynamics (AIMD) simulations26 in explicit water solution, together with static geometry optimizations, both based on Hubbard U corrected density functional theory (DFT+U) in a restricted open-shell Kohn–Sham approach, as implemented in the Quantum ESPRESSO package.27 Co–O molecular clusters have been used to simulate the properties of the interface between water and the amorphous CoCat. The clusters have been saturated by H atoms and surrounded by a quite large amount of water molecules within periodic boundary conditions. Such a setup has been proven to provide an accurate description of the structural properties of the building blocks of the catalyst, as detailed in our previous contribution.21 The clusters have been built in order to satisfy several constraints: (i) X-ray absorption measurements indicate that all the Co atoms are surrounded by six O atoms.18–20 This implies the occurrence of several terminal O atoms (labeled “T” in Figure 1) at the CoCat/water interface. Terminal Co–O species, as well as Co–OH3 ones, are unstable in resting conditions of the catalyst. The former are supposed to be first intermediates of the oxygen evolution reaction24 and to be formed only when the catalyst undergoes a high positive potential. The latter have been neither observed nor suggested by previous theoretical calculations.21,22 Co–OH and Co–OH2 species have been therefore considered to be present at the catalyst/water interface, in agreement with the catalyst models mentioned above.4,24 (ii) A fine comparison between XAS measurements and theoretical calculations21 indicates that O atoms placed as μ2-O bridges between Co(III) ions (labeled “2” in Figure 1) are likely to be protonated under working conditions of the CoCat, at variance with the μ3-O bridges (labeled “3” in Figure 1). (iii) H atoms play actually a role in the saturation of the CoCat, as confirmed by the fact that active catalyst films are obtained from a starting solution containing K+ and Cl− only as counterions, both unable to act as ligands at the catalyst boundaries.23 This is not a prerogative of Co-based catalysts: the properties of a similar catalyst, containing Mn(IV), O, and H atoms only, have been reported.8 (iv) The total number of H atoms distributed at the cluster/water interface has been chosen to ensure a Co(III) oxidation state, indicated as the resting state of Co atoms in the catalyst by XANES and EPR measurements.24,25 Simulations with different numbers of H atoms have been also performed in order to show that our results do not depend (within reasonable limits) on such a number of H atoms; the corresponding results are discussed in the Supporting Information. (v) Co(III)–OH2 and Co(III)–OH species at the CoCat boundaries have been detected by preliminary IR measurements.28
A Hubbard U correction has been applied to the Hamiltonian. Such an approach has proven successful in improving the DFT description of electron correlation in TM oxides and related compounds, in particular when the localization of charge carriers (electrons and holes) is involved.9,29,30 In detail, a U correction for the 3d electrons of Co atoms was set to the average value of 5.9 eV calculated by using a self-consistent linear response approach described in refs 31 and 32, which was applied to all the nonequivalent Co atoms belonging to the clusters. The calculated U values are in agreement with similar calculations performed in the case of the LiCoO2 crystal;33 the low spread found (5.9 ± 0.2 eV) is not expected to affect the achieved results. In addition to the Co 3d correction, an Hubbard U correction was applied also to the 2p electrons of O atoms, since Coulomb interactions between p electrons of the ligands have to be considered comparable to those between the d electrons of the metals.34,35 The strong coupling between Co 3d and O 2p shells can indeed induce a spurious charge transfer from O atoms to metal atoms when the U correction is applied only to the metal d shell.30 For O atoms, a U value of 5.9 eV, identical by chance to the Co 3d value, has been estimated by founding on experimental results.9,30 This approach has proven to be useful to reproduce the strong p–d coupling reported on the ground of XANES33 and PES36 investigations in the case of the LiCoO2 crystal,21 also found in the case of the CoCat models, as shown by the total and projected DOS plots in Figure 1. Further, in-depth analyses of the effects of the U correction on several Co- and O-based systems, also including the joint effects of the application of U and dispersion corrections, have been performed to ensure a complete reliability of our theoretical setup. The results are reported in the Supporting Information.
Several DFT+U-based AIMD simulations, employing several CoCat molecular models differing for their protonation states and other details, have been carried out to investigate the properties of our catalytic complexes as well as to assess the reliability of our results with respect to the details of our computational protocols. In these simulations, electrons (e−) have been gradually removed from the systems to simulate the behavior of a CoCat-based electrolytic cell. All the simulations have been performed without imposing any external constraint. In detail, the H-saturated c1 (Co6O23) and c2 (Co7O24) clusters (see Figures 1 and 2, respectively) have been accommodated into periodic cubic supercells and surrounded by a fairly large amount of water molecules (an example is shown in Figure 2). AIMD simulations have been performed by using the Γ point for the k-point sampling of the Brillouin zone, ultrasoft pseudopotentials,37 and the Hubbard U corrected PBE exchange–correlation functional.38 Kohn–Sham orbitals have been expanded into plane waves up to energy cutoffs of 40 and 320 Ry for the wave functions and the charge density, respectively, in order to achieve satisfactorily converged results. Such very strict convergence criteria on the plane wave basis set as well as the inclusion of Co semicore 3s and 3p shells among the valence electrons have proven to be necessary in order to estimate with high-accuracy interatomic distances.21 Car–Parrinello equations of motion have been integrated using a time step of 0.073 fs (3 au). Parrinello–Rahman NPT simulations39 were performed until the internal pressure reached values close to ambient pressure, the cubic simulation cells fluctuating around average values of 24.4 au3 (c1) and 25.2 au3 (c2). Then NVT simulations were carried on for about 5 ps using the Nosé-Hoover thermostat at 300 K, before starting the removal of electrons. Such a removal of electrons (i. e., ignition of oxidizing holes) has been carried on at a rate of one e− per ps, by using an “on-the-fly” procedure which allows one to change the total number of electrons without affecting the nuclear velocities. Since in our simulations the O2 molecule is initially formed in its highest energy singlet state, an instantaneous switch of the total magnetization of the system to triplet is performed to observe the O2 release preserving the nuclear velocities along the AIMD trajectories. To estimate the energy barriers along the oxygen evolution pathways, we calculate the minimum energy path connecting reactants to products by using a nudged elastic band (NEB) scheme40,41 at the DFT+U level of theory. A detailed analysis of the electronic structure has been also performed on selected snapshots along the reaction path using the DFT+U approach.
Figure 2.
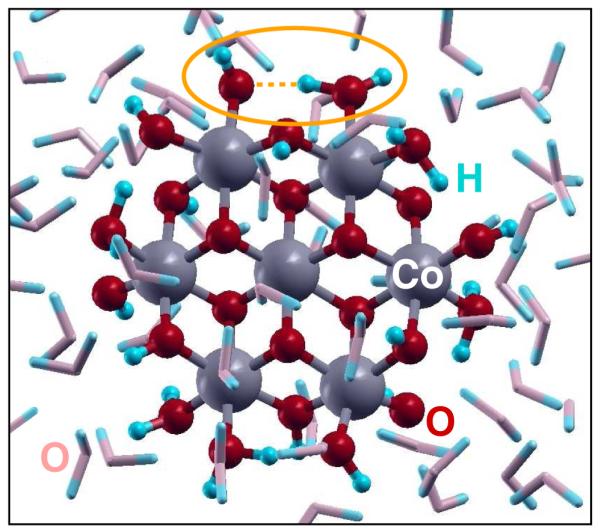
Snapshot from the AIMD simulation of the H saturated Co7O24H24 c2 cluster in water solution. An orange ellipse identifies one of the H–O–H⋯O–H structures undergoing a fast proton exchange during the AIMD simulation (see the text for details).
The electrochemical properties of the above CoCat models have been simulated by using a robust technique, generally employed to estimate energy levels of dopant and defects in semiconductors,42 which has been already extended to the investigation of catalytic processes like, e.g., the O2 photo-reduction at the TiO2/water interface.43 In this approach, first, the formation energy Ωf of a q-charged species M, embedded in a dielectric host matrix H, has to be estimated, which is defined as
| (3) |
where E[H] and E[Mq] are the total energies of supercells containing the undoped host matrix, and the dopant agent (molecule, metal atom) surrounded by the host, respectively; nM is the number of dopant agents inserted in (or subtracted by) the defected supercell and μM is the chemical potential of the same species; εF is the Fermi level of the system, corresponding to the chemical potential of electrons, and referenced to εVBM, i. e., the energy of the highest occupied electronic level, or the maximum of the valence band (VBM) of the host. In the present case, we have considered the CoCat models as dopant agents and the water solution as host matrix. The calculation of formation energies permits to estimate transition energy levels, εq/q+1, corresponding to the position of the Fermi level where the q and q + 1 charge states of the dopant agent have the same formation energy, i.e., the species Mq and Mq+1 are in equilibrium. More specifically, the εq/q+1 value is an estimate of the chemical potential at which electrons can be exchanged with an external reservoir in contact with the system. When the CoCat acts as an electrode, such a chemical potential corresponds to the applied external bias up to an additive constant. Its alignment to the standard hydrogen electrode (SHE) has been achieved by using the Cu(I)/Cu(II), Fe(II)/Fe(III), and Co(II)/Co(III) oxidation potentials vs SHE as a reference, as detailed in the Supporting Information.
RESULTS AND DISCUSSION
Catalyst Activation and Reactivity
Unconstrained AIMD simulations provide an effective tool to investigate room temperature chemical processes occurring within a rather fast time scale (1–10 ps) and without crossing very high potential energy barriers (few tenths of eV). On the basis of our previous results,21 which closely agree with XAS measurements, we have performed such simulations by considering two different configurations of fully solvated CoCat cluster models, labeled c1 and c2 in Figure 3. Both clusters have been considered to simulate the behavior of CoCat samples exposed to positive electrode potentials. The corresponding oxygen evolving pathways observed along the dynamics are sketched in Figure 3.
Figure 3.

Reaction paths for oxygen evolution promoted by the c1 and c2 CoCat models. Details of the path A, including Co–O and O–O distances and spin-switching effects related to the formation of the O2 molecule, are shown in the lower panel.
Preliminarily, some special features of proton motion at the CoCat/water interface should be considered. All the atomistic models proposed for the building blocks of the CoCat are expected to contain terminal O atoms, labeled “T” in Figure 1, saturated by one or two H atoms (OHx in Figure 3). We have shown in a previous contribution that such T atoms tend to exchange protons among them (either by means of direct exchange, or through double proton exchange involving a nearby water molecule) quite frequently.21 In particular, Co–Co nearest-neighbors carrying pairs of parallel Co–O bonds (see Figure 2) are expected to be present in all of the CoCat structures proposed since the first catalyst synthesis.11,18–20,22,24 The occurrence of such Co–Co pairs permits the formation of stable H–O–H⋯O–H structures (an example is shown in Figure 2) characterized by low-barrier H bonds.21 Such a proton mobility plays a role in the present AIMD simulations, where the effects of positive electrode potentials are simulated by removing electrons from the clusters. As a general feature of the AIMD results, the displacement of terminal Co–OH groups at the cluster/water interface results indeed to be strongly correlated with the displacement of charge density within the clusters. In Figure 3, the high proton mobility is schematically represented by the OHx notation.
Let us focus now on the most significant features of the CoCat-promoted oxygen evolution process, sketched in Figure 3. First, all the processes leading to the formation of an O2 molecule are triggered by the removal of four electrons from the clusters (we discuss all the implications of such a procedure in terms of the oxidation potential vs SHE of the CoCat model in the next section). The removal of electrons induces the release of a proton from one of the Co–OH terminal groups with the ensuing formation of a Co═O oxyl species. The formation of a metal-oxyl M═O species agrees with eq 2 and with the reaction scheme suggested for the CoCat4,24 as well as with different reaction pathways suggested for other Mn- or Ru-based metal–oxygen complexes.7,44,45 A second significant feature is that the Co═O group evolves always toward the formation of an O–O bond, crucial step of the oxygen evolution process, by means of a geminal coupling with an O atom, either inner (paths B and D in Figure 3) or terminal (paths A and C in Figure 3), bonded to the same Co. This finding is in agreement with the results of isotopic labeling experiments indicating the significant extraction of O atoms from the catalyst during the oxygen evolution process.24 The possible occurrence of a different mechanism, involving the nucleophilic attack of an external water molecule to the Co═O intermediate, has been explored and ruled out on the ground of results discussed in detail below. A more detailed analysis of path A, involving both the time scale and the most significant interatomic distances related to the reaction, is reported in the lower panel of Figure 3 to illustrate the above features in a more quantitative way. Apart from the proton motion at the cluster/water interface, no significant structural changes were observed before the removal of the 4th electron from the c1 cluster. 0.8 fs after such removal, one of the terminal Co–OH groups of the complete cubane unit releases its H+ into the solution leaving back a Co═O oxyl radical. The Co═O species is not stable and evolves after 1.1 ps toward a Co(OOH) hydroperoxo intermediate by means of geminal coupling with a neighboring terminal Co–OH group. The Co(OOH) releases a further H+ after 1.4 fs, thus forming a Co(O2) peroxo intermediate, which after 1.8 ps breaks one of the Co–O bonds resulting in a Co–O–O superoxo group. This last configuration is highly stable (up to 10 ps dynamics) unless the total spin state of the system is switched from the restricted open shell framework to an unrestricted open shell framework in a triplet configuration. This is sufficient to promote an almost immediate breaking of the Co–O bond and the release of O2. The analysis of such a process will be completed by discussing the electronic properties of all of the above intermediates in a following section. Further details on all the AIMD simulations underlying the reaction schemes sketched in Figure 3, including also additional calculations on a small Co4-oxo cluster investigated in a previous theoretical contribution,46 are given in the Supporting Information.
Finally, two details are worth mentioning in a tentative comparison between CoCat and PS(II): As a main difference, in the artificial CoCat system the changes in the proton network occur within the cluster itself and involve a local reshuffling of the protons that can easily jump at water/CoCat interface from one site to another. Other surrounding ligands can, at opposite, participate in this rearrangement in the case of PS(II). On the other hand, it has been recently suggested that in the case of the natural system the oxygen evolution reaction should proceed through an hexa-coordinated TM configuration, in a close similarity between PS(II) and CoCat.47
Oxidation Potential of the Catalyst
The equilibrium process indicated by eq 2 favors the product Co═O when the oxidation potential of the catalyst reaches a certain level (1.18 V vs SHE),24 or, in other words, when a certain amount of electron equivalents is drained from the catalyst, as indicated by an estimate of about 3% of Co(III) atoms oxidized to Co(IV) during the O2 evolution process.24 In the case of the Mn4Ca-oxo core of PS(II), a well-defined, stoichiometric cluster, an accumulation of oxidizing equivalents has been also proposed, although the change in metal oxidation state is supposed to be assisted by a series of proton-coupled electron-transfer steps.1,11 In the present case, four electrons have been removed in all the investigated AIMD simulations before observing the formation of a Co═O oxyl species. As we are modeling an extended electrode by using molecular-like clusters, there are two crucial questions that must be answered to assess the reliability of our model as representative of the electrochemical properties of the CoCat: at what “external” potential the electrons are removed from the cluster, and to what extent the results of AIMD simulations are affected by such a potential level.
In order to answer the first question, we have estimated the oxidation potentials vs SHE at which electrons are removed from the solvated c1 cluster. As introduced in the Theoretical Methods section, the transition energy level related to the removal of one electron from the cluster can be directly compared with the oxidation potential vs SHE of the cluster/solution system. In the case of the solvated c1 cluster, oxidation potentials of 1.07, 1.49, 1.64, and 1.87 V have been calculated at which a first, second, third, and fourth electron are removed from the system, respectively. These oxidation potentials favorably compare with the measured one if it is taken into account that their differences decrease with increasing cluster size. More specifically, when the cluster size increases and approaches macroscopic dimensions, a discrete succession of molecular orbitals of the cluster tends to a continuous density of states (DOS), thus leading to the shrinking of the above calculated potential values.
Regarding the second question, in order to follow the evolution of the oxidation potential across the reaction mechanism as well as to evaluate if its high initial value used in the AIMD simulations introduces artificial effects in the calculations, we have estimated the effect of the external potential on the energy barriers along the path A of Figure 3 after the progressive removal of one, two, three and four electrons from the c1 cluster. The results achieved by using the above introduced NEB formalism are displayed in Figure 4. The removal of the first electron, corresponding to a calculated oxidation potential of 1.07 V vs SHE, does not promote the formation of a Co═O species, as indicated by a quite high 1.7 eV potential energy barrier. Moreover, an overall huge barrier of more than 3 eV is needed to the formation of the hydroperoxo and peroxo species, which are also energetically unfavored with respect to the starting Co–OH species. The removal of a second electron has a 2-fold significant effect: (i) the potential energy barrier to the formation of the Co═O species is lowered to the more reasonable value of 0.7 eV; (ii) the formation of the hydroperoxo and peroxo species becomes largely favored with respect to the starting Co–OH species. Notwithstanding, the formation of a Co═O species is still not observed in the simulated AIMD time upon removal of the second electron. In this regard, we have estimated the lifetime τ of the Co–OH species by using the Arrhenius relation
| (4) |
If we use a pre-exponential factor A of 3000 cm−1, roughly corresponding to the stretching frequency of the O–H bond of the Co–OH group, and an activation energy Ea of 0.7 eV, we obtain a lifetime τ of about 100 μs at 300 K, much larger than the typical duration of AIMD simulations. The removal of two further electrons shifts downward the barrier to the formation of a Co═O species to the value of 0.05 eV, leading to an estimate of τ of about 10 fs at 300 K, fully compatible with the AIMD time scale, and does not alter the shape of the reaction path following the formation of the Co═O intermediate. More specifically, the potential energy barriers characterizing the formation of the hydroperoxo intermediate by geminal coupling of the Co═O species do not depend anymore on the external potential, as clearly shown by the −2, −3, and −4 curves of Figure 4. These results answer to the above second question by showing that at least the first two electrons need to be drained from the catalyst in order to proceed up to the formation of the Co(OO) peroxo and, therefore, in the AIMD simulations, the initial removal of four electrons is required only for inducing a starting oxidation potential level in the system which accelerates the formation of the Co═O oxyl species without altering the reaction mechanism.
Figure 4.

Potential energy curves along the minimum energy path for the formation of a Co(OO) peroxo intermediate after removal of one (brown curve), two (dark red curve), three (red curve), four (orange curve) electrons from the solvated c1 cluster. Step 1: optimized resting conditions of the solvated cluster; Step 2: formation of a Co═O oxyl intermediate; Step 3: formation of a Co(OOH) hydroperoxo intermediate; Step 4: formation of a Co(OO) peroxo species, anticipating the release of an O2 molecule.
Moreover, the calculation of total energies values along all the reaction paths sampled in Figure 4 allows for an estimate of transition levels εq/q+1 across all the curves and for all the reaction intermediates. As such levels are directly connected with the oxidation potential, they provide information on the removal of a third and fourth electrons from the O atoms involved in the formation of the O2 molecule. In this regard, a value of 1.45 V vs SHE has been calculated for the removal of a third electron (i.e., across the −2 and −3 curves in Figure 4) after the formation of the Co(OO) species. This is lower than the 1.49 V value needed to extract the second electron from the Co–OH species that permits the formation of the first Co═O intermediate and, in turn, of the O–O bond. This supports the indication that the two further electrons, needed to the stoichiometric oxidation of two O2− atoms to an O2 molecule, can be actually drained from the catalyst along the reaction path after the formation of the O–O bond. We anticipate that an analysis of the local rearrangements of electronic charge within the cluster, discussed in detail in the next section, shows that the formation of the O–O bond induces the raising of an occupied electronic level, localized on the Co(OO) species, above the Fermi level of the catalyst, thus promoting the removal of the third and fourth electrons.
Electronic Properties of the CoCat along the Oxygen Evolution Pathways
The evolution of electronic properties of the solvated clusters during the AIMD simulations gives further significant indications on the functioning of the CoCat, which complete the above investigation of the structural changes and of the oxidation potential of the catalyst. The removal of electrons from the CoCat models, corresponding to the application of a positive potential to the catalyst, can be also considered as the ignition of positively charged, oxidizing holes in the catalyst, in agreement with the picture of positive and negative charge carriers typical of extended semiconductor systems.48 In general, there is a known tendency of wide band gap metal oxides to the trapping (also referred to as “self-trapping”) of such holes as small polarons in quite localized states which have the substantial character of O 2p atomic orbitals.49 The strong mixing between Co 3d and O 2p orbitals in the case of the CoCat, clearly shown by the DOS plots in Figure 1, suggests a slightly different model where the holes produced by the removed electrons are localized on both Co atoms and on their O nearest neighbors. As already pointed out,24 this implies that the oxidation state of Co atoms involved in the reaction mechanism of oxygen evolution cannot be assigned in a quantitative, conventional way. However, previous XAS,18 EPR,25 and voltammetric24 measurements indicate the Co(III) oxidation state as a lower limit related to the catalyst resting conditions and the Co(IV) state as an upper limit reached by about 3% of the Co atoms during O2 evolution.
In order to provide a better clarification of the Co oxidation states as well as to elucidate how the removal of electrons triggers the formation of the Co═O species, selected snapshots of the path A in Figure 3 have been used again to investigate the electronic properties of the most important intermediates of the process. The achieved results are summarized in Figure 5. Four electrons have been removed from all the solvated clusters. The removal of a first and a second e− from the c1 cluster lefts back two holes accommodated in the cluster HOMO. This orbital is mainly localized on one of the terminal Co–OH groups belonging to the complete cubane unit shown in Figure 1, upper panel. This indicates a tendency to the localization of holes at complete cubane units,50 thus strengthening the suggestion of a major catalytic role of such structural motifs of the CoCat in the oxygen evolution mechanism, together with an interesting parallelism between a complete cubane unit and the Mn-oxo core of PS(II). The removal of two further electrons from the system does not affect the localization of the first two holes: the four holes are indeed accommodated in two nonoverlapping electronic states (configuration A in Figure 5). More specifically, the higher energy one (the HOMO of the cluster) is still localized on the same terminal Co–OH of the complete cubane unit, while the lower energy one (HOMO-1) is mainly localized on the three Co atoms and on the μ3-O atom belonging to the incomplete cubane unit (see Figure 5; compare also the right and left parts of the sticks and balls model of Figure 1). It should be noted that the removal of the first electron pair from the Co(III)–OH group can be considered as equivalent to the formation of a terminal Co(IV)-O•H group, preceding the formation of a Co(IV)═O• oxyl radical. This may be regarded as a refinement of the equilibrium process suggested by eq 2. After the formation of the Co═O• oxyl radical (configuration B in Figure 5), the HOMO-1 remains quite unaffected and always localized on the incomplete unit (not shown in the figure). The HOMO is instead now wholly localized on the Co–O pair, thus fully justifying the assignment of a +4 and −1 valence state to the Co and O atoms of the Co═O species, respectively. The Co═O intermediate evolves then toward the formation of a Co(OOH) hydroperoxo species (configuration C in Figure 5) by means of geminal coupling with a neighboring terminal OH group. In other words, the removal of the first two electrons is sufficient for inducing the formation of a terminal Co(IV)-O•H group on the complete cubane unit as well as for triggering the formation of the Co(IV)═O• species and its evolution toward the formation of the Co(OOH) one.
Figure 5.

∣ψ∣2 plots of the HOMO-1 and HOMO orbitals in selected snapshots of the AIMD trajectory (Path A in Figure 3), related to the following reaction intermediates: (A) terminal Co–OH; (B) Co═O oxyl radical; (C) Co(OOH) hydroperoxo; (D) Co(OO) peroxo; (E) Co-OO superoxo; (F) solvated O2 molecule. The surrounding water molecules have not been displayed for the sake of clarity.
As anticipated in the previous section, the formation of the O–O bond is accompanied by a significant rearrangement of electronic charge inside the cluster which involves, in particular, one of the electron pairs localized on the O atom indicated by a red arrow in Figure 5. In detail, the O–O formation is accompanied by a raising in energy of the molecular orbital hosting such a pair above the HOMO-1 and by the consequent shift of the electron pair to that orbital, i.e., to the incomplete cubane unit, in order to avoid the presence of five electron pairs around the O atom. From the specular point of view, two holes (the third and fourth ones left back by the removal of two further electrons) are transferred from the incomplete cubane unit to the Co(OOH) group belonging to the complete one. The formation of the O–O bond is therefore able to induce the capture and localization of the two holes still needed to the complete oxidation of two O2− atoms, yielding the O2 molecule. The occurrence of this charge-transfer process from the active site of the catalyst to the incomplete cubane unit suggests an extrapolation beyond the limited extension of the investigated models: the active site can be considered indeed as able to exchange further electrons with a close incomplete unit as well as with a reservoir placed at a chemical potential of electrons defined as the Fermi level of the catalyst, dependent on the external bias only.
After its formation, the Co(OOH) group loses its H+ ion and forms a Co(OO) peroxo intermediate (configuration D in Figure 5). The features of an O2 antibonding LUMO orbital start to emerge, still partially involving the Co atom. Such features become clearly appreciable when the Co-OO superoxo group replaces the Co(OO) peroxo one (configuration E in Figure 5) and the Co atom recovers its initial Co(III) oxidation state. Finally, all the holes left back in the catalyst are replenished by electrons coming from the two O atoms forming the O2 molecule (compare A and F configurations in Figure 5).
In summary, the CoCat induces the localization of holes (left back by the removal of electrons) at the boundaries between the catalyst and the water solution and provides a path along which they are physically transferred to the O–O bond of the oxygen molecule, where they are irreversibly stored as the O2 antibonding LUMO orbitals. The O atoms involved in the formation of the molecule are therefore formally oxidized from −2 to 0.
Geminal Coupling vs Nucleophilic Attack
All the above-discussed results support the idea that the formation of the O–O bond by geminal coupling represents the suggested chemical step in the catalysis of oxygen evolution promoted by the CoCat. We provide here a further validation of such findings by performing a comparison between two alternative mechanisms which could be expected to compete after the formation of the first Co═O intermediate: the geminal coupling of the Co═O species with terminal and internal O atoms or the nucleophilic attack of external H2O molecules to the same species. We have therefore performed further NEB calculations to shed more light on the potential energy landscape surrounding the Co═O oxyl radicals formed in both the c1 and c2 models under removal of electrons. In the case of the c1 cluster, the left branch of the related curve in Figure 6 shows that the Co═O intermediate is not stable along the explored path, and spontaneously evolve toward the formation of a Co(OOH) hydroperoxo species, followed by the formation of a Co(O2) peroxo species, with a large (2.7 eV) energy gain. The right branch of the same curve is characterized instead by a significant potential energy barrier (1.0 eV) which acts to prevent the nucleophilic attack of a water molecule to the Co═O intermediate. The same Co═O species is characterized by slightly different features in the case of the c2 cluster. It is a more stable intermediate of the O2 evolution reaction, separated by a 0.25 eV energy barrier from the Co(OOH) hydroperoxo intermediate formed by geminal coupling with a neighboring Co–OH species. A significant 1.0 eV barrier characterizes again the right branch of the curve, thus hindering the nucleophilic attack of an external water molecule to the Co═O oxyl radical.
Figure 6.

Potential energy curves along the minimum energy path for oxygen evolution for different reaction pathways, all starting with Co═O oxyl species (framed in color) and leading to the formation of O–O bonds. The upper red (lower blue) curve is related to the c1 (c2) cluster model. Figure insets schematically represent reaction intermediates.
Further AIMD simulations, in which external H2O molecules have been forced to the formation of an O–O bond with the oxyl radical, have been also performed and are discussed in detail in the Supporting Information. However, on the ground of the above NEB results and of the previously discussed AIMD simulations, in which a spontaneous nucleophilic attack has never been observed, the geminal coupling can be considered as largely favored in the case of Co═O species formed at the boundaries of complete and incomplete cubane units. Proton accepting electrolytes, as PO43−, are suggested to allow the catalyst for a higher activity for the oxygen evolution reaction, within an optimal 7–9 pH range.14,24,51 This may also suggest a direct role for a stronger nucleophilic species like OH−, which may reduce the high 1 eV potential energy barrier calculated in the case of a water molecule approaching the Co═O oxyl radical. We have therefore performed a parallel calculation of the right branch curve related to the c2 cluster, involving the nucleophilic attack of an hydroxide anion to the stable Co═O species. Not even an appreciable lowering of the barrier (0.3 vs 1.0 eV), seems sufficient to overrule the indications favoring the geminal coupling with respect to the nucleophilic attack, at least when the CoCat operates within its most favorable 7–9 pH range, and a low concentration of OH− is expected.
Finally, the difference between the stability and evolution of the Co═O species formed at the boundaries of a complete cubane unit, as in the case of the c1 cluster, or at the boundaries of an incomplete unit, as in the case of the c2 cluster, indicates that there can be room for improvement of the catalyst performance. Close similarities have been suggested, on the ground of X-ray absorption and diffraction measurements, between the CoCat structure and the layered LiCoO2 and heterogenite CoO(OH) crystals, both characterized by extended Co–O sheets formed by connecting incomplete cubane units.52–54 Complete cubane units may be considered indeed as out-of-plane defects of such sheets,20,22 possibly behaving as the most active catalytic centers of the CoCat structure. This suggests, in turn, that synthetic routes of the catalyst aimed to raise the concentration of complete cubane units may improve the CoCat activity.
Revised Reaction Mechanism
On the grounds of all the findings discussed above, we propose a revision of the previously suggested reaction mechanism,24 in which a one proton, one electron equilibrium illustrated by eq 2 is followed by a single chemical step. Our results indicate that the removal of two electrons from a terminal Co–OH species actually precedes the release of protons and is sufficient to promote the formation of a Co═O species, first intermediate of the oxygen evolution reaction. This equilibrium process, following the localization of the injected holes and strongly dependent on the applied external potential, can be formalized by a revised equation
| (5) |
corresponding to the evolution of the system from the configuration A to the configuration B shown in Figure 5.
Moreover, the formation of a Co═O species is followed by a single chemical step, in agreement with the results cited above,24 whose barrier does not depend significantly on the external potential, as shown in Figure 4. Such a step is identified as the geminal coupling of the [Co(IV)═O•] species with one of the neighboring O atoms, resulting in the formation of an O–O bond belonging to a hydroperoxo Co(IV)-OOH or to a peroxo Co(IV)-OO species. Such intermediates are able to host the remaining two holes and to evolve spontaneously toward the final release of an O2 molecule. Finally, although four electrons are needed for the final release of an oxygen molecule, the proposed reaction mechanism implies that at least two electrons play an active role in the formation of the O–O bond. Due to the subtle processes driving the transfer of electrons between active sites and electrode, a firm indication of the number of electrons actually involved in the formation of the bond is rather problematic and deserves further investigation.
CONCLUSIONS
The oxygen evolution reaction promoted by a cobalt-based catalyst (CoCat) has been investigated by means of ab initio DFT+U based molecular dynamics simulations. Several simulations, accompanied by in-depth analyses of energetic and electronic features, have been performed in order to provide a proposal of the reaction mechanism. The achieved results can be framed into a sound, coherent picture of the catalyst activity, which suggests interesting similarities with the oxygen evolving complex of photosystem (II) and gives indications for designing improved catalyst architectures. In the proposed mechanism, the removal of electrons from the catalyst corresponds to the ignition of positively charged, oxidizing holes in the catalyst (in agreement with the picture of positive and negative charge carriers typical of solid-state extended systems). The fast mobility of protons at the CoCat/water interface leads to an optimal distribution of terminal Co(III)–OH species which favors the localization of holes at such sites, thus playing a role similar to the intramolecular proton-coupled electron-transfer processes proposed in the case of the OEC. Vertexes of complete cubane-like units at the CoCat/water interface are preferred sites for hole localization, thus emerging as the most active sites of the CoCat and indicating a possible route to the design of more active catalysts. Co(IV)=O• oxyl radicals, forming in an equilibrium process involving terminal Co(IV)-O•H species when the catalyst reaches a sufficient positive potential, represent the first intermediate in the oxygen evolution process which spontaneously leads to the formation of O–O bonds by means of a geminal coupling with O atoms bonded to the same Co. Concurrent nucleophilic attack processes of external water molecules to the oxyl radicals seem to be discouraged by high potential energy barriers.
Supplementary Material
Assessment of theoretical methods (LiCoO2 properties, [Co(II)(H2O)6] properties, liquid water properties, structural and dynamical properties of solvated clusters in DFT+U and DFT-D2 AIMD simulations, optimized geometries of the c1 and c2 clusters and details of the employed Co, O, and H pseudopotentials); details of AIMD simulations; oxidation potential of the CoCat model. This material is also available free of charge via the Internet at http://pubs.acs.org.
ACKNOWLEDGMENTS
We acknowledge computational resources provided by the CINECA consortium (Casalecchio di Reno, Italy), the CASPUR consortium (Roma, Italy), and the Caliban-HPC computer center of the University of L’Aquila. L.G. acknowledges funding provided by the European Research Council project no. 240624 “MultiscaleChemBio” within the VII Framework Program of the European Union. G.M. acknowledges financial support by the Italian Institute of Technology (IIT) under Project SEED “POLYPHEMO”.
Footnotes
Notes: The authors declare no competing financial interest.
REFERENCES
- (1).Barber J. Chem. Soc. Rev. 2009;38:185. doi: 10.1039/b802262n. [DOI] [PubMed] [Google Scholar]
- (2).Hambourger M, Moore GF, Kramer DM, Gust D, Moore AL, Moore TA. Chem. Soc. Rev. 2009;38:25. doi: 10.1039/b800582f. [DOI] [PubMed] [Google Scholar]
- (3).Reece SY, Hamel JA, Sung K, Jarvi TD, Esswein AJ, Pijpers JJH, Nocera DG. Science. 2011;334:645–648. doi: 10.1126/science.1209816. [DOI] [PubMed] [Google Scholar]
- (4).Nocera DG. Acc. Chem. Res. 2012;45:767. doi: 10.1021/ar2003013. [DOI] [PubMed] [Google Scholar]
- (5).Sartorel A, Carraro M, Toma FM, Prato M, Bonchio M. Energy Environ. Sci. 2012;5:5592–5603. [Google Scholar]
- (6).Dismukes GC, Brimblecombe R, Felton GAN, Pryadun RS, Sheats JE, Spiccia L, Swiegers GF. Acc. Chem. Res. 2009;42:1935. doi: 10.1021/ar900249x. [DOI] [PubMed] [Google Scholar]
- (7).Liu X, Wang F. Coord. Chem. Rev. 2012;256:1115. [Google Scholar]
- (8).Zaharieva I, Chernev P, Risch M, Klingan K, Kohlhoff M, Fischer A, Dau H. Energy Environ. Sci. 2012;5:7081. [Google Scholar]
- (9).Cao C, Hill S, Cheng H-P. Phys. Rev. Lett. 2008;100:167206. doi: 10.1103/PhysRevLett.100.167206. [DOI] [PubMed] [Google Scholar]
- (10).Kwabena Bediako D, Lassalle-Kaiser B, Surendranath Y, Yano J, Yachandra VK, Nocera DG. J. Am. Chem. Soc. 2012;134:6801. doi: 10.1021/ja301018q. [DOI] [PubMed] [Google Scholar]
- (11).Dau H, Limberg C, Reier T, Risch M, Roggan S, Strasser P. ChemCatChem. 2010;2:724. [Google Scholar]
- (12).Duan L, Bozoglian F, Mandal S, Stewart B, Privalov T, Llobet A, Sun L. Nat. Chem. 2012;4:418–423. doi: 10.1038/nchem.1301. [DOI] [PubMed] [Google Scholar]
- (13).Baruah T, Pederson MR. Chem. Phys. Lett. 2002;360:144. [Google Scholar]
- (14).Kanan MW, Nocera DG. Science. 2008;321:1072. doi: 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- (15).McAlpin JG, Stich TA, Ohlin CA, Surendranath Y, Nocera DG, Casey WH, Britt RD. J. Am. Chem. Soc. 2011;133:15444. doi: 10.1021/ja202320q. [DOI] [PubMed] [Google Scholar]
- (16).Berardi S, La Ganga G, Natali M, Bazzan I, Puntoriero F, Sartorel A, Scandola F, Campagna S, Bonchio M. J. Am. Chem. Soc. 2012;134:11104–11107. doi: 10.1021/ja303951z. [DOI] [PubMed] [Google Scholar]
- (17).Kanan MW, Surendranath Y, Nocera DG. Chem. Soc. Rev. 2009;38:109–114. doi: 10.1039/b802885k. [DOI] [PubMed] [Google Scholar]
- (18).Risch M, Khare V, Zaharieva I, Gerencser L, Chernev P, Dau H. J. Am. Chem. Soc. 2009;131:6936. doi: 10.1021/ja902121f. [DOI] [PubMed] [Google Scholar]
- (19).Kanan MW, Yano J, Surendranath Y, Dinca M, Yachandra VK, Nocera DG. J. Am. Chem. Soc. 2010;132:13692. doi: 10.1021/ja1023767. [DOI] [PubMed] [Google Scholar]
- (20).Du P, Kokhan O, Chapman KW, Chupas PJ, Tiede DM. J. Am. Chem. Soc. 2012;134:11096. doi: 10.1021/ja303826a. [DOI] [PubMed] [Google Scholar]
- (21).Mattioli G, Risch M, Amore Bonapasta A, Dau H, Guidoni L. Phys. Chem. Chem. Phys. 2011;13:15437. doi: 10.1039/c1cp21776c. [DOI] [PubMed] [Google Scholar]
- (22).Hu XL, Piccinin S, Laio A, Fabris S. ACS Nano. 2012;6:10497–10504. doi: 10.1021/nn3044325. [DOI] [PubMed] [Google Scholar]
- (23).Risch M, Klingan K, Ringleb F, Chernev P, Zaharieva I, Fischer A, Dau H. ChemSusChem. 2012;5:542. doi: 10.1002/cssc.201100574. [DOI] [PubMed] [Google Scholar]
- (24).Surendranath Y, Kanan MW, Nocera DG. J. Am. Chem. Soc. 2010;132:16501. doi: 10.1021/ja106102b. [DOI] [PubMed] [Google Scholar]
- (25).McAlpin JG, Surendranath Y, Dinca M, Stich TA, Stoian SA, Casey WH, Nocera DG, Britt RD. J. Am. Chem. Soc. 2010;132:6882. doi: 10.1021/ja1013344. [DOI] [PubMed] [Google Scholar]
- (26).Car R, Parrinello M. Phys. Rev. Lett. 1985;55:2471–2474. doi: 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- (27).Giannozzi P, et al. J. Phys.: Condens. Matter. 2009;21:395502. doi: 10.1088/0953-8984/21/39/395502. [DOI] [PubMed] [Google Scholar]
- (28).Unpublished results by Holger Dau. Dept. of Physics, Freie Universität Berlin; Germany: [Google Scholar]
- (29).Hsu H, Umemoto K, Cococcioni M, Wentzcovitch R. Phys. Rev. B. 2009;79:125124. [Google Scholar]
- (30).Mattioli G, Alippi P, Filippone F, Caminiti R, Amore Bonapasta A. J. Phys. Chem. C. 2010;114:21694. [Google Scholar]
- (31).Cococcioni M, de Gironcoli S. Phys. Rev. B. 2005;71:035105. [Google Scholar]
- (32).Kulik HJ, Cococcioni M, Scherlis DA, Marzari N. Phys. Rev. Lett. 2006;97:103001. doi: 10.1103/PhysRevLett.97.103001. [DOI] [PubMed] [Google Scholar]
- (33).Juhin A, de Groot F, Vankò G, Calandra M, Brouder C. Phys. Rev. B. 2010;81:115115. [Google Scholar]
- (34).Norman MR, Freeman A. J. Phys. Rev. B. 1986;33:8896. doi: 10.1103/physrevb.33.8896. [DOI] [PubMed] [Google Scholar]
- (35).McMahan AK, Martin RM, Satpathy S. Phys. Rev. B. 1988;38:6650. doi: 10.1103/physrevb.38.6650. [DOI] [PubMed] [Google Scholar]
- (36).Galakhov VR, Kurmaev EZ, Uhlenbrock S, Neumann M, Kellerman DG, Gorshkov VS. Solid State Commun. 1996;99:221. [Google Scholar]
- (37).Vanderbilt D. Phys. Rev. B. 1990;41:7892–7895. doi: 10.1103/physrevb.41.7892. [DOI] [PubMed] [Google Scholar]
- (38).Perdew JP, Burke K, Ernzerhof M. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- (39).Parrinello M, Rahman A. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- (40).Henkelman G, Jònsson H. J. Chem. Phys. 1999;110:7010–7022. [Google Scholar]
- (41).Weinan E, Ren W, Vanden-Eijnden E. Phys. Rev. B. 2002;66:052301. [Google Scholar]
- (42).Van de Walle CG, Neugebauer J. J. Appl. Phys. 2004;95:3851–3879. [Google Scholar]
- (43).Mattioli G, Filippone F, Amore Bonapasta A. J. Am. Chem. Soc. 2006;128:13772–13780. doi: 10.1021/ja062145x. [DOI] [PubMed] [Google Scholar]
- (44).Lundberg M, Blomberg MRA, Siegbahn PE. M. Inorg. Chem. 2004;43:264–274. doi: 10.1021/ic0348188. [DOI] [PubMed] [Google Scholar]
- (45).McAlpin JG, Stich TA, Casey WH, Britt RD. Coord. Chem. Rev. 2012;256:2445–2452. [Google Scholar]
- (46).Wang L-P, Van Voorhis T. J. Phys. Chem. Lett. 2011;2:2200. [Google Scholar]
- (47).Bovi D, Narzi D, Guidoni L. Angew. Chem., Int. Ed. 2013 doi: 10.1002/anie.201306667. DOI: 10.1002/anie.201306667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ashcroft NW, Mermin ND. Solid State Physics. Thomson Learning, Inc.; Stamford, CT: 1976. [Google Scholar]
- (49).Varkey JB, Janotti A, Franchini C, Van de Walle CG. Phys. Rev. B. 2012;85:081109(R). [Google Scholar]
- (50).Positively charged holes, at variance with electrons, are more stable when accommodated in higher energy levels.
- (51).Surendranath Y, Dinca M, Nocera DG. J. Am. Chem. Soc. 2009;131:2615. doi: 10.1021/ja807769r. [DOI] [PubMed] [Google Scholar]
- (52).Risch M, Ringleb F, Khare V, Chernev P, Zaharieva I, Dau H. J. Phys.: Conf. Series. 2009;190:012167. [Google Scholar]
- (53).Shao-Horn Y, Hackney SA, Kahaian AJ, Thackeray MM. J. Solid State Chem. 2002;168:60. [Google Scholar]
- (54).Delaplane RG, Ibers JA, Ferraro JR, Rush JJ. J. Chem. Phys. 1969;50:1920–1927. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Assessment of theoretical methods (LiCoO2 properties, [Co(II)(H2O)6] properties, liquid water properties, structural and dynamical properties of solvated clusters in DFT+U and DFT-D2 AIMD simulations, optimized geometries of the c1 and c2 clusters and details of the employed Co, O, and H pseudopotentials); details of AIMD simulations; oxidation potential of the CoCat model. This material is also available free of charge via the Internet at http://pubs.acs.org.