

Abstract
Vitiligo is an autoimmune disease characterized by destruction of melanocytes, leaving 0.5% of the population with progressive depigmentation. Current treatments offer limited efficacy. We report that modified inducible heat shock protein 70 (HSP70i) prevents T cell–mediated depigmentation. HSP70i is the molecular link between stress and the resultant immune response. We previously showed that HSP70i induces an inflammatory dendritic cell (DC) phenotype and is necessary for depigmentation in vitiligo mouse models. Here, we observed a similar DC inflammatory phenotype in vitiligo patients. In a mouse model of depigmentation, DNA vaccination with a melanocyte antigen and the carboxyl terminus of HSP70i was sufficient to drive autoimmunity. Mutational analysis of the HSP70i substrate-binding domain established the peptide QPGVLIQVYEG as invaluable for DC activation, and mutant HSP70i could not induce depigmentation. Moreover, mutant HSP70iQ435A bound human DCs and reduced their activation, as well as induced a shift from inflammatory to tolerogenic DCs in mice. HSP70iQ435A-encoding DNA applied months before spontaneous depigmentation prevented vitiligo in mice expressing a transgenic, melanocyte-reactive T cell receptor. Furthermore, use of HSP70iQ435A therapeutically in a different, rapidly depigmenting model after loss of differentiated melanocytes resulted in 76% recovery of pigmentation. Treatment also prevented relevant T cells from populating mouse skin. In addition, ex vivo treatment of human skin averted the disease-related shift from quiescent to effector T cell phenotype. Thus, HSP70iQ435A DNA delivery may offer potent treatment opportunities for vitiligo.
INTRODUCTION
Vitiligo is an autoimmune disease that affects about 0.5% of the world population; patients with vitiligo present with progressive skin depigmentation (1). About 25% more women than men develop the disease (2). Current treatment modalities seldom induce lasting repigmentation. Ablation of autoimmunity is the most commonly prescribed approach, through the use of systemic or topical corticosteroids or topical application of calcineurin inhibitors (tacrolimus, pimecrolimus), often supplemented by ultraviolet (UV) phototherapy (3). Treatment responses are frequently inadequate, and complications from steroid therapy or calcineurin inhibition can occur (4). Melanocyte-protective pseudocatalase treatment does not appear to offer superior efficacy over UV treatment alone (5). Transplantation can remedy local depigmentation only in patients with already stable disease (6). Accordingly, a safe and effective approach that induces immune tolerance to melanocyte differentiation antigens may offer a superior alternative. This prompted us to identify a molecular entity critically involved in depigmentation and design an intervention method.
Expanding vitiligo lesions are consistently infiltrated with T cells in the areas lining the lesional borders; these infiltrating T cells are composed primarily of CD8+ cytotoxic T cells reactive with melanocyte-specific antigens (7). Because melanocyte-specific antigens are also targeted by T cells in deadly melanoma skin cancers, patients with vitiligo may develop immune responses that affect their odds of developing melanoma. Autoimmune vitiligo patients develop immune responses mediated by T cells expressing high-affinity T cell receptors (TCRs) that may be exploited for melanoma treatment (8). Indeed, melanoma and other skin cancers appear to occur less frequently in established vitiligo patients (8). Moreover, increased numbers of CD11c+ dendritic cells (DCs) have been observed in vitiligo perilesional skin, which may lend to an increased vitiligo/antimelanoma response (9).
Inducible heat shock protein 70 (HSP70i) has recently gained attention for its role in precipitating vitiligo (10). In an initial screening of potential stress protein involvement, HSP70i in nonlesional and lesional skin biopsies provided the most apparent example of differential expression (11). Depigmentation in mice vaccinated with DNA encoding melanocyte antigens was markedly enhanced in response to the presence of HSP70i-encoding DNA, suggesting the immune-enhancing role of the stress protein (12). In knockout mice, the absence of HSP70i greatly affected their ability to develop progressive depigmentation, which correlated with reduced activation of a CD8+ T cell–mediated response to melanocytes (10). In addition, the antigen-presenting cell (APC) profile is driven toward an inflammatory repertoire in vitiligo-prone mice after vaccination with HSP70i (10).
HSP70i can be secreted by live cells of neuronal ancestry (13). Soluble HSP70i can activate DCs, leading to more efficient uptake, processing, and presentation of antigens (14). Thus, the interaction between extracellular, soluble HSP70i and recipient DCs will need to be compromised to intervene with depigmentation. A strategy involving targeting of the DC surface is limited by the existence of multiple different receptors for HSP70i on DCs (15–18), and functionally blocking all receptors will have consequences well beyond preventing HSP70i activity. Blocking HSP70i itself at a site otherwise involved in binding DCs may be more successful.
Here, we identified an HSP70i variant that prevents the vitiligo-associated inflammatory DC phenotype and averts depigmentation in mice. We conclude that mutant HSP70i is not only an inactive variant but also binds DCs and alters their function to interfere with subsequent T cell activation. The implications of this study are that HSP70iQ435A DNA delivery may be used for the treatment of vitiligo patients.
RESULTS
Vitiligo patients present with inflammatory DCs
DC subsets can be identified on the basis of surface expression of CD11b and CD11c markers and by the cytokines they generate. Inflammatory DCs (CD11b+CD11c+) produce interleukin-17 (IL-17) and are associated with inflammation, whereas an anti-inflammatory CD11b+CD11c− population will generate cytokines including IL-10 and transforming growth factor–β to support regulatory T cell differentiation (19, 20). Conventional CD11b−CD11c+ DCs are particularly supportive of T helper 1 responses and secrete IL12p70 (19, 20). Differential expression of HSP70 in vitiligo skin is accompanied by DC infiltrates (9, 11). Exposure of mice to HSP70i results in increased abundance of inflammatory (CD11b+CD11c+) DCs (10). To establish whether the same elevated inflammatory DC content is also found in vitiligo patients (see table S1 for patient information), we stained peripheral blood mononuclear cells (PBMCs) from vitiligo and non-vitiligo controls for CD11b and CD11c expression (after gating out T and B cells). A marked increase in CD11b+CD11c+ cells (R2) was observed among PBMCs from the vitiligo patients (Fig. 1, A and B; P = 0.04). No differences were found between the two groups among the percent of cells staining for CD11b+ or CD11c+ alone.
Fig. 1.
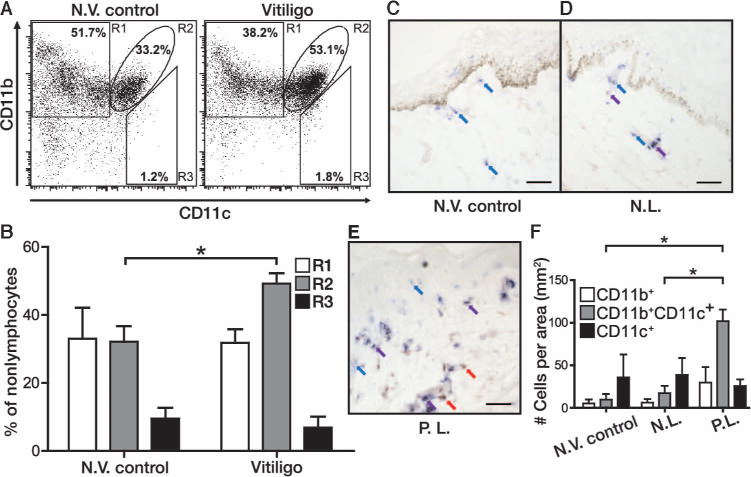
APCs in progressive human vitiligo reflect the phenotype observed in response to HSP70i. (A) PBMCs from non-vitiligo (N.V.) control (left flow plot) and vitiligo (right flow plot) patients were gated for expression of CD11b and CD11c after excluding T and B cells. (B) Quantification of the percentage of cells among R1 to R3 indicates an increase in R2 (CD11b+CD11c+) cells among the vitiligo patient samples. (C to E) Non-vitiligo (N.V.) patient (C) and vitiligo patient (D) nonlesional (N.L.) and (E) perilesional (P.L.) skin were stained for expression of APC markers CD11b (red staining and arrows) and CD11c (blue staining and arrows) with colocalization observed in purple (purple staining and arrows). (F) Quantification of double positive–stained cells indicates a significant increase in CD11b+CD11c+ cells within perilesional skin. Scale bars, 25 μm. n = 3. *P < 0.05, **P < 0.01.
To assess whether this inflammatory DC subset is present in skin, we stained biopsies from non-vitiligo controls and nonlesional and perilesional tissue from vitiligo patients for CD11b and CD11c expression to identify different subsets of APCs. A significant increase was observed in inflammatory CD11b+CD11c+ cells in perilesional skin versus nonlesional (P = 0.0495) and non-vitiligo skin (P = 0.0495) (Fig. 1, C to F). The inflammatory DC population was observed mainly in the dermis and toward the dermo-epidermal junction (Fig. 1, C to E). These data reveal a markedly increased abundance of inflammatory CD11b+CD11c+ DCs in human vitiligo.
Autoimmune stimulatory activity resides in the C terminus of HSP70i
HSP70i is necessary for autoimmune vitiligo in mice (10). Therefore, we hypothesized that manipulating HSP70i could locally alter the APC profile and affect the resulting immune response. Thus, it was important to identify the DC-activating region in HSP70i. We cloned overlapping sequence fragments of HSP70i encoding either the N-terminal (amino acids 1 to 377) or C-terminal (amino acids 320 to 641) regions. Protein expression was confirmed by Western blot with HSP70i-reactive antibodies (fig. S1). C57BL/6 mice were then vaccinated with DNA plasmids encoding human melanocyte antigen TRP-2 (hTRP-2), which is capable of inducing temporary, minor depigmentation, combined with DNA encoding full-length HSP70i, N- or C-terminal HSP70i fragments, or empty vector DNA (Fig. 2A). Significant depigmentation was observed in mice vaccinated with full-length HSP70i, which accounted for >14% depigmentation compared to ~2% in the empty vector–vaccinated mice (P = 0.0088; Fig. 2A). The HSP70i C terminus alone achieved markedly greater levels of depigmentation (>40%) than the full-length sequence when compared to empty vector–vaccinated mice (P = 0.0139; Fig. 2A) and N terminus–vaccinated mice (P = 0.0275; Fig. 2A). By contrast, the N terminus did not induce depigmentation (<5%, similar to empty vector–vaccinated mice) (Fig. 2A). The overlapping N- and C-terminal regions are depicted in Fig. 2B.
Fig. 2.
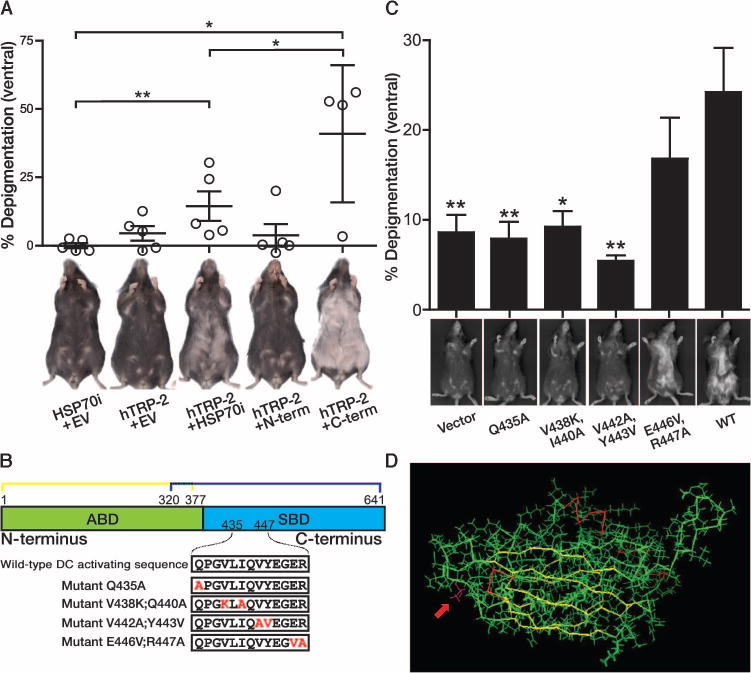
A peptide within the C terminus of HSP70i is required for inducing depigmentation. (A) To identify the region of HSP70i critical for inducing depigmentation, mice were vaccinated with DNA plasmids encoding wildtype (WT) N terminus, C terminus, or full-length HSP70i. Mice vaccinated with HSP70i N terminus (residues 1 to 377) expressed depigmentation similar to the empty vector control–treated animals. Mice vaccinated with the HSP70i C terminus displayed far greater levels of depigmentation than those exposed to full-length HSP70i plus hTRP-2 vaccination (n = 5). Representative images of C57BL/6 mice imaged 4 weeks after vaccination with DNA-encoding melanosomal antigen hTRP-2 and either full-length HSP70i, N-terminal (N-term) or C-terminal (C-term) HSP70i fragments, or empty vector (EV) are shown. Mice were vaccinated five times, every 7 days, with 5.6 μg of total DNA. (B) Schematic of the N-terminal ATP-binding domain (ABD) and C-terminal substrate–binding domain (SBD) of HSP70i and fragments (HSP70i1–377 and HSP70i320–641) and mutations introduced within the putative DC-activating region. (C) No depigmentation was observed in mice receiving vaccinations with hTRP-2 and several mutant versions of HSP70i compared to hTRP-2 and WT HSP70i, supporting the crucial involvement of the 11-mer with the exceptions of amino acids 446 and 447. Representative images of mice 5 weeks after vaccination (n = 10) are shown. Mice were vaccinated four times, every 7 days, with 4 μg of total DNA. (D) Three-dimensional representation showing the peripheral location of the mutant Q435A (magenta) residue (red arrow) ideally located at the interface of the ATP- and substrate-binding domains. *P < 0.05, **P < 0.01.
The DnaK peptide QPSVQIQVYQGEREIAAHNK (DnaK407–426) was shown to drive DC activation during inflammation in response to infection, inducing DCs to produce tumor necrosis factor–α and IL-12 (18). However, the corresponding DC-activating region within human HSP70i remains unidentified to date. To further define the region within the C terminus of human HSP70i required for inducing depigmentation, we identified the peptide sequence QPGVLIQVYEGER as maximally homologous with the proposed DC-activating region of microbial DnaK (18). To assess this peptide’s actual contribution to autoimmune activation, we generated mutant constructs using site-directed mutagenesis (Fig. 2B). Protein expression from mutant sequences was demonstrated by Western blot with monoclonal antibody SPA-810, generated against a 67-mer partially overlapping the peptide of interest, and polyclonal antibody SPA-811 recognizing a C-terminal peptide downstream (fig. S1A).
We next determined the depigmenting effects of mutant HSP70i in vivo. Wild-type C57BL/6 mice were vaccinated with plasmids encoding wild-type or mutant HSP70i and melanosomal antigen hTRP-2, and depigmentation was assessed 5 weeks after the final vaccination (Fig. 2C). Depigmentation was significantly decreased in the presence of variants HSP70iQ435A (P = 0.0055), HSP70iV438K, I440A (P = 0.0178), and HSP70iV442A, Y443V (P = 0.0015) compared to wild-type HSP70i. This further confirms a peptide of interest consisting of amino acids QPGVLIQVYEG as crucial to immune activation [and excludes amino acids ER (amino acids 446 to 447) as being necessary for depigmentation].
Of interest, antibody SPA-810 demonstrated markedly reduced reactivity toward mutants HSP70iV438K, I440A, HSP70iV442A, Y443V (>60% reduced reactivity), but not HSP70iE446V, R447A compared to the wild-type sequence (fig. S1, A and B). In concordance with the immunizing peptide used to generate SPA-810, recognition of HSP70iQ435A was not affected (fig. S1, A and B). Together, the data indicate that SPA-810 recognizes an epitope that overlaps with our DC activation sequence of interest, yet antibody binding is unaffected by the HSP70iQ435A mutation. Because SPA-810 can inhibit DC activation in vitro, these data provide further support for the involvement of its target sequence in DC activation (10). Because the HSP70iQ435A mutation does not affect binding of SPA-810, this mutation is well suited for further studies. Further, a three-dimensional representation of HSP70i demonstrates that the Q435A mutation is peripherally located, and thus suggests that it is uniquely positioned to interact with DCs independent of its chaperone function (Fig. 2D, arrow). Therefore, a single amino acid difference in the QPGVLIQVYEG peptide is sufficient to inactivate the depigmentation accelerating effects of HSP70i and supports the crucial involvement of the moiety in inducing autoimmune depigmentation.
Mice vaccinated with HSP70i develop a humoral response to the protein (12). The exact sequence with HSP70i bound by these circulating antibodies may affect the ability of HSP70i to activate DCs. To determine whether the N or C terminus of HSP70i was the target of humoral responses in mice, we prepared protein homogenates from HSP70i-transfected COS7 cells and electrophoresed them on an SDS-polyacrylamide gel. In serum from the empty vector– and HSP70i1–377–vaccinated mice, antibodies toward HSP70i were not detected, whereas mice vaccinated with either full-length HSP70i or the C terminal (HSP70i320–642) developed antibodies toward the C terminal of HSP70i (fig. S2A). Further Western blotting revealed that humoral responses were directed toward residues 494 to 641, rather than residues 262 to 551, recognizing C-terminal amino acids beyond amino acid 551 (fig. S2B). The data are congruent with the proposed location of the DC-activating region within HSP70i proposed above and explain why endogenous antibodies to HSP70i may not affect DC binding and activation mediated by HSP70i. The C terminus is thus required for inducing depigmentation and is more immunogenic than the full-length HSP70i.
Mutant HSP70i interferes with DC activation
To determine the role of the QPGVLIQVYEG moiety in DC activation, we vaccinated wild-type C57BL/6 mice and vitiligo-prone TCR transgenic Pmel-1 mice (21) using plasmids encoding either wild-type HSP70i, HSP70iQ435A, or empty vector DNA. The Q435A mutation was used because of its position potentially affecting DC binding and activation (Fig. 2D). Splenocytes were stained for expression of markers CD11b and CD11c after excluding T and B cells. No additional gating for monocytes was performed. We observed marked differences in the abundance of monocyte-derived subpopulations based onCD11b and CD11c expression levels shown as R1 to R3 (Fig. 3, A and B). The R1 population is characterized by expression of CD11b+CD11c−, typical for regulatory DCs (20). Compared to HSP70i-based vaccination, we observed significant skewing toward a greater R1 population after HSP70iQ435A vaccination (P = 0.022; Fig. 3B). There was also a trend toward increasing activation after HSP70iQ435A vaccination compared to empty vector (P = 0.0618; Fig. 3B). By contrast, the DC populations R2 (CD11b+CD11c+), representative of proinflammatory DCs, and R3 (CD11b−CD11c+), representative of classic inflammatory DCs (20), showed opposing changes (Fig. 3B; P = 0.028 and P = 0.0029, respectively). Overall, these data indicate that wild-type HSP70i and HSP70iQ435A oppositely drive the relative abundance of immune cell subpopulations associated with autoimmune depigmentation in mice.
Fig. 3.
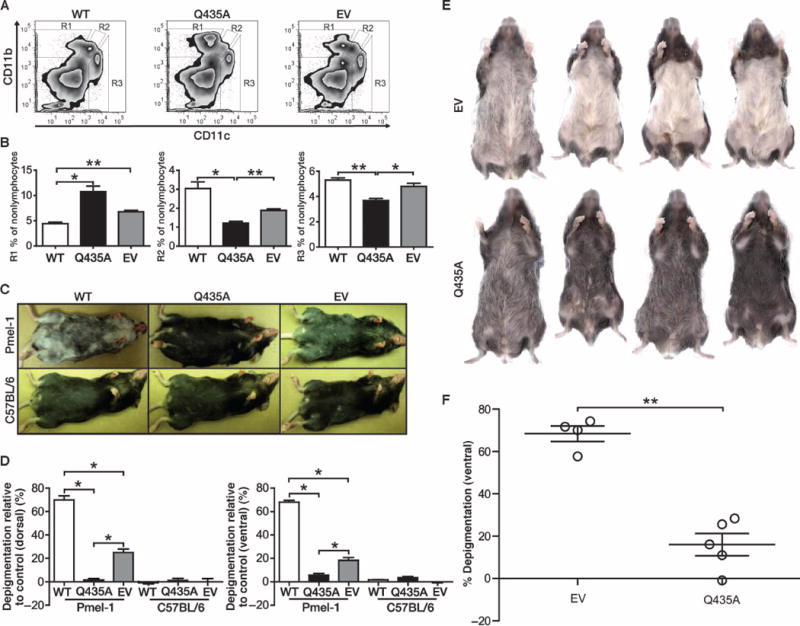
WT HSP70i accelerates and mutant HSP70i prevents depigmentation in vitiligo-prone mice. (A) Tolerogenic DCs are induced in response to HSP70iQ435A. (B) Representative flow cytometry plots and cumulative graphs of mice vaccinated three times in total, every 7 days, with 4 μg of WT, HSP70iQ435A (Q435A), or empty vector (EV) control DNA. Splenocytes from Pmel-1 mice (n = 3) 6 months after vaccination were analyzed by flow cytometry. Gating for CD11b and CD11c expression among nonlymphocytes, we observed three distinct populations that vary in abundance in response to the treatments. Differential expression of CD11b and CD11c was found after vaccination with WT or HSP70iQ435A plasmids. In contrast to WT HSP70i, the anti-inflammatory population (R1) displays increased, and the inflammatory population (R2) displays decreased, abundance in response to mutant HSP70i-encoding DNA. (C) Images were obtained 6 months after treatment. Pmel-1 mice depigmented within and distal to the site of vaccination. (D) Cumulative graphs of mice in (C). Pmel-1 mice receiving mutant HSP70i displayed significantly less depigmentation compared to WT HSP70i or empty vector–exposed C57BL/6 mice, confirming inhibition by mutant HSP70i. (E) To test the therapeutic effects of HSP70iQ435A in rapidly depigmenting vitiligo-prone mice, tyrosinase-responsive TCR transgenic h3TA2 mice were vaccinated five times, every 6 days, with 5.6 μg of either empty vector– or HSP70iQ435A-encoding DNA plasmid. Mice were imaged 4 weeks after the final vaccination at just over 9 weeks of age. (F) Cumulative data from (E) demonstrating that empty vector–vaccinated mice had significantly more depigmentation than the HSP70iQ435A-vaccinated mice (n = 4). *P < 0.05, **P < 0.01.
Mutant HSP70i inhibits depigmentation in vitiligo-prone mice
Six months after vaccination, Pmel-1 mice exhibited spontaneous depigmentation, yet animals exposed to wild-type HSP70i displayed significantly increased ventral and dorsal depigmentation compared to HSP70iQ435A or empty vector (P = 0.0495; Fig. 3, C and D, and fig. S3). As expected, in the absence of a targetable antigen, no depigmentation was observed in vaccinated C57BL/6 mice (Fig. 3, C and D). Depigmentation was significantly prevented in Pmel-1 mice vaccinated with HSP70iQ435A DNA compared to mice vaccinated with empty vector (P = 0.0495; Fig. 3, C and D, and fig. S3). Thus, a single amino acid variation in the putative DC binding region of HSP70i is sufficient to actively interfere with immune activation and subsequent depigmentation in vivo.
On the basis of our observations that HSP70iQ435A can prevent depigmentation in mice prone to delayed and slowly progressing depigmentation, we next assessed whether HSP70iQ435A can be therapeutic in the context of existing vitiligo. We treated the early and rapidly depigmenting mouse strain h3TA2, which expresses T cells bearing a human tyrosinase-reactive TCR transgene and HLA-A2.1 (22). Depigmentation initiates by about 4 weeks of age, accompanied by a marked loss of melanocytes in 5-week-old animals (fig. S4). Treatment with HSP70iQ435A or empty vector DNA was initiated at 5 weeks of age. Images of representative mice comparing pre- and postvaccination are shown in fig. S4. Eight weeks later, the pelage of h3TA2 mice unvaccinated or vaccinated with empty vector DNA was 86% depigmented (Fig. 3, E and F). By contrast, an astonishing 76% of depigmentation in the pelage of mice was restored with mutant HSP70iQ435A and the pelage returned almost fully pigmented (P = 0.0143; Fig. 3, E and F, and fig. S4).Thus, HSP70iQ435A can reverse the depigmenting phenotype, critical for treatment of active disease.
Mutant HSP70i prevents accumulation of antigen-specific T cells in the skin
T cell activity mediates the loss of melanocytes in patients with vitiligo (23). To analyze the local immune-modulating effects of wild-type HSP70i and HSP70iQ435A DNA exposure, we probed the skin of the Pmel-1, C57BL/6, and h3TA2 mice for T cell infiltration. Skin from Pmel-1 mice contained significantly more CD3+ T cells in response to wild-type HSP70i compared to either empty vector– or HSP70iQ435A-vaccinated mice, and skin-infiltrating T cells were primarily located in the dermis and hair follicles (P = 0.0495; Fig. 4, A and E). By contrast, a trend toward decreased T cell infiltration was observed in response to HSP70iQ435A vaccination (Fig. 4, B and E). Skin from the h3TA2 mice contained significantly fewer CD3+ T cells in response to HSP70iQ435A (P = 0.03; Fig. 4, C, D, and F). We next examined cytotoxic T lymphocyte infiltrates held responsible for melanocyte killing by staining for CD8. HSP70iQ435A vaccination significantly decreased this population in Pmel-1 mice compared to HSP70i (P = 0.0495; Fig. 4, G, H, and K) and showed a decreasing trend compared to empty vector–exposed mice. The number of melanocyte-reactive antigen-specific T cells was also significantly reduced in HSP70iQ435A-treated h3TA2 skin (P = 0.03; Fig. 4, I, J, and L) corresponding to pigment retention. Together, these data reveal a direct effect between skin exposure to HSP70iQ435A and decreased T cell infiltration resulting in pigment retention.
Fig. 4.
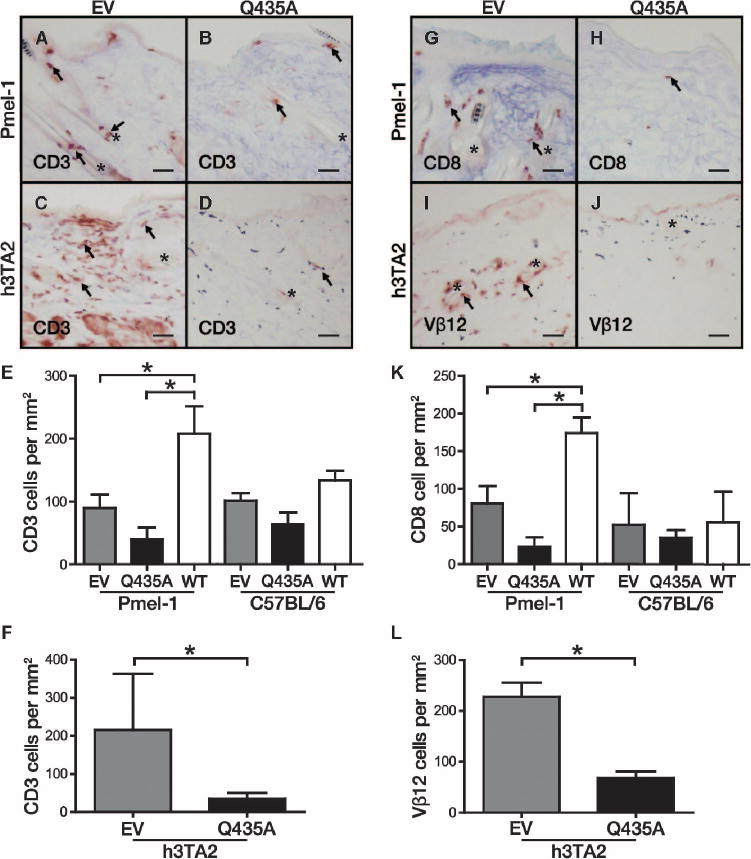
Decreased T cell infiltration is observed after HSP70iQ435A vaccination. (A) Pmel-1 mice vaccinated with empty vector (EV) DNA showed abundant skin infiltration by CD3+ T cells (arrows) localized mostly to hair follicles (asterisks). (B) CD3+ T cell infiltrates seen in (A) were decreased after HSP70iQ435A (Q435A) administration. (C and D) T cell infiltrates in skin of h3TA2 mice in response to empty vector and (D) HSP70iQ435A. (E) Cumulative data showing changes in skin-infiltrating T cells in treated mice. (F) Quantification of T cell infiltrates in h3TA2 mice. (G) Most of the skin-infiltrating T cells were cytotoxic. (H) In the Pmel-1 mice, CD8+ infiltrates in response to empty vector decreased after HSP70iQ435A exposure. (I) Reduced number of melanocyte-reactive T cells was observed in response to HSP70iQ435A. T cells were quantified on the basis of expression of the tyrosinase transgene–reactive TCR Vβ12 subunit. (J) A decrease in Vβ12+ cells was observed after vaccination with HSP70iQ435A. (K) Quantification of cytotoxic T cell infiltrates in C57BL/6 and Pmel-1 mice. (L) Quantification of melanocyte-reactive T cells in h3TA2 mice. Arrows mark stained cells. Scale bars, 50 μm. *P < 0.05.
To further demonstrate a role for T cells in mediating depigmentation, we vaccinated CD4 and CD8 knockout mice using DNA plasmids encoding either wild-type HSP70i, HSP70iQ435A, or empty vector in combination with antigenic mouse Tyrp1ee (optimized TRP-1) (24). Wild-type HSP70i significantly supported depigmentation only in C57BL/6 control mice compared to empty vector (P = 0.0088; fig. S5) and HSP70iQ435A (P = 0.0493; fig. S5). By contrast, depigmentation was not observed in the CD4 or CD8 knockout mice, demonstrating the requirement for both helper and cytotoxic T cells in depigmentation (fig. S5).
Mutant HSP70i interferes with T cell activation
The relative expression of the transcription factors Eomes and T-bet has been shown in T cells to define a quiescent versus effector status (25). T cells from the HSP70iQ435A-treated Pmel-1 mice were analyzed for these transcription factors. Although T-bet expression remained similar among the mouse groups, Eomes expression was lower among CD8+ T cells from HSP70i-vaccinated Pmel-1 mice (versus empty vector–vaccinated mice, P = 0.039; Fig. 5A) and showed a decreasing trend compared to HSP70iQ435A vaccination (P = 0.05; Fig. 5A), reflecting a more apparent effector phenotype. By contrast, treatment with HSP70i induces an effector phenotype resulting in a significant depression of the Eomes to T-bet ratio despite minimal changes in T-bet alone (P = 0.035 compared to HSP70iQ435A, and P = 0.02 compared to empty vector; Fig. 5, B and C).
Fig. 5.
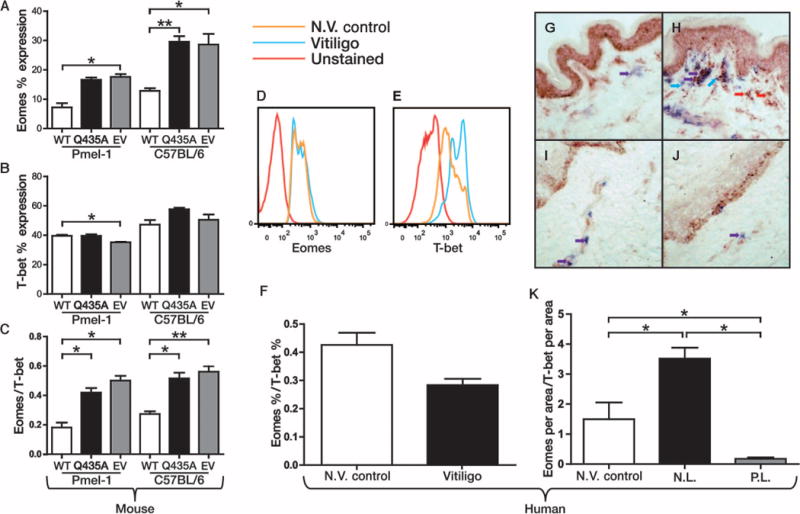
HSP70i-treated mice display a similar effector T cell phenotype as vitiligo patient tissue. (A) A decreased percentage of splenocytes from the WT HSP70i-exposed Pmel-1 mice (Fig. 3) expresses the T cell transcription factor Eomes, supporting an enhanced effector T cell phenotype. (B and C) Combined with T-bet expression (C), the resulting Eomes/T-bet ratio further indicates increased effector responses in mice exposed to HSP70i (n = 3). (D and E) Representative histograms of human PBMCs from non-vitiligo (N.V.) control and vitiligo patients similarly analyzed for Eomes and T-bet expression. (F) The Eomes/T-bet ratio indicates an effector response in vitiligo peripheral blood compared to non-vitiligo control patient samples. (G and H) Nonlesional (N.L.) (G) and perilesional (P.L.) (H) vitiligo skin was stained for T-bet (red staining and arrows) and CD8 (blue staining and arrows) expression, with the cytotoxic T cells shown in purple (purple staining and arrows). A significantly larger number of CD8+/T-bet+–expressing cells were observed in the dermis of perilesional skin. (I and J) Nonlesional (I) and perilesional (J) skin stained for Eomes (red staining and arrows) and CD8 (blue staining and arrows). Eomes-expressing CD8 cells were significantly increased in non-vitiligo control and nonlesional skin from vitiligo patients. (K) The ratio of Eomes/T-bet–expressing CD8 cells indicates an effector response in perilesional vitiligo skin, similar to that seen in the periphery. n = 3. *P < 0.05, **P < 0.01.
In the previous experiment, we demonstrated that the Eomes/T-bet ratio in mice reflects an effector T cell status after vaccination with HSP70i. Because the immune profiles in vitiligo patients appear to reflect HSP70i-induced changes, we next examined T cell profiles in vitiligo patient tissues. T cells from non-vitiligo and vitiligo patients were stained for Eomes and T-bet, and analyzed by flow cytometry (Fig. 5, D and E). We observed a decreasing trend in the Eomes/T-bet ratio, again indicating an effector response in the periphery of vitiligo patients (P = 0.06; Fig. 5F). Similarly, perilesional skin from vitiligo patients displays decreased Eomes expression by CD8+ T cells (P = 0.0495; Fig. 5, G and H) as well as increased T-bet expression (P = 0.0495; Fig. 5, I and J) compared to nonlesional and non-vitiligo control skin. As with the HSP70i-vaccinated mice, reduced Eomes/T-bet ratios were observed in the skin of vitiligo patients (P = 0.0495; Fig. 5K). We also determined that human vitiligo perilesional skin contains more cells expressing the homing receptor CXCR3 cells than nonlesional skin (P = 0.02; fig. S6), again correlating with the role assigned to this gene product in a mouse model of vitiligo (26).
Wild-type, but not mutant, HSP70i activates human DCs in vitro
Next, we compared the DC-stimulatory effects of wild-type and mutant HSP70i in vitro. Human PBMCs were magnetically sorted and driven toward immature DCs by addition of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 cytokines (27). After 1 week, His-labeled wild-type HSP70i or HSP70iQ435A full-length protein, or commercially obtained wild-type HSP70i was added. Twenty-four hours later, the DCs were analyzed for CD11c, CD80, CD83, CD86, and HLA-ABC expression by flow cytometry (Fig. 6, A to E). Among the entire CD11c+ population, increases in expression of all DC activation markers were observed in presence of HSP70i or lipopolysaccharide (LPS) (Fig. 6, A to E). By contrast, addition of HSP70iQ435A decreased expression of the activation markers (Fig. 6, A to E).
Fig. 6.
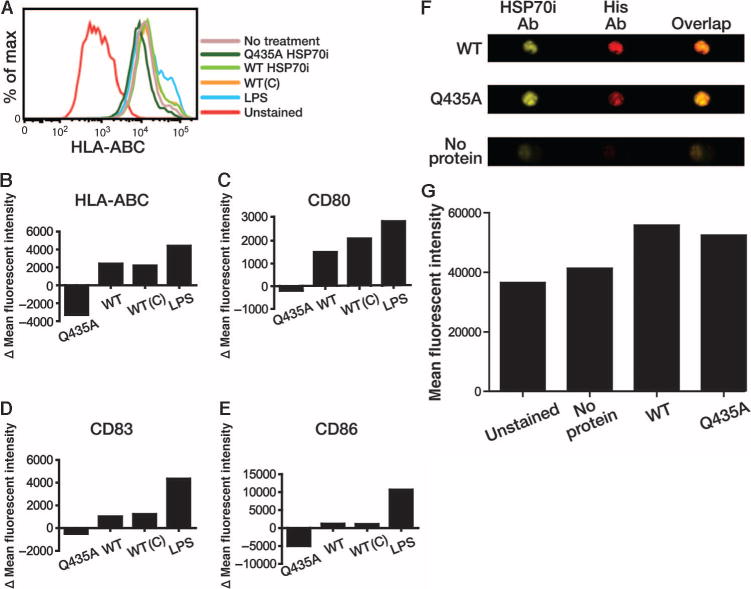
Mutant HSP70i suppresses human DC activation. (A) Representative histogram displaying HLA-ABC expression from human PBMCs differentiated into immature DCs via GM-CSF and IL-4 and exposed to His-tagged WT or HSP70iQ435A (Q435A) protein (1 μg/ml). Commercially purchased HSP70i [WT(C)], LPS (1 μg/ml), and complete media served as controls. CD11c+ cells were analyzed for expression of CD80, CD83, CD86, or HLA-ABC and analyzed by flow cytometry. (B to E) Changes in mean fluorescence intensity (MFI) between treatments indicating increased expression of activation markers after addition of His-tagged isolated WT HSP70i and commercially purchased HSP70i [WT(C)] and reduced expression of all activation markers after addition of HSP70iQ435A compared to control media. (F) ImageStream images of DCs after the addition of His-tagged WT or HSP70iQ435A (Q435A), or media alone. Cells were stained with antibodies (Ab) toward HSP70i (SPA-820) or the His tag. Single and overlaid channel images are displayed. (G) Similar MFIs using the anti-His antibody show that DCs bind both mutant and WT HSP70i.
The previous findings show that HSP70iQ435A protein directly affects DC activity. To determine whether HSP70iQ435A can bind DCs despite the mutation, we used ImageStream analysis, allowing us to quantify expression of a marker by fluorocytometry and localize its expression by confocal analysis within the same set of cells. Antibodies toward HSP70i (SPA-820) and the His tag were used to determine relative binding of wild-type and HSP70iQ435A to DCs (Fig. 6E). Indeed, we detected labeling by both antibodies in the wild-type and also HSP70iQ435A-treated samples, whereas media alone samples contained background levels of fluorescence (Fig. 6, E and F). Together, these data confirm that HSP70iQ435A binds to human DCs and actively interferes with activation marker expression.
HSP70i activates transcriptional and homing factors in human skin
To translate responses to HSP70iQ435A toward treatment of human disease, we exposed human skin samples ex vivo and followed the profiles of collected immune cells by fluorocytometry. Skin explants were treated with or without wild-type or mutant HSP70i protein added to the media. Skin T cells were analyzed by flow cytometry 6 days later. Eomes expression was up-regulated in response to HSP70iQ435A (P = 0.02; Fig. 7A) and wild-type HSP70i (P = 0.0027; Fig. 7A). By contrast, a trend toward decreased T-bet expression was visible only in response to HSP70iQ435A (Fig. 7B). As we have shown in treated mice (Fig. 5C), HSP70iQ435A resulted in a shift of the Eomes/T-bet ratio from an effector to a quiescent phenotype (P= 0.0154 compared to HSP70i, and P = 0.02 compared to media only; Fig. 7C). In addition, the T cell skin-homing marker cutaneous lymphocyte-associated antigen (CLA) was up-regulated in response to wild-type HSP70i (P = 0.0147 compared to HSP70iQ435A, and P = 0.0327 compared to media only; Fig. 7D), congruent to observations in vitiligo donors (28).
Fig. 7.
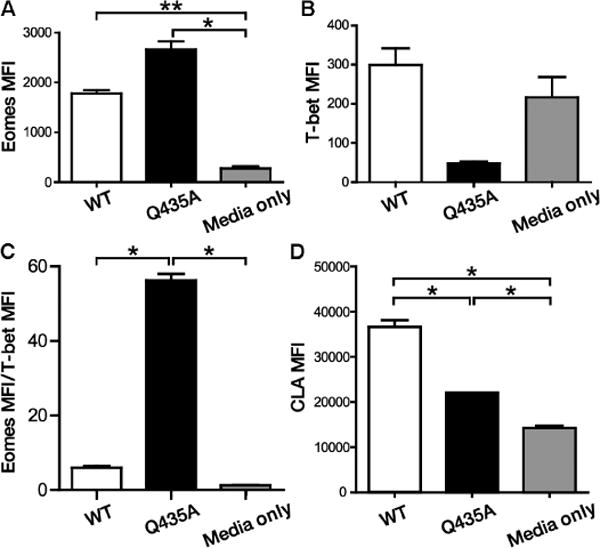
WT and mutant HSP70i differentially activate human skin T cells. Skin explants were maintained submerged in control media supplemented with or without 4 μg of WT or HSP70iQ435A (Q435A) protein for 6 days, and T cell profiles were analyzed after gating for CD3 expression. (A) Eomes expression was significantly up-regulated in T cells from WT and mutant HSP70i-treated skin T cells. (B) T-bet expression is reduced among T cells in response to HSP70iQ435A. (C) Together, the Eomes/T-bet ratios reveal an effector versus memory phenotype in response to WT and mutant HSP70i, respectively. (D) The T cell–homing receptor CLA is highly expressed among T cells in response to WT HSP70i. n = 2. *P < 0.05, **P < 0.01.
We also examined whether T cell activation can be abrogated by HSP70iQ435A in human peripheral blood samples. To measure antigen-specific T cell responses, we took advantage of preexisting cytomegalovirus (CMV)–reactive T cells from CMV+ HLA-A2+ donors. PBMCs were pulsed with CMVpp65 peptide and cocultured with wild-type or HSP70iQ435A. After 48 hours, the cells were analyzed by flow cytometry. As in the skin explant model (Fig. 7C), Eomes expression was up-regulated in response to HSP70iQ435A (P = 0.0041; fig. S7A) and wild-type HSP70i (P = 0.0244; fig. S7A). By contrast, T-bet was significantly decreased in response to HSP70iQ435A (P = 0.0095; fig. S7B) and increased in response to HSP70i (P = 0.0299 compared to media only, and P = 0.027 compared to HSP70iQ435A; fig. S7B). HSP70iQ435A resulted in a notable shift of the Eomes/T-bet ratio from an effector to a quiescent status (P = 0.0262 compared to HSP70i, and P = 0.0216 compared to media only; fig. S7C). Likewise, expression of the homing receptor CLA was significantly up-regulated in response to wild-type HSP70i (P = 0.0147 compared to HSP70iQ435A, and P = 0.0327 compared to media only; fig. S7D).
We also demonstrated that human skin cells express both wildtype and mutant HSP70i proteins after biolistic transfection (Fig. 8), indicating that DNA vaccination can affect human skin. This may be an important step in bringing HSP70iQ435A to the clinic. Together, these data reflect that the immune response in human tissues can be differentially driven by HSP70i, which may provide a crucial step in treating vitiligo.
Fig. 8.
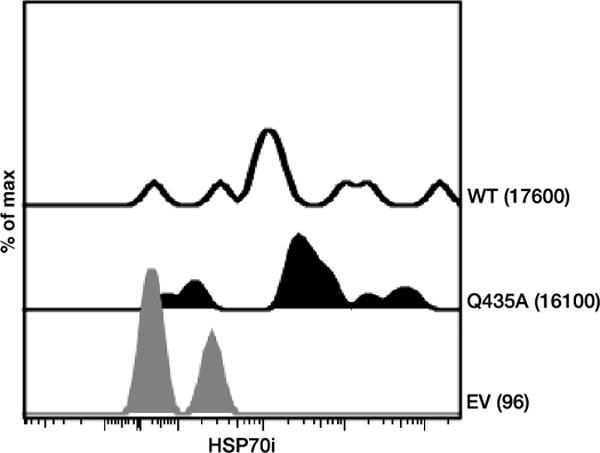
Transfected human skin expresses HSP70i. Skin biopsies were biolistically DNA-transfected with WT HSP70i (WT), HSP70iQ435A (Q435A), or empty vector and analyzed by flow cytometry 4 days later for expression of HSP70i. Histograms display the relative expression and MFI for each vaccination. Note marked expression of HSP70i as well as its mutant variant among cells collected from adult skin with the respective constructs. n = 3.
DISCUSSION
Differential expression of HSP70i has been observed among non-lesional and lesional human skin samples from vitiligo patients, which prompted us to investigate the phenotype of infiltrating DCs in perilesional vitiligo patient skin. A prevalence of CD11b+CD11c+ DCs as observed here is congruent with our hypothesis that HSP70i secreted under stress will activate local DCs and induce an inflammatory phenotype as previously observed in mice in response to the HSP70i vaccine (10), and this inflammatory CD11b+CD11c+ DC subset may provide a target for therapy. To better understand the role of HSP70i in autoimmune activation, and identify a region that may be blocked from binding and activating DCs, we generated DNA constructs containing either the N or the C terminus of the molecule, in effect comprising the adenosine triphosphate (ATP)– and substrate-binding domains. Indeed, there is discussion in the field as to the region within stress proteins most important to immune activation. Although protein fusion molecules have been generated that include only the substrate-binding region, others have suggested that the ATP-binding domain is required and sufficient for immune activation (29, 30). In part, this will be defined by the necessity to include an antigenic moiety that provides direction for the immune response to follow (31). Here, the C terminus, including the substrate-binding domain and some residues from the ATP-binding region, had even more pronounced effects than the full-length molecule. Subject to further analysis, it is possible that the compact size or folding of the resulting molecule, optimally exposing its immune-activating region, positively affects its DC binding ability.
The DC-activating peptide identified within HSP70i was derived by aligning the molecule to DnaK, looking for a sequence with high homology (18). Among mutant plasmids, some residues proved more critical than others, ultimately supporting the importance of our 11-mer for DC activation. The QPGVLIQVYEG sequence is shared among HSP70 isoforms and is highly conserved among species from protists to mice to humans (32, 33).
Among the HSP70i mutations that affected immune function, we initially selected HSP70iQ435A to further examine using in vivo effects in vitiligo-prone mice. Its position within the periphery of the molecule, strategically positioned to affect DC binding independent of bound substrate, together with the change from a charged, hydrophilic moiety (Q) to a smaller, hydrophobic one (A), suggests a potential role for enhanced aggregation or preferential receptor binding in the observed effects. This is important particularly as this moiety can affect the preferred receptor for wild-type versus mutant HSP70i. It stands to reason that engaging and disengaging DCs involves activation and possibly blocking of cell surface receptors. DC activation has been related to the relative engagement of C-type lectin receptors (CLRs) and Toll-like receptors (TLRs), with autoimmune pathology resulting from simultaneous activation of both and tolerance resulting from CLR activation in the absence of TLR engagement (34). Knowing that HSP70i can bind CLRs, including LOX1, as well as TLRs, including TLR2 and TLR4 (35), we can infer that the differential effects observed for mutant and wild-type HSP70i in these studies may be explained at least in part by differential binding to these receptors, resulting in a different DC phenotype and, in turn, in differential T cell activation and recruitment to the skin. This implies that both wild-type and mutant HSP70i bind DCs, which we showed here.
In Pmel-1 mice, T cells do not become spontaneously activated early on, possibly partly due to differences between the human epitope used to generate the model and the mouse epitope targeted in autoimmunity (21). These animals are thus ideal to study depigmentation after activation of melanocyte-reactive T cells, much as this occurs in humans. In this model, HSP70i is sufficient to induce depigmentation (10). However, natural depigmentation did not develop in animals vaccinated with HSP70iQ435A. Instead, we observed persistent skewing of the DC phenotype toward the inflammatory subset in response to wild-type HSP70i and encountered this subset among PBMCs and in skin of human patients with progressive disease. Conversely, we observed persistent skewing of the DC phenotype toward the tolerogenic subset in animals vaccinated with HSP70iQ435A. Also, HSP70iQ435A exposure similarly affected human DC activation in vitro. Our data support the concept that the direct effect of HSP70iQ435A is on DCs. The Q435A mutation may affect the ability of the resulting stress protein to bind and activate DCs based on differential engagement of receptors described for HSP70i (36).
We postulate that the resulting DC phenotypes support different cytokine profiles, necessary to activate or suppress relevant T and B cell subsets. Indeed differential Eomes/T-bet ratios as observed are likely to affect the effector phenotype and quiescent potential of resulting cytotoxic T cells (25). It has been shown that T-bet is the master regulator of type I effector differentiation, whereas low T-bet expression (and high Eomes expression) promotes memory formation (25). Exposure to wild-type HSP70i was accompanied by enhanced cytotoxic T cell responses and suppressed responses after exposure to HSP70iQ435A. The same shift toward increased Eomes/T-bet ratios found in mice treated with HSP70iQ435A was also observed in human skin explants. Among cells collected from HSP70iQ435A-treated human skin, Eomes/T-bet ratios were markedly increased. In addition, wild-type HSP70i was shown to up-regulate expression of both CXCR3 and the T cell surface marker CLA implicated in increased homing to vitiligo patient skin observed in earlier studies (28).
HSP70i vaccination stimulated humoral responses in mice, directed to the C terminus, leaving the question why naturally occurring antibodies do not deplete HSP70i and prevent the stress protein from inducing autoimmunity. Further analysis of the targeted sequence within HSP70i revealed that circulating antibodies bind downstream of our peptide of interest, offering an explanation for the coexistence of HSP70i overexpression and vitiligo development. Elevated serum levels of HSP70i have, in fact, been reported in coronary artery disease patients, where it was likewise determined that such antibodies do not affect disease expression (37).
Another vitiligo model included in our studies expresses a transgenic TCR to tyrosinase (22). The advantage of including this model is that spontaneous depigmentation initiates even before animals become available for vaccination. The ears and tail are then fully depigmented, and hair depigmentation initiates by 4 weeks of age. When DNA vaccination was initiated at 5 weeks of age, the pelage remained black yet melanocytes were lost from the hair follicles. Animals untreated or vaccinated with empty vector DNA at that age show a completely depigmented pelage after the hair has regrown. However, in animals treated with HSP70Q435A, the pelage returned pigmented; thus, melanocyte precursors must have migrated from the hair bulge to the follicle and differentiated to express tyrosinase, which is both the antigen recognized by the transgenic TCR as well as the enzyme responsible for pigmentation (38).
Using knockout mouse models, we demonstrated that both helper and cytotoxic T cells are required for depigmentation. Moreover, in both sets of vitiligo-prone mice, depigmentation was accompanied by relevant T cell infiltration to the skin. In the Pmel-1 model, CD8+ T cells were observed infiltrating the hair follicles where depigmentation takes place. In the h3TA2 model, we observed infiltration by transgenic TCR-expressing T cells that can react with remaining melanocytes (22). In both models, responses were muted by HSP70iQ435A, attesting to its amazing ability to deactivate an ongoing autoimmune response. Several lines of evidence support a particular function for stress proteins in DC activation. Similarly, an immature phenotype among DCs supports a regulatory function (39). Several recent studies suggest that immunizing autoimmune patients with tolerogenic DCs as a means of interfering with immune responses to self-antigens can be successful, and CD11b-expressing myeloid-derived suppressor cells have been widely implicated in tumor immune suppression (40, 41).
Last but not the least, we were able to induce elevated HSP70i expression, both wild-type and mutant, among lymphocytes collected from gene gun–vaccinated human skin. This provides a platform to translate our findings to a human setting; however, this is also a limitation of the study. Whereas this study suggests that gene gun vaccination may be effective in humans as well, current DNA applications in humans do not make use of a gene gun. We have yet to find whether other means of introducing DNA that are more readily supported by the U.S. Food and Drug Administration will show similar results in human skin, for example, tattooing or electroporation (42). Also, we do not yet know whether dampening immune responses by skin application of mutant HSP70i will have consequences for other, more desirable immune responses and perhaps affect systemic immunity as well. It is also possible that additional peptides within HSP70i affect DC activation and currently remain undetected, although this will not necessarily affect our proposed use of HSP70iQ435A in a therapeutic setting. Before performing an actual clinical trial, we cannot be certain that the impact observed on immunocyte profiles in human skin will likewise lead to improved skin pigmentation in treated human patients. Such studies can, however, be further designed following the results presented in our current article. Here, we provide evidence for the positive impact of HSP70iQ435A on DC and T cell profiles and show in vivo data demonstrating that HSP70iQ435A can affect depigmentation and repigmentation in mice, strongly suggesting that the same HSP70i variant will support repigmentation of lesional skin in human patients.
In summary, the current study gives rise to a potential treatment opportunity for vitiligo by introducing HSP70i with a single amino acid modification. Preclinical data obtained in mouse models reflecting the effector phase of the autoimmune depigmentation response support the use of HSP70iQ435A in patients. We have demonstrated that HSP70iQ435A DNA can be transfected in human skin and can alter the immune profile toward an anti-inflammatory phenotype similar to results observed in preclinical mouse models. The in vitro and in situ data support the potential significance for human vitiligo; however, it remains to be determined whether the same construct will be effective when treating patients with active disease. DNA vaccination has shown limited overall efficacy when applied to human subjects, yet when applied toward dermatologic conditions, the use of electroporation, for example, enables the next move to a clinical trial in perilesional vitiligo skin using existing constructs (43). Should protein or peptide-based constructs ultimately prove more effective than DNA-based applications, the next step will be to define the minimal sequence to exert an effect upon topical application. With the current strategy, the therapeutic value of HSP70iQ435A can offer a potential treatment for vitiligo.
MATERIALS AND METHODS
Mice
C57BL/6, B6.129S2-Cd4tm1Mak/J (CD4 knockout), B6.129S2-Cd8atm1Mak/J (CD8 knockout), and the vitiligo-prone 78B6.Cg-Thy1a/CyTg(TcraTcrb)8Rest/J (Pmel-1) gp100-reactive TCR transgenic mice were purchased from Jackson Laboratories. Vitiligo-prone h3TA2 tyrosinase-reactive TCR transgenic mice were generated in the laboratory of S.M. In Pmel-1 mice, the melanocyte-reactive, transgenic TCR is CD8-dependent and is thus expressed on most of the CD8+ T cells. In these mice, relevant T cells are followed by virtue of CD8 expression. In h3TA2 mice, however, the major histocompatibility complex class I–restricted, transgenic TCR was originally isolated from human CD4+ tumor-infiltrating lymphocytes and functions independent of CD8 co-receptor expression. Most of the transgenic T cells are CD4−CD8− in this model (22). In these mice, transgenic T cells were followed by CD3 expression and detection of the TCR transgene. Mice were 5 to 6 weeks old when included in experiments. Animals were gender-matched to wild-type controls where relevant. Experiments included 10 mice per group and are repeated twice unless stated otherwise in the figure legends. Mice were maintained in facilities approved by Loyola University Institutional Animal Care and Use Committee regulations and adhered to guidelines provided by the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Human tissues
Blood samples (30 ml) were obtained from patients with active vitiligo, defined as noticeable progression over the past 3 months. PBMCs were purified over a Ficoll density gradient, washed in phosphate-buffered saline, and stored in 10% dimethyl sulfoxide under liquid nitrogen until further use. Four-millimeter punch biopsies were obtained from the border of actively progressing vitiligo lesions in volunteer patients attending the Loyola Dermatology outpatient clinic or from patients undergoing unrelated surgeries at Rush University Medical Center. Tissues and blood samples were obtained from three or more patients per group, and staining was performed on 10,000 cells per fluorocytometry sample. Immunohistochemical analysis was performed at least in triplicate for each staining and on each tissue sample, on at least three sections for each staining. Actual sample sizes are listed in the figure legends. Informed consent was obtained from patients, and all samples were obtained with approval from the Institutional Review Board at Loyola University and at Rush University Medical Center, adopting the principles described in the Declaration of Helsinki.
Gene gun vaccinations
Vaccinations for all experiments were prepared as described previously (12) unless stated otherwise. Briefly, bullets were prepared by precipitating endotoxin-free plasmid DNA onto spermidine-coated gold beads (Fluka BioChemika and Sigma-Aldrich) and used to coat silicone tubing (Bio-Rad). All bullets were used within 14 days of preparation and maintained under vacuum before use. Mice were prepared for vaccination by biweekly abdominal hair removal with Nair (Church and Dwight Co.) and vaccinated with a Helios Gene Gun (Bio-Rad) under isoflurane anesthesia (E-Z Euthanex gas chamber, E-Z Systems Corp.). Unless otherwise stated, mice were ventrally vaccinated weekly for four consecutive weeks with a 4.8-μg dose of total DNA. Where combinations of plasmids were used, such DNA was mixed 1:1 to the same total dose of DNA and combined to prepare bullets.
Skin explant transfections and culture
For ex vivo skin explant gene transfections, skin from two volunteer patients was biopsied and 1 mm × 2 mm skin pieces (three) were placed on each Statamatrix matrix (Cytomatrix Pty Ltd.; a gift from the laboratory of R. A. Clark, Brigham and Women’s Hospital, MA). In one experiment, on day 10, the skin/matrix was transfected (4.8-μg dose of total DNA) with wild-type or mutant (Q435A) HSP70i. On day 14, media were collected and cells were stained for HSP70i expression. For another experiment (as indicated), the skin/matrix was cultured in media only or with wild-type or mutant (Q435A) HSP70i protein (4 μg/ml). Half of the skin T media were refreshed every 2 to 3 days. On day 6, media were collected and cells were stained for transcription factors and homing markers.
Evaluating depigmentation
Depigmentation was quantified by flatbed scanning of the ventral and dorsal sides of mice under anesthesia immediately before the initial vaccination and 4 weeks after final vaccination (6 months after the final vaccination in Pmel-1 mice), and subsequent image analysis was performed with Adobe Photoshop software (Adobe Systems Inc.). The percent depigmentation was calculated from the largest evaluable area as the percentage of pixels among >150,000 evaluated with a luminosity above the cutoff level set to include 95% of pixels for untreated mice (12). Mice were also photographed with a DSC-S950 SteadyShot digital camera (Sony) as indicated.
Immunohistology
After euthanasia, mouse skin biopsies from vaccinated sites were snap-frozen in OCT compound (Sakura Finetek). Cryostat sections (8 μm) were fixed in cold acetone, and indirect immunoperoxidase staining procedures were performed essentially as described previously (44). Tissue sections were treated with Super Block (ScyTek) to prevent nonspecific antibody binding. Primary antibodies Ta99 to mouse TRP-1 (mouse monoclonal; Covance), biotinylated 145-2C11 to CD3ɛ (Armenian hamster monoclonal; BD Pharmingen), biotinylated 53-6.7 to CD8α (mouse monoclonal; BioLegend), and 511 to Vβ12 (mouse monoclonal; Thermo Scientific) followed by horseradish peroxidase–conjugated secondary antibodies [streptavidin, goat anti-mouse immunoglobulin 2a (IgG2a), or goat anti-rat; Santa Cruz Biotechnology Inc.] were used. Enzymatic detection was finalized with aminoethylcarbazole as a substrate (Sigma). Staining was quantified as the number of cells per square millimeter of skin observed by light microscopy (Nikon) and Adobe Photoshop software (Adobe Systems Inc.).
Human skin sections were prepared as above. Primary antibodies EP1345Y to human CD11b (rabbit monoclonal; Novus Biologicals), B-ly6 to CD11c (mouse monoclonal; BD Pharmingen), 16H4L5 to T-bet (rabbit monoclonal; Invitrogen), rabbit serum to Eomes452–618 (rabbit polyclonal; Novus Biologicals), and 49801 to CXCR3 (mouse monoclonal; R&D Systems) followed by horseradish peroxidase–conjugated secondary antibodies (streptavidin, goat anti-mouse IgG2a, or goat anti-rabbit; Santa Cruz Biotechnology Inc.). Enzymatic detection and quantification was performed as described above.
Site-directed mutagenesis
Human HSP70i and microbial DnaK were aligned in a BLAST search to identify a 13–amino acid stretch as the putative DC-activating peptide region within human HSP70. To test this, we next created vectors with single- or double-nucleotide mutations in the putative DC-binding region of human HSP70i (HSP70i435–447) using appropriate primers to induce mutations in the sequence QPGVLIQVYEGER. As a template for the site-directed mutagenesis, our expression vector encoding HSP70i was used as described (12). In vitro mutagenesis of HSP70 residues was performed on a double-stranded template with polymerase chain reaction as previously described (45). Mutant sequences were verified by DNA sequencing (Sequenase version 2.0, U.S. Biochemical Corp.). To verify protein expression, we transfected COS7 cells (Invitrogen) with individual plasmids encoding human HSP70i or mutant HSP70i plasmids using Lipofectamine (Invitrogen), and we verified protein expression by Western blotting. Blots were probed with anti-HSP70 antibody (rabbit polyclonal antibody SPA-811 or mouse monoclonal antibody SPA-810; Assay Designs) and horseradish peroxidase–conjugated secondary antibodies (goat anti-rabbit or goat anti-mouse; Santa Cruz Biotechnology Inc.). Blots were developed with aminoethylcarbazole as a substrate (Sigma).
Activating immature DCs and in vitro cell cultures
Generation of DCs from human PBMCs was prepared from a modified protocol (27). Briefly, PBMCs were isolated on different occasions from whole blood of healthy volunteers by Ficoll-Paque (GE Healthcare) density gradient centrifugation. The experiment was performed three times with samples from different donors. Subsequently, monocytes were enriched with the EasySep Human Monocyte Enrichment Kit (Stemcell Technologies) according to the manufacturer’s instructions. Data from a representative experiment are shown. To generate DCs, we maintained the cells in Teflon containers (Savillex) to prevent adherence in RPMI medium (Mediatech Inc.) supplemented with 10% fetal bovine serum, penicillin (100 U/ml), streptomycin (100 mg/ml), recombinant human GM-CSF (100 ng/ml) (Leukine, Berlex), and recombinant human IL-4 (25 ng/ml) (R&D Systems). On day 7, either LPS (Sigma) or HSP70i (1 μg/ml) (BioVision) was added. To assess the wild-type and HSP70iQ435A isoforms, we isolated His-tagged protein from transfected COS7 cells with His-Select Spin Columns (Sigma) and added it at a final concentration of 1 μg/ml to the DCs. After 24 hours, expression of activation markers was assessed as described under flow cytometry.
Flow cytometry
For experiments including the Pmel-1 mice, splenocytes were recovered 25 weeks after the final vaccination and stained for the following surface markers: CD3, CD8, CD11b, CD11c, CLA, CXCR3, F4/80, and IgM-Thy1.2 (BD Biosciences). Initial gating was performed on live nondebris singlets, with subsequent gating toward CD11c+ versus CD11b+ cells using FACS LSR-II equipment (BD Biosciences). IgM+, Thy1.2+, and CD3+ cells were excluded from the final gating. For intracellular profiles, antibodies to CD62L, T-bet, Eomes, FoxP3, CD44, and CD25 (eBioscience) were used. For experiments using in vitro–generated human DCs from PBMCs, the cells were gently spun and stained with mouse anti-human CD11c, CD80, CD83, CD86, and HLA-ABC antibodies labeled with allophycocyanin, phycoerythrin, phycoerythrin-cyan, fluorescein isothiocyanate, and V450 fluorochromes, respectively (BD Biosciences). Initial gating was performed on live nondebris singlets, with subsequent gating toward CD11c+ cells using FACSCanto II and FACS LSR-II equipment (BD Biosciences). For ImageStream X (Amnis) analysis, cells were incubated with primary antibodies SPA-820 toward HSP70i (Assay Designs) and 3D5 toward 6× His (histidine tag; Invitrogen) followed by phycoerythrin goat anti-mouse IgG1 (BD Biosciences) and allophycocyanin goat anti-mouse IgG2b (BD Biosciences) secondary antibodies, respectively.
Statistical analyses
All data are presented as means ± SEM, unless otherwise indicated. Distributions of data and means were compared with Wilcoxon’s rank sum test for depigmentation and immunohistochemistry data throughout, and t tests allowing for unequal variance for fluorocytometry, and Western blot data, except when skewness of the data suggested that the rank test would be more appropriate. Degrees of freedom for two-sample t tests were calculated with Satterthwaite’s formula. Two-sided tests were performed except for two data sets where we are confirming a previously reported increase or decrease only (Western blot probed with SPA-810 and CXCR3 immunostaining of skin). Wilcoxon’s rank sum and t tests were calculated with Stata version 11 (StataCorp). Graphs were made with Prism software (GraphPad). P values less than 0.05 were considered statistically significant.
Supplementary Material
Materials and Methods
Fig. S1. SPA-810 antibody recognizes the DC-activating region of HSP70i.
Fig. S2. The C terminus induces a humoral response to HSP70i in vaccinated mice.
Fig. S3. Dorsal images of Pmel-1 mice after HSP70i vaccination.
Fig. S4. Skin melanocytes are lost before treatment in h3TA2 mice.
Fig. S5. T cells are required for autoimmune depigmentation.
Fig. S6. The T cell–homing antigen CXCR3 is overexpressed in actively depigmenting vitiligo skin.
Fig. S7. Wild-type and mutant HSP70i differentially activate antigen-challenged human peripheral blood cells.
Table S1. Vitiligo patient information.
Acknowledgments
We thank the vitiligo and non-vitiligo patients who contributed tissue samples; P. Simms (Loyola, IL) for her skillful assistance with flow cytometric data; and R. A. Clark and J. Teague (Brigham and Women’s Hospital, MA) for supplying the Statamatrix matrices and skin culture protocol.
Funding: This work was supported by NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases grant RO1AR054749 to I.C.L.P and NIH Grants R21 AR56524 to S.M., and R01 CA104947 as well as PO1 CA154778 to M.I.N.
Footnotes
Author contributions: J.A.M., A.Z., A.T.L., J.M.E., and F.J.K. performed the experiments in TCR transgenic and knockout mice, including analytical experiments and manuscript preparation. E.J.H. and A.A. performed human tissue matrix and PBMC culture experiments. J.D.N., P.K., and C.J.D. were engaged in mutagenesis, subcloning, and associated wild-type mouse experiments. A.O. and C.H. were engaged in the delivery and interpretation of human tissue data. S.M. and M.I.N. provided, promoted, and helped interpret data for the h3TA2 TCR transgenic strain. T.H., S.D.B., and H.L.K. were engaged in the acquisition and processing of non-vitiligo human skin samples. E.G.-M. performed statistical data analysis. J.A.G.-P. and I.C.L.P. provided overall interpretation and oversight, and finalized the manuscript.
Competing interests: Provisional patent applications U.S. Serial No. 60/904,550 “Domain in human heat shock protein 70 (HSP70) involved in the induction of autoimmune diseases such as vitiligo” (J.D.N. and I.C.L.P.) and International PCT patent application no. PCT/US12/053139 “Mutant HSP70i to prevent autoimmune disease” (A.Z., J.A.G.-P., and I.C.L.P.) were filed related to the work in this manuscript. The authors declare no other competing interests.
REFERENCES AND NOTES
- 1.Das PK, van den Wijngaard RM, Wankowicz-Kalinska A, Le Poole IC. A symbiotic concept of autoimmunity and tumour immunity: Lessons from vitiligo. Trends Immunol. 2001;22:130–136. doi: 10.1016/s1471-4906(00)01844-5. [DOI] [PubMed] [Google Scholar]
- 2.Cedercreutz K, Denman CJ, Klarquist J, Vaitla R, Boissy RE, Westerhof W, Hernandez C, Le Poole IC. Vitiligo etiology and treatment: Parameters derived from a patient survey. J Dermatol Nurs Assoc. 2010;2:265–272. [Google Scholar]
- 3.Colucci R, Lotti T, Moretti S. Vitiligo: An update on current pharmacotherapy and future directions. Expert Opin Pharmacother. 2012;13:1885–1899. doi: 10.1517/14656566.2012.712113. [DOI] [PubMed] [Google Scholar]
- 4.Teraki Y, Hitomi K, Sato Y, Izaki S. Tacrolimus-induced rosacea-like dermatitis: A clinical analysis of 16 cases associated with tacrolimus ointment application. Dermatology. 2012;224:309–314. doi: 10.1159/000338693. [DOI] [PubMed] [Google Scholar]
- 5.Hossani-Madani AR, Halder RM. Topical treatment and combination approaches for vitiligo: New insights, new developments. G Ital Dermatol Venereol. 2010;145:57–78. [PubMed] [Google Scholar]
- 6.Fongers A, Wolkerstorfer A, Nieuweboer-Krobotova L, Krawczyk P, Tóth GG, van der Veen JP. Long-term results of 2-mm punch grafting in patients with vitiligo vulgaris and segmental vitiligo: Effect of disease activity. Br J Dermatol. 2009;161:1105–1111. doi: 10.1111/j.1365-2133.2009.09367.x. [DOI] [PubMed] [Google Scholar]
- 7.Wańkowicz-Kalińska A, van den Wijngaard RM, Tigges BJ, Westerhof W, Ogg GS, Cerundolo V, Storkus WJ, Das PK. Immunopolarization of CD4+ and CD8+ T cells to type-1–like is associated with melanocyte loss in human vitiligo. Lab Invest. 2003;83:683–695. doi: 10.1097/01.lab.0000069521.42488.1b. [DOI] [PubMed] [Google Scholar]
- 8.Oyarbide-Valencia K, van den Boorn JG, Denman CJ, Li M, Carlson JM, Hernandez C, Nishimura MI, Das PK, Luiten RM, Le Poole IC. Therapeutic implications of autoimmune vitiligo T cells. Autoimmun Rev. 2006;5:486–492. doi: 10.1016/j.autrev.2006.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kroll TM, Bommiasamy H, Boissy RE, Hernandez C, Nickoloff BJ, Mestril R, Le Poole I Caroline. 4-Tertiary butyl phenol exposure sensitizes human melanocytes to dendritic cellmediated killing: Relevance to vitiligo. J Invest Dermatol. 2005;124:798–806. doi: 10.1111/j.0022-202X.2005.23653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosenson JA, Zloza A, Klarquist J, Barfuss AJ, Guevara-Patino JA, Poole IC. HSP70i is a critical component of the immune response leading to vitiligo. Pigment Cell Melanoma Res. 2012;25:88–98. doi: 10.1111/j.1755-148X.2011.00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Poole IC, Luiten RM. Autoimmune etiology of generalized vitiligo. Curr Dir Autoimmun. 2008;10:227–243. doi: 10.1159/000131485. [DOI] [PubMed] [Google Scholar]
- 12.Denman CJ, McCracken J, Hariharan V, Klarquist J, Oyarbide-Valencia K, Guevara-Patiño JA, Le Poole IC. HSP70i accelerates depigmentation in a mouse model of autoimmune vitiligo. J Invest Dermatol. 2008;128:2041–2048. doi: 10.1038/jid.2008.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asea A. Mechanisms of HSP72 release. J Biosci. 2007;32:579–584. doi: 10.1007/s12038-007-0057-5. [DOI] [PubMed] [Google Scholar]
- 14.Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: Chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- 15.Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: Role of Toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 16.Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- 17.Becker T, Hartl FU, Wieland F. CD40, an extracellular receptor for binding and uptake of Hsp70-peptide complexes. J Cell Biol. 2002;158:1277–1285. doi: 10.1083/jcb.200208083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Whittall T, McGowan E, Younson J, Kelly C, Bergmeier LA, Singh M, Lehner T. Identification of stimulating and inhibitory epitopes within the heat shock protein 70 molecule that modulate cytokine production and maturation of dendritic cells. J Immunol. 2005;174:3306–3316. doi: 10.4049/jimmunol.174.6.3306. [DOI] [PubMed] [Google Scholar]
- 19.Pulendran B, Tang H, Denning TL. Division of labor, plasticity, and crosstalk between dendritic cell subsets. Curr Opin Immunol. 2008;20:61–67. doi: 10.1016/j.coi.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17–producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 21.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mehrotra S, Al-Khami AA, Klarquist J, Husain S, Naga O, Eby JM, Murali AK, Lyons GE, Li M, Spivey ND, Norell H, Martins da Palma T, Onicescu G, Diaz-Montero CM, Garrett-Mayer E, Cole DJ, Le Poole IC, Nishimura MI. A coreceptor-independent transgenic human TCR mediates anti-tumor and anti-self immunity in mice. J Immunol. 2012;189:1627–1638. doi: 10.4049/jimmunol.1103271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Poole IC, van den Wijngaard RM, Westerhof W, Das PK. Presence of T cells and macrophages in inflammatory vitiligo skin parallels melanocyte disappearance. Am J Pathol. 1996;148:1219–1228. [PMC free article] [PubMed] [Google Scholar]
- 24.Guevara-Patiño JA, Engelhorn ME, Turk MJ, Liu C, Duan F, Rizzuto G, Cohen AD, Merghoub T, Wolchok JD, Houghton AN. Optimization of a self antigen for presentation of multiple epitopes in cancer immunity. J Clin Invest. 2006;116:1382–1390. doi: 10.1172/JCI25591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gregg RK, Nichols L, Chen Y, Lu B, Engelhard VH. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J Immunol. 2010;184:1909–1917. doi: 10.4049/jimmunol.0902778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Neill DW, Bhardwaj N. Current Protocols in Immunology. John Wiley & Sons Inc.; New York: 2001. [Google Scholar]
- 28.van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, Tigges B, Westerhof W, Das P. Local immune response in skin of generalized vitiligo patients. Destruction of melanocytes is associated with the prominent presence of CLA+ T cells at the perilesional site. Lab Invest. 2000;80:1299–1309. doi: 10.1038/labinvest.3780138. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Kelly CG, Singh M, McGowan EG, Carrara AS, Bergmeier LA, Lehner T. Stimulation of Th1-polarizing cytokines, C-C chemokines, maturation of dendritic cells, and adjuvant function by the peptide binding fragment of heat shock protein 70. J Immunol. 2002;169:2422–2429. doi: 10.4049/jimmunol.169.5.2422. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Wang W, Li Q, Huang W. Fusion protein of ATPase domain of Hsc70 with TRP2 acting as a tumor vaccine against B16 melanoma. Immunol Lett. 2006;105:167–173. doi: 10.1016/j.imlet.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Javid B, MacAry PA, Lehner PJ. Structure and function: Heat shock proteins and adaptive immunity. J Immunol. 2007;179:2035–2040. doi: 10.4049/jimmunol.179.4.2035. [DOI] [PubMed] [Google Scholar]
- 32.Germot A, Philippe H. Critical analysis of eukaryotic phylogeny: A case study based on the HSP70 family. J Eukaryot Microbiol. 1999;46:116–124. doi: 10.1111/j.1550-7408.1999.tb04594.x. [DOI] [PubMed] [Google Scholar]
- 33.Adams E, Britton WJ, Morgan A, Goodsall AL, Basten A. Identification of human T cell epitopes in the Mycobacterium leprae heat shock protein 70-kD antigen. Clin Exp Immunol. 1993;94:500–506. doi: 10.1111/j.1365-2249.1993.tb08225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geijtenbeek TBH, van Vliet SJ, Engering A, ’t Hart BA, van Kooyk Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu Rev Immunol. 2004;22:33–54. doi: 10.1146/annurev.immunol.22.012703.104558. [DOI] [PubMed] [Google Scholar]
- 35.Delneste Y, Magistrelli G, Gauchat J, Haeuw J, Aubry J, Nakamura K, Kawakami-Honda N, Goetsch L, Sawamura T, Bonnefoy J, Jeannin P. Involvement of LOX-1 in dendritic cellmediated antigen cross-presentation. Immunity. 2002;17:353–362. doi: 10.1016/s1074-7613(02)00388-6. [DOI] [PubMed] [Google Scholar]
- 36.Zitzler S, Hellwig A, Hartl FU, Wieland F, Diestelkötter-Bachert P. Distinct binding sites for the ATPase and substrate binding domain of human Hsp70 on the cell surface of antigen presenting cells. Mol Immunol. 2008;45:3974–3983. doi: 10.1016/j.molimm.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 37.Kocsis J, Veres A, Vatay A, Duba J, Karádi I, Fust G, Prohászka Z. Antibodies against the human heat shock protein hsp70 in patients with severe coronary artery disease. Immunol Invest. 2002;31:219–231. doi: 10.1081/imm-120016242. [DOI] [PubMed] [Google Scholar]
- 38.Watabe H, Valencia JC, Yasumoto K, Kushimoto T, Ando H, Muller J, Vieira WD, Mizoguch M, Appella E, Hearing VJ. Regulation of tyrosinase processing and trafficking by organellar pH and by proteasome activity. J Biol Chem. 2004;279:7971–7981. doi: 10.1074/jbc.M309714200. [DOI] [PubMed] [Google Scholar]
- 39.Diao J, Mikhailova A, Tang M, Gu H, Zhao J, Cattral MS. Immunostimulatory conventional dendritic cells evolve into regulatory macrophage-like cells. Blood. 2012;119:4919–4927. doi: 10.1182/blood-2011-11-392894. [DOI] [PubMed] [Google Scholar]
- 40.Ioanno M, Alissafi T, Lazarisdis I, Deraos G, Matsoukas J, Gravani A, Mastorodemos V, Plaitakis A, Sharpe A, Boumpas D, Verginis P. Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J Immunol. 2012;188:1136–1146. doi: 10.4049/jimmunol.1101816. [DOI] [PubMed] [Google Scholar]
- 41.Rezzonico R, Chicheportiche R, Imbert V, Dayer JM. Engagement of CD11b and CD11c β2 integrin by antibodies or soluble CD23 induces IL-1β production on primary human monocytes through mitogen-activated protein kinase–dependent pathways. Blood. 2000;95:3868–3877. [PubMed] [Google Scholar]
- 42.Bolhassani A, Safaiyan S, Rafati S. Improvement of different vaccine delivery systems for cancer therapy. Mol Cancer. 2011;10(3) doi: 10.1186/1476-4598-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murakami T, Sunada Y. Plasmid DNA gene therapy by electroporation: Principles and recent advances. Curr Gene Ther. 2011;11:447–456. doi: 10.2174/156652311798192860. [DOI] [PubMed] [Google Scholar]
- 44.Le Poole IC, van den Wijngaard RM, Westerhof W, Dutrieux RP, Das PK. Presence or absence of melanocytes in vitiligo lesions: An immunohistochemical investigation. J Invest Dermatol. 1993;100:816–822. doi: 10.1111/1523-1747.ep12476645. [DOI] [PubMed] [Google Scholar]
- 45.Jones DH, Sakamoto K, Vorce RL, Howard BH. DNA mutagenesis and recombination. Nature. 1990;344:793–794. doi: 10.1038/344793a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods
Fig. S1. SPA-810 antibody recognizes the DC-activating region of HSP70i.
Fig. S2. The C terminus induces a humoral response to HSP70i in vaccinated mice.
Fig. S3. Dorsal images of Pmel-1 mice after HSP70i vaccination.
Fig. S4. Skin melanocytes are lost before treatment in h3TA2 mice.
Fig. S5. T cells are required for autoimmune depigmentation.
Fig. S6. The T cell–homing antigen CXCR3 is overexpressed in actively depigmenting vitiligo skin.
Fig. S7. Wild-type and mutant HSP70i differentially activate antigen-challenged human peripheral blood cells.
Table S1. Vitiligo patient information.