

Abstract
Loss of cochlear hair cells in mammals is currently believed to be permanent, resulting in hearing impairment that affects more than 10% of the population. Here, we developed two genetic strategies to ablate neonatal mouse cochlear hair cells in vivo. Both Pou4f3DTR/+ and Atoh1-CreER™; ROSA26DTA/+ alleles allowed selective and inducible hair cell ablation. After hair cell loss was induced at birth, we observed spontaneous regeneration of hair cells. Fate-mapping experiments demonstrated that neighboring supporting cells acquired a hair cell fate, which increased in a basal to apical gradient, averaging over 120 regenerated hair cells per cochlea. The normally mitotically quiescent supporting cells proliferated after hair cell ablation. Concurrent fate mapping and labeling with mitotic tracers showed that regenerated hair cells were derived by both mitotic regeneration and direct transdifferentiation. Over time, regenerated hair cells followed a similar pattern of maturation to normal hair cell development, including the expression of prestin, a terminal differentiation marker of outer hair cells, although many new hair cells eventually died. Hair cell regeneration did not occur when ablation was induced at one week of age. Our findings demonstrate that the neonatal mouse cochlea is capable of spontaneous hair cell regeneration after damage in vivo. Thus, future studies on the neonatal cochlea might shed light on the competence of supporting cells to regenerate hair cells and on the factors that promote the survival of newly regenerated hair cells.
Keywords: Lgr5, Direct transdifferentiation, Mitotic regeneration, Diphtheria toxin, Atoh1, Fate mapping
INTRODUCTION
Hair cells (HCs) regenerate in both the auditory and vestibular systems of non-mammalian vertebrates, leading to restoration of hearing and balance (Balak et al., 1990; Corwin and Cotanche, 1988; Lombarte et al., 1993; Ryals and Rubel, 1988). This process occurs by two mechanisms: direct transdifferentiation and mitotic regeneration. Direct transdifferentiation refers to a cell fate change when neighboring supporting cells (SCs) convert into HCs without cell division. Mitotic regeneration occurs when a SC first divides and, subsequently, one or both daughter cells becomes a HC (Adler and Raphael, 1996; Baird et al., 1996; Corwin and Cotanche, 1988; Jones and Corwin, 1996; Ryals and Rubel, 1988; Warchol and Corwin, 1996).
In mammals, limited HC regeneration occurs in the vestibular system (Burns et al., 2012; Forge et al., 1993; Golub et al., 2012; Kawamoto et al., 2009; Warchol et al., 1993), yet no spontaneous regeneration has been observed in the mature auditory system (Bohne et al., 1976; Hawkins et al., 1976; Oesterle et al., 2008). Recent studies demonstrate that SCs isolated from the neonatal cochlea are competent to form new HCs in culture (Chai et al., 2012; Doetzlhofer et al., 2006; Oshima et al., 2007; Savary et al., 2007; Shi et al., 2012; Sinkkonen et al., 2011; White et al., 2006). In addition, neonatal SCs can be induced to generate supernumerary HCs upon inhibition of the Notch pathway (Doetzlhofer et al., 2009; Yamamoto et al., 2006), ectopic expression of Atoh1 (Kelly et al., 2012; Liu et al., 2012a; Zheng and Gao, 2000) or overexpression of β-catenin (Shi et al., 2013). Similar manipulations failed to coerce a HC fate in the undamaged, adult cochlea, suggesting that the neonatal cochlea is a more permissive environment for the formation of new HCs.
To investigate possible HC regeneration in the embryonic cochlea, Kelley and colleagues laser ablated HCs in cultured explants and found rare regenerated HCs (Kelley et al., 1995). Whether the postnatal cochlea can regenerate lost HCs and the source of potential regenerated cells have not been clearly defined, in part because HCs in the neonatal cochlea are insensitive to damage in vivo. Aminoglycoside antibiotics are widely used to damage HCs in vitro but preferentially inflict damage in the basal turn and are ineffective in vivo.
Here, we present two strategies to kill neonatal HCs in vivo using mouse genetics. After HC death was induced at birth, fate-mapping studies showed that SCs acquire a HC fate. We also observed mitotic regeneration, with regenerated cells expressing five markers of HCs and exhibiting immature stereocilia bundles, although most new HCs failed to survive. In addition, we defined the time period when HC regeneration can occur, finding it to be limited to the first postnatal week. Together, these findings demonstrate that neonatal SCs have the intrinsic capacity to regenerate HCs after damage.
RESULTS
Hair cell ablation in the neonatal cochlea
The neonatal cochlea is resistant to HC damage caused by exposure to noise or ototoxic drugs in vivo. To circumvent this limitation, we developed two genetic methods to damage neonatal HCs in vivo. First, we used a knock-in mouse in which expression of the human diphtheria toxin receptor (DTR) is driven by the Pou4f3 promoter (Pou4f3DTR/+) (Golub et al., 2012; Mahrt et al., 2013; Tong et al., 2011) and selective HC death is induced by injection of diphtheria toxin (DT), as Pou4f3 is exclusively expressed by HCs in the inner ear (Erkman et al., 1996; Xiang et al., 1998). Progressive HC death was observed in Pou4f3DTR/+ mice after DT injection at P1 (Fig. 1), consistent with previous reports (Golub et al., 2012; Mahrt et al., 2013; Tong et al., 2011).
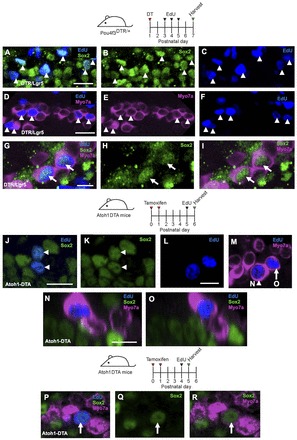
Fig. 1.
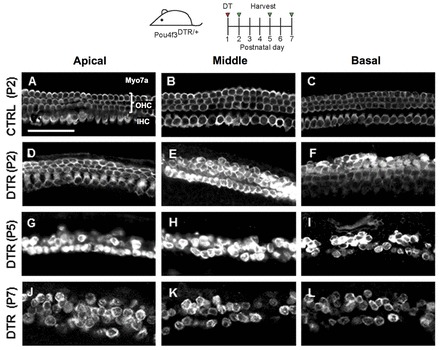
Progressive HC death in the Pou4f3DTR/+ model. Projection images of Myo7a immunofluorescence in cochlear whole-mounts of control wild-type mice at P2 (A-C) and Pou4f3DTR/+ mice at P2 (D-F), P5 (G-I) and P7 (J-L) after diphtheria toxin (DT) injection at P1. Repopulation of HCs was most robust in the apical turn at P7 (J). OHC, outer hair cells; IHC, inner hair cells. Scale bar: 50 μm.
Second, we used a HC-specific inducible Cre line, Atoh1-CreER™, to drive expression of diphtheria toxin fragment A (DTA). When tamoxifen was administrated at postnatal day (P) 0 and P1, Cre recombinase was activated in ∼80-90% of HCs (Chow et al., 2006; Weber et al., 2008). In other organ systems, Cre-mediated excision of the floxed stop sequence in the ROSA26-loxP-stop-loxP-DTA (ROSA26DTA) allele causes cell-autonomous ablation of Cre+ cells (Abrahamsen et al., 2008; Ivanova et al., 2005). Beginning 2 days after tamoxifen administration, Atoh1-CreER™; ROSA26DTA/+ (Atoh1DTA) mice show rapid and reproducible loss of both inner and outer HCs (Fig. 2A-O). There is also considerable disorganization in the organ of Corti, with Sox2+ nuclei detected in the HC layer (Fig. 2P,Q).
Fig. 2.
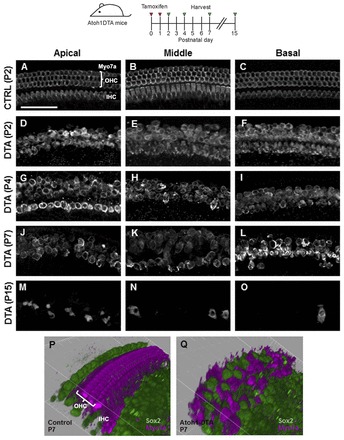
Progressive HC death in the Atoh1DTA model. (A-O) Projection images of Myo7a immunofluorescence in cochlear whole-mounts from control mice (lacking either the Cre or DTA allele) at P2 (A-C) and Atoh1DTA mice at P2 (D-F), P4 (G-I), P7 (J-L) and P15 (M-O). Repopulation of HCs was most robust in the apical turn at P4 (G). (P,Q) 3D reconstruction of confocal z-stack images with SC nuclei labeled by Sox2 (green) and HCs labeled by Myo7a (magenta) in the middle turn of control (P) and Atoh1DTA (Q) cochleae at P7. Scale bar: 50 μm.
Supporting cells acquire a hair cell fate
To determine how SCs respond to HC damage induced at birth and whether they could acquire a HC fate, we generated Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ transgenic mice. This strategy was designed to fate map SCs using the Lgr5-EGFP-IRES-CreERT2 allele (Lgr5CreER) (Barker et al., 2007) and the ROSA26CAG-tdTomato reporter line (Madisen et al., 2010) after HC ablation. Lgr5 is expressed in a subset of SCs, so when control animals (Pou4f3+/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice) were given tamoxifen at P1, tdTomato expression was detected at P7 in several SC subtypes, including cells in the greater epithelial ridge (GER). Specifically, tdTomato expression was detected in Deiters’ cells (first row, 4.0±1.8%; second row, 5.0±0.3%; third row, 98.3±0.4%), pillar cells (outer pillar, 3.7±2.0%; inner pillar, 67.0±1.6%) and inner phalangeal/border cells (87.3±0.8%) (n=3). Also, we detected occasional tdTomato+/Myosin VIIa+ (Myo7a+) cells at P7 (apex, 12±1.5; middle, 2±0.7; base, 0.5±0.3; n=4; Fig. 3A,H-J; supplementary material Table S2A). These results are consistent with the previous report (Chai et al., 2012). Lgr5 expression decreased and remained limited to SCs in Pou4f3DTR/+; Lgr5CreER/+ mice 8, 24 and 48 hours after DT injection at P1 (supplementary material Fig. S1).
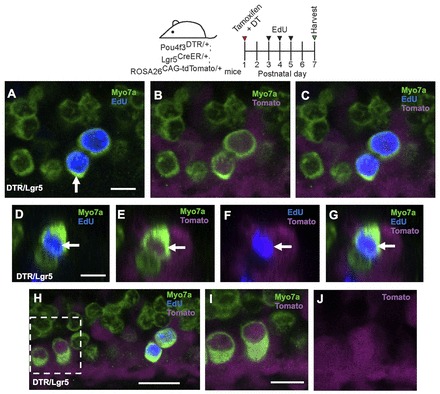
Fig. 3.
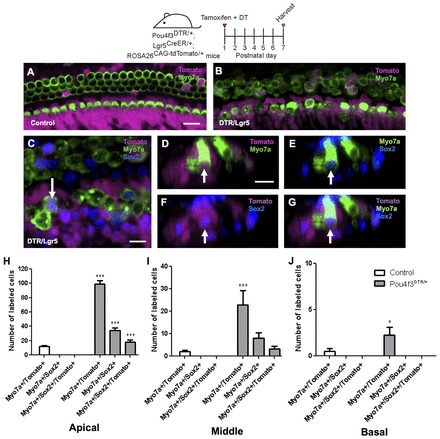
Fate mapping of SCs in the Pou4f3DTR/+ model. Confocal images of tdTomato+ (magenta) HCs (Myo7a, green) in the apical cochlear turn of control (Lgr5CreER/+; ROSA26CAG-tdTomato/+) (A) and Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ (B) mice at P7. (C) Confocal image of tdTomato+/Myo7a+ HCs that also express Sox2 (blue) in the apical turn of Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice at P7. (D-G) Cross-section focused on the tdTomato+/Sox2+ HC indicated by the arrow in C. Note that GFP expression from the Lgr5CreER/+ allele is much weaker than Sox2 labeling. Number of double (Myo7a+/tdTomato+ or Myo7a+/Sox2+) or triple (Myo7a+/Sox2+/tdTomato+) labeled cells in the apical (H), middle (I) and basal (J) turns of Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice and control littermates. Data are expressed as mean ± s.e.m., n=3. *P<0.05, ***P<0.001 compared with control number of the corresponding turn as determined by a two-way ANOVA followed by a Student’s t-test with a Bonferroni correction. Scale bars: A,B, 20 μm; C-G, 10 μm.
In Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice, tamoxifen was given at P1 to label Lgr5+ SCs and DT ∼8 hours later to kill HCs. At P7, we found a significant increase of tdTomato+/Myo7a+ cells in all three cochlear turns, with a nearly 10-fold increase in the apical and middle turns and a 5-fold increase in the basal turn (apex, 99.0±4.6; middle, 22.8±6.5; base, 2.3±0.9; n=4; Fig. 3B,H-J; supplementary material Table S2A) compared with undamaged controls lacking the Pou4f3DTR/+ allele (P<0.001 for apical and middle turns and P<0.05 for base). In this experiment, we also stained for Sox2, which is transiently expressed in nascent HCs in the embryonic cochlea (Dabdoub et al., 2008; Hume et al., 2007; Kiernan et al., 2005; Mak et al., 2009) and becomes restricted to SCs after P1 (Hume et al., 2007; Oesterle et al., 2008). In addition, Myo7a+ cells that formed from isolated SCs in vitro expressed Sox2 (Sinkkonen et al., 2011). Therefore, Sox2 and Myo7a co-expression can be viewed as a marker of immature HCs. After HC ablation, we found many Myo7a+/Sox2+ cells and Myo7a+/Sox2+/tdTomato+ cells in the apical and middle turns of Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice at P7 (Fig. 3C-J; supplementary material Table S2A), whereas neither were observed in control samples lacking the Pou4f3DTR/+ allele (n=4). In summary, these findings demonstrate that newly regenerated Myo7a+ cells are derived from adjacent SCs after HC ablation in the neonatal mouse cochlea. This regenerative capacity was most robust in the apex and decreased towards the base.
Similarly, we performed fate-mapping studies of SCs in the Atoh1DTA model. Although fate mapping is primarily performed using the Cre/loxP system, we were already using this system to kill HCs and thus needed another mouse genetic tool to trace SCs (Stern and Fraser, 2001). Since Hes5 is expressed in SCs of the postnatal cochlea (Hartman et al., 2009; Lanford et al., 2000; Li et al., 2008; Zine et al., 2001), we characterized a recently generated Hes5-nlsLacZ knock-in allele (Imayoshi et al., 2010). lacZ was strongly expressed throughout the P1 cochlea (Fig. 4A,B) and robustly labeled Deiters’ cells and outer pillar cells, with mosaic weak labeling of inner phalangeal cells/border cells (Fig. 4C-H). lacZ expression was not detected in inner pillar cells or HCs. For fate mapping, we generated Atoh1DTA; Hes5-nlsLacZ+/- mice. After tamoxifen injection at P0/P1, we first observed Myo7a+/lacZ+ cells at P2 (Fig. 4I-L) in an apical-basal gradient (apex, 58.3±30.2; middle, 9.3±7.9; base, 1.3±1.3; n=3), whereas no lacZ+ HCs were found in control samples that were lacking either the Cre or DTA allele (Fig. 4M; supplementary material Table S1B) (n=3) or at P1 in experimental or control cochleae (n=3). This finding is consistent with the fate-mapping experiments performed in the Pou4f3DTR/+ model indicating that SCs have changed cell fate and differentiated into Myo7a+ cells in vivo.
Fig. 4.
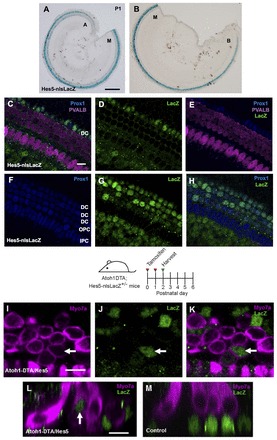
Fate mapping of SCs in the Atoh1DTA model. (A,B) X-Gal staining (blue) in Hes5-nlsLacZ cochlea at P1. Cochlear turns are labeled as apical (A), middle (M) and basal (B). (C-H) Confocal images of the apical turn of Hes5-nlsLacZ mice at P1. lacZ expression is detected with anti-β-gal antibody (green) and is specific to SCs. HCs are labeled by parvalbumin (PVALB; magenta). No β-gal+ cells were detected in control samples lacking the Hes5-nlsLacZ allele. Deiters’ cells (DC), outer pillar cells (OPC) and inner pillar cells (IPC) are labeled by Prox1 (blue). (I-K) Confocal images of β-gal+ (green) HCs (Myo7a, magenta) in the apical turn of Atoh1DTA; Hes5-nlsLacZ+/- mice at P2. (L) Cross-section focused on the β-gal+ HC labeled by the arrow in I-K. (M) Transverse section of a littermate control (lacking either the Cre or DTA allele) at P2, in which all β-gal+ cells are in the SC nuclear layer. Scale bars: 200 μm in A,B; 10 μm in C-M.
Hair cell damage stimulates proliferation
Since mitotic HC regeneration has been described in non-mammalian vertebrates, we investigated whether cell division can occur after HC damage in the neonatal mouse cochlea. When the mitotic tracer 5-ethynyl-2′-deoxyuridine (EdU) was administered once per day from P3-P5 to control mice lacking the Pou4f3DTR/+ allele (and DT given at P1), no EdU+ cells were observed in the organ of Corti at P7 (n=4), confirming previous findings that postnatal HCs and SCs are mitotically quiescent (Lee et al., 2006; Ruben, 1967). In Pou4f3DTR/+ mice identically treated with DT and EdU, EdU+/Sox2+ cells were observed in the P7 organ of Corti (apex, 13.2±3.7 cells per 225 μm cochlear length; middle, 5.8±1.6; base, 3.5±1.5; n=6; Fig. 5A-C; supplementary material Table S2C). We also observed EdU+/Myo7a+ cells restricted to the apical turn, averaging 11.0±1.8 in this whole region (n=6) and suggesting mitotic HC regeneration (Fig. 5D-F; supplementary material Table S2C). Moreover, some cells that were double positive for EdU and Myo7a also had Sox2+ nuclei (4.7±1.3, n=6; Fig. 5G-I; supplementary material Table S2C), suggesting that they were immature, regenerated HCs.
Fig. 5.
Mitotic HC regeneration in the neonatal mouse cochlea. Confocal images of EdU (blue) incorporation in Sox2+ SCs (green, A-C) and Myo7a+ cells (magenta, D-F) in the apical turn of Pou4f3DTR/+ mice at P7 after EdU injections at P3-P5. Some EdU+ HCs (Myo7a+) were also co-labeled with Sox2 (G-I). EdU+ SCs (Sox2, J,K) and EdU+/Myo7a+ cells (L,M) were also observed in the apical turn of Atoh1DTA mice 24 hours after EdU injection at P5. (N-O) Cross-section focused on the EdU+ nucleus of the cells indicated by the arrowhead and arrow in M. (P-R) Confocal images of EdU+ (blue) HCs (Myo7a, magenta) co-labeled with Sox2 (green) in the apical turn of Atoh1DTA mice 24 hours after EdU injection at P4. Scale bars: 20 μm in A-F; 10 μm in G-O.
In parallel, we probed for proliferative cells after HC loss in Atoh1DTA mice by injecting EdU once at ages ranging from P2 to P5. Cochlear tissues were analyzed 24 hours after each injection when one round of cell division was presumably complete. Similar to the results obtained with Pou4f3DTR/+ mice, this regimen also yielded no EdU+ cells in the organ of Corti from control mice that lacked either the Cre or DTA allele (n=3). By contrast, EdU+/Sox2+ cells were found at the SC nuclear level in the damaged organ of Corti in all three turns between P2 and P5 (Fig, 5J,K; supplementary material Table S2D). In addition, we observed EdU+/Myo7a+ cells in the apical turn only from P2-P5 (P2, 2.0±2.0; P3, 3.3±0.9; P4, 3.7±2.3; P5, 1.0±0.6; Fig. 5L,M; supplementary material Table S2D; n=3). EdU+/Myo7a+ cells were found at the lumenal surface of the sensory epithelium where HCs normally reside (Fig. 5N,O). As in the Pou4f3DTR/+ model, we also detected EdU+/Myo7a+/Sox2+ cells (Fig. 5P-R). These data show that HC ablation results in proliferation in the normally mitotically quiescent organ of Corti in both the Pou4f3DTR/+ and Atoh1DTA models. The majority of proliferation and all EdU+/Myo7a+ cells were observed in the apical turn, where the progression of cell cycle arrest first occurs (Lee et al., 2006; Ruben, 1967) and where HC differentiation is last observed during development (Lumpkin et al., 2003; Montcouquiol and Kelley, 2003).
Mitotic hair cell regeneration
To determine whether SCs can proliferate and differentiate into HCs, we next treated Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice with tamoxifen and DT at P1, followed by the mitotic tracer EdU at P3-P5. At P7, we observed 6.7±0.6 EdU+/Myo7a+/tdTomato+ cells in the whole apical turn and none in the middle and basal turns (Fig. 6A-G; supplementary material Table S2E; n=3). In the same organs, there were also EdU-/Myo7a+/tdTomato+ cells (apex, 106.3±14.8; middle, 25.7±5.5; base, 2.7±1.5; Fig. 6H-J). Control animals (Pou4f3+/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+) receiving the same drug regimen did not exhibit any EdU+ cells and significantly fewer EdU-/Myo7a+/tdTomato+ cells than in damaged organs (apex, 9.8±2.1; middle, 1.3±0.5; base, 0.3±0.5; n=6; supplementary material Table S2E), which is similar to the number of Myo7a+/tdTomato+ cells found in controls without EdU injection (Fig. 3A,H-J; supplementary material Table S2A). The finding of EdU+/Myo7a+/tdTomato+ cells likely indicate that Lgr5+ SCs proliferated prior to acquiring a HC fate, consistent with mitotic HC regeneration described previously in non-mammalian species (Baird et al., 1996; Corwin and Cotanche, 1988; Jones and Corwin, 1996; Ryals and Rubel, 1988; Warchol and Corwin, 1996). Although substantially more EdU+/Sox2+ cells were found in all three cochlear turns (supplementary material Table S2C), mitotic HC regeneration (EdU+/Myo7a+/tdTomato+ cells) appears limited to the apical turn and represents 5.9% of total fate-mapped, regenerated HCs in this region.
Fig. 6.
Both mitotic regeneration and direct transdifferentiation occur in the neonatal mouse cochlea. (A-C) Confocal images of tdTomato+ (magenta) HCs (Myo7a, green) that are labeled by EdU (blue) in the apical turn of Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice at P7 after EdU injections at P3-P5. (D-G) Cross-section focused on the tdTomato+/EdU+ HC indicated by the arrow in A. Note that GFP expression from the Lgr5CreER/+ allele is much weaker than EdU labeling. In the apical turn of the same organs, there were also EdU-/Myo7a+/tdTomato+ cells (H-J). (I,J) Higher magnification of the boxed region in H. Scale bars: 20 μm.
We next sought to investigate whether original, differentiated HCs could also contribute to the observed EdU+/Myo7a+ cells. Based on reporter assays using the ROSA26lacZ allele, ∼80-90% of HCs in Atoh1-CreER™ mice were lacZ+ at P6 after tamoxifen injection at P0/P1 (Chow et al., 2006; Weber et al., 2008). Since the ROSA26DTA allele uses the same promoter as the ROSA26lacZ allele, ∼80-90% of HCs in our model probably expressed DTA and thus ∼10-20% of original, differentiated HCs might remain. To explore the possibility that these surviving HCs serve as a source of EdU+/Myo7a+ cells in the Atoh1DTA cochleae, we traced HCs using the ROSA26CAG-tdTomato allele, which labels 99.5±0.2% of HCs at P6 in Atoh1-CreER™ mice after tamoxifen injection at P0/P1 (Fig. 7A,B). We injected tamoxifen at P0/P1 and then EdU at P3 or P4 in Atoh1-CreER™; ROSA26DTA/CAG-tdTomato mice and analyzed the cochlea 24 hours later. All EdU+/Myo7a+ cells (12 cells from six mice) were tdTomato- (Fig. 7C-E), suggesting that EdU+/Myo7a+ cells are likely to be derivatives of surrounding SCs and not surviving HCs, since they were not present when tamoxifen was injected at P0/P1 to turn on the tdTomato reporter.
Fig. 7.
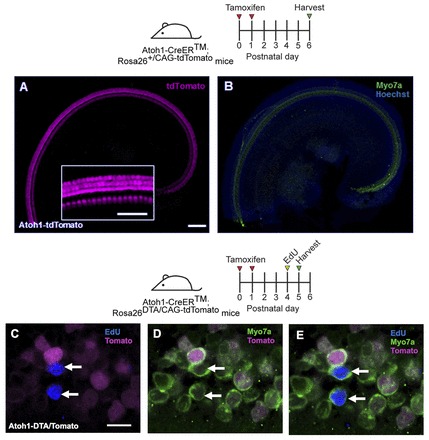
Regenerated HCs are not derived from original, differentiated HCs. (A,B) Confocal images of tdTomato+ (magenta) HCs (Myo7a, green) in the apical turn of Atoh1-CreER™; ROSA26CAG-tdTomato mice at P6 after tamoxifen injection at P0/P1. Nuclei are labeled by Hoechst (blue). Inset is a high-magnification image of tdTomato-labeled HCs. (C-E) Confocal images of EdU (blue) incorporation in Myo7a+ cells in the apical turn of Atoh1-CreER™; ROSA26DTA/CAG-tdTomato mice 24 hours after EdU injection at P4. DTA- HCs were traced with tdTomato (magenta). All EdU+/Myo7a+ cells were tdTomato-. Scale bars: 100 μm in A,B, 50 μm in inset; 10 μm in C-E.
During development, mitosis of HC precursors terminates by E14.5 (Lee et al., 2006; Ruben, 1967), and HC differentiation and maturation is a dynamic process that occurs over 3 perinatal weeks in mice. Based on our findings that HC loss stimulates SC proliferation, we investigated whether newly generated Myo7a+ cells could also be active in the cell cycle. We analyzed cochleae just 4 hours after EdU injection at P4 in Atoh1DTA mice, as the mammalian cell cycle usually takes ∼24 hours to complete (Alberts et al., 2002). We found 2±0.5 EdU+/Myo7a+ cells (n=4) in the apical turn (Fig. 8A-D). Moreover, we observed several Myosin VI+ (Myo6+) cells that were co-labeled with the M-phase marker phospho-histone H3 (pH3) (Fig. 8E-H) and an EdU+/calbindin+ cell with mitotic figures (Fig. 8I-M), providing further evidence that rare Myo7a+ cells can be active in the cell cycle. Likewise, in the Pou4f3DTR/+ model, we observed rare pH3+/Myo7a+ cells (Fig. 8N,O). pH3 labeling only marks cells that are active in M phase [which lasts ∼1 hour (Alberts et al., 2002)] at the time of tissue collection, and therefore most likely underestimates the total number of dividing cells over the several days following HC ablation. To exclude the possibility that EdU+/Myo7a+ cells are in the process of dying, we performed terminal dUTP nick end labeling (TUNEL) staining in the Atoh1DTA model and failed to detect degenerating cells (Kuan et al., 2004) in any of the nine EdU+/Myo7a+ cells from seven mice (data not shown).
Fig. 8.
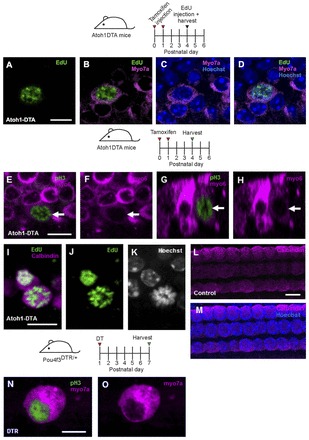
Myo7a+ cells are active in the cell cycle. (A-D) Confocal image of EdU incorporation (green) in Myo7a+ cells (magenta) in the Atoh1DTA model 4 hours after EdU injection at P4. (E-H) A pH3+ (green) HC (Myo6, magenta) was observed at P4 in the Atoh1DTA model. (G-H) Cross-section focused on the pH3+ HC indicated by the arrow in E. Note that Myo6 expression of this cell is less robust than in adjacent cells. (I-K) An EdU+ (green) HC (calbindin, magenta) with mitotic figures (Hoechst, grayscale) was observed in the Atoh1DTA model 24 hours after EdU injection at P4. (L,M) Control tissue (lacking either the Cre or DTA allele) shows expression of calbindin (magenta) in HC nuclei in tissues that were processed for EdU staining. (N,O) A pH3+ (green) HC (Myo7a, magenta) was observed at P7 in the Pou4f3DTR/+ model. Scale bars: 10 μm.
Taken together, these findings from the Atoh1DTA and Pou4f3DTR/+ models suggest that it is possible for SCs acquiring a HC fate to be active in the cell cycle. Considering that the frequency of EdU labeling was higher in Sox2+ cells than in Myo7a+ cells, it is plausible that the proliferative capacity of SCs gradually diminishes as they convert to a HC fate.
Maturation of regenerated hair cells
To elucidate whether regenerated HCs can survive and mature, we used EdU to trace newly generated Myo7a+ cells at longer recovery time points in the Atoh1DTA model. EdU+/Myo7a+ cells expressed other markers of HCs, including calbindin (Fig. 9A-C), parvalbumin (Fig. 9D-F) and prestin (Slc26a5 - Mouse Genome Informatics) (Fig. 9G-I). As precursor cells acquire a HC fate these markers are expressed in a stepwise fashion, with Myo7a and calbindin among the first proteins to be expressed (Dechesne and Thomasset, 1988; Montcouquiol and Kelley, 2003), followed by parvalbumin (Zheng and Gao, 1997) and finally prestin, a marker of terminal differentiation of outer HCs (Belyantseva et al., 2000; Legendre et al., 2008; Zheng et al., 2000). Myo7a and calbindin were detected as early as 4 hours after EdU injection, whereas cells double positive for EdU and parvalbumin were not found until 2 days after EdU injection. At 6 days post EdU injection, 46.7±13.3% of EdU+/Myo7a+ cells also expressed prestin. We did not detect the inner HC marker VGlut3 (Slc17a8 - Mouse Genome Informatics) in any EdU+/Myo7a+ cells at P10 or P15 (supplementary material Fig. S2), suggesting that regenerated HCs preferentially feature an outer HC phenotype. Under scanning electron microscopy, all stereocilia bundles in the apical turn of P15 Atoh1DTA cochleae appeared short and tightly packed and thus immature (Fig. 9M-O). Some bundles contained a kinocilium (Fig. 9O), which normally regresses by P10 (Sobkowicz et al., 1995). We confirmed that newly formed HCs have stereocilia bundles using EdU as a tracer and detected EdU+/Myo7a+ cells with espin+ stereocilia bundles at P15 (Fig. 9J-L). Together, these data support the notion that EdU+/Myo7a+ cells are regenerated HCs that acquire features of endogenous HCs. Furthermore, the timing of expression for these HC markers in EdU+ cells closely follows that of developing HCs, supporting the notion that regenerated HCs mature in a similar pattern to that seen in development.
Fig. 9.
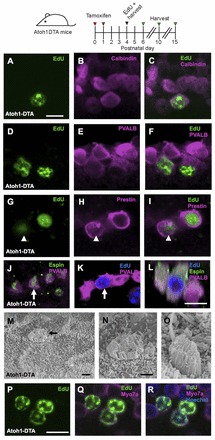
Regenerated HCs are similar to endogenous HCs. Confocal images of EdU+ (green) cells co-labeled with HC markers in the apical turn of Atoh1DTA mice. (A-C) Four hours after EdU injection at P5, EdU+ cells also express calbindin (magenta). (D-F) Two days after EdU injection at P4 (at P6), EdU+ cells express parvalbumin (PVALB; magenta). (G-I) Six days after EdU injection at P4 (at P10), EdU+ cells express prestin (magenta). Note that prestin is expressed in the cytoplasm of the EdU+ HC indicated by the arrowhead, which is characteristic of a young HC (Mahendrasingam et al., 2010). (J-L) Eleven days after EdU injection at P4 (at P15), EdU+ cells (blue) have espin+ (green) stereocilia bundles. (L) Cross-section focused on the EdU+ HCs indicated by the arrow in J and K. (M-O) Scanning electron micrographs of the apical turn in Atoh1DTA mice at P15. (N) High-magnification image of M, showing an immature stereocilia bundle. The immature bundle in O still has a kinocilium. (P-R) Confocal images of EdU+ (green) HCs (Myo7a, magenta) in the apical turn of Atoh1DTA mice at P10 after EdU injection at P4. Scale bars: 10 μm in A-L,P-R; 1 μm in M-O.
Some regenerated HCs survived for more than a week, as EdU+/Myo7a+ cells were still found at P10 and P15 after EdU injection at P4 (Fig. 9P-R). However, the majority of regenerated HCs died progressively, with only a small number remaining at P15 (Fig. 2M-O).
We investigated potential factors that might be linked to the death of regenerated HCs. Pou4f3 is crucial for HC survival. In mice with germline deletion of Pou4f3, HCs initially form but die 1-2 weeks later (Erkman et al., 1996; Xiang et al., 1998). In the apical turn of P6 Atoh1DTA mice, out of 81.3±4.5 Myo7a+ HCs (per 200 μm region), only 11.0±1.8 expressed Pou4f3 (13.9±4.4%), whereas all 104.0±8.3 HCs in control mice (lacking either the Cre or DTA allele) were Myo7a+/Pou4f3+ (100%) (supplementary material Fig. S3; n=3). Since the Atoh1DTA model targets ∼80-90% of HCs, our data suggest that the majority of regenerated HCs lack a key intrinsic survival factor.
Because a mosaic loss of SCs (<40% loss) also leads to HC death (Mellado Lagarde et al., 2013), we quantified fate-mapped SCs after HC ablation in Pou4f3DTR/+ mice to determine whether SC loss also occurs. After tamoxifen and DT were administrated to P1 Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice, we noted at P7 significantly fewer tdTomato-labeled Sox2+ SCs in the organ of Corti in comparison with undamaged controls (Pou4f3+/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice) (supplementary material Table S2F; P<0.01). There were ∼28-35% fewer SCs in all three cochlear turns and also a net loss of total traced organ of Corti cells (supplementary material Table S2F; P<0.05 in apex and P<0.01 in middle and base). As an additional control, we compared tdTomato+ cell counts in the GER, a region more remote from HC ablation, and did not observe any significant change (supplementary material Table S2F).
Last, we made auditory measurements of P30 Atoh1DTA mice and found them to exhibit elevated thresholds for auditory brainstem response across all frequencies tested (supplementary material Fig. S4). Prior work on HC ablation (at P2) using the Pou4f3DTR/+ allele similarly found elevated auditory thresholds in adult animals (Mahrt et al., 2013). In summary, the absence of Pou4f3 in HCs and/or the degeneration of surrounding SCs might have contributed to the poor survival of regenerating HCs and, consequently, to hearing loss.
Spontaneous hair cell regeneration can no longer occur after hair cell damage at 1 week
To determine when the neonatal cochlea loses the ability to spontaneously regenerate HCs, we ablated HCs and performed fate mapping of SCs using Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice that were given tamoxifen at P1 and DT at P6, and found no significant difference in the number of Myo7a+/tdTomato+ cells at P9 (apex, 15.3±0.9; middle, 0.3±0.3; base, 0±0; n=3) compared with control that lacked the Pou4f3DTR/+ allele (apex, 13.8±0.9; middle, 0.3±0.3; base, 0±0; n=4) (Fig. 10A-K).
Fig. 10.
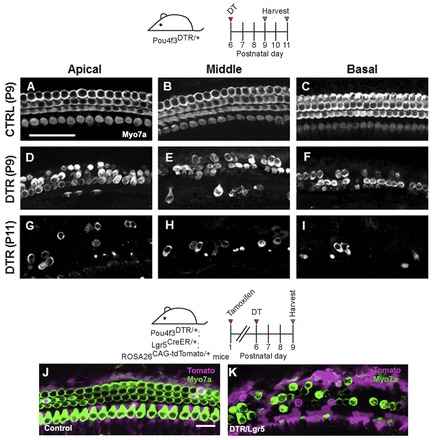
No signs of HC regeneration when HC loss occurs 1 week after birth in the Pou4f3DTR/+ model. Projection images of Myo7a immunofluorescence in cochlear whole-mounts of control wild-type mice at P9 (A-C) and Pou4f3DTR/+ mice at P9 (D-F) and P11 (G-I) after DT injection at P6. Confocal images of tdTomato+ (magenta) HCs (Myo7a, green) in the apical turn of control (Lgr5CreER/+; ROSA26CAG-tdTomato/+) (J) and Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ (K) mice that were given tamoxifen at P1, DT at P6, and analyzed at P9. Scale bars: 50 μm in A-I; 20 μm in J,K.
In parallel, we induced DTA expression in HCs at P6. Since Atoh1 is rapidly downregulated after birth (Lumpkin et al., 2003), we instead used Prestin-CreERT2 mice (Fang et al., 2012). When injected with tamoxifen (once daily from P6-8), all outer HCs in Prestin-CreERT2; ROSA26CAG-ZsGreen/+ mice expressed the reporter ZsGreen (Fig. 11A-C). When tamoxifen was administered in the same fashion, Prestin-CreERT2; ROSA26DTA/+ (PrestinDTA) mice had progressive outer HC loss, whereas inner HCs remained intact (Fig. 11D-L). We also gave one EdU injection to PrestinDTA mice at ages ranging from P7 to P11 and analyzed the cochlea 24 hours after each injection (n=3). No EdU+ SCs or EdU+/Myo7a+ cells were observed in the organ of Corti in any cochlear turn. Together, these data support the notion that proliferation and HC regeneration after HC loss only take place within a limited time window in the neonatal mouse cochlea.
Fig. 11.
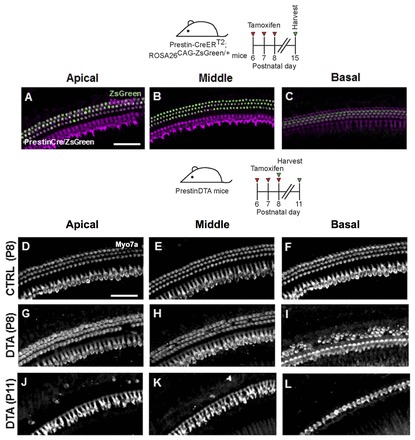
No signs of HC regeneration when HC loss occurs 1 week after birth in the PrestinDTA model. (A-C) Prestin-CreERT2; ROSA26CAG-ZsGreen/+ mice induced with tamoxifen from P6-P8 and analyzed at P15 have robust Cre activity in all outer HCs. HCs are labeled with Myo7a (magenta). Projection images of Myo7a immunofluorescence in cochlear whole-mounts of control mice (lacking the Cre or DTA allele) at P8 (D-F) and PrestinDTA mice at P8 (G-I) and P11 (J-L) given tamoxifen from P6-P8. Scale bars: 50 μm.
DISCUSSION
Previous studies in mice have reported that isolated, postmitotic SCs can divide and generate HCs in vitro (Chai et al., 2012; Doetzlhofer et al., 2006; Savary et al., 2007; Shi et al., 2012; Sinkkonen et al., 2011; White et al., 2006) and that HC regeneration occurs in embryonic cochlear explants in vitro after laser ablation of HCs, with regeneration ending ∼48 hours after HC fate commitment (Kelley et al., 1995). In other studies, HCs were damaged in the neonatal rat cochlea using antibiotic treatment in vivo and in vitro and transient ‘atypical cells’ were found that resembled immature HCs, with tufts of microvilli in the damaged regions (Daudet et al., 1998; Lenoir and Vago, 1997; Parietti et al., 1998; Romand et al., 1996; Zine and de Ribaupierre, 1998). However, it is unclear whether these cells expressed HC markers or if mitotic regeneration occurred. Moreover, the ototoxic antibiotic treatments were unable to damage HCs in the apical turn of the cochlea and most analyses were undertaken many days after HC damage. In our experiments using an effective method to eliminate neonatal HCs throughout the cochlea, we observed spontaneous HC regeneration during the first postnatal week. Fate-mapping experiments using a SC-specific CreER line and the Hes5-nlsLacZ allele reveal SCs as the source of newly regenerated HCs.
In experiments in which the Hes5-nlsLacZ allele was used to fate map SCs, the lacZ+/Myo7a+ cells observed are most likely to be regenerated HCs derived from Hes5-expressing SCs, particularly when interpreted in conjunction with the fate-mapping results obtained using the Cre-loxP system in the Pou4f3DTR model. It is possible, but unlikely, that Myo7a+ cells de-differentiated and upregulated Hes5, considering the antagonistic effect of Hes5 on the HC differentiation factor Atoh1 (Doetzlhofer et al., 2009; Kelley, 2006).
HC regeneration in non-mammalian vertebrates occurs by two mechanisms: mitotic regeneration and direct transdifferentiation. During mitotic regeneration, a SC first divides and then several days later one or both daughter cells changes fate to become a HC (Adler and Raphael, 1996; Baird et al., 1996; Corwin and Cotanche, 1988; Jones and Corwin, 1996; Ryals and Rubel, 1988; Warchol and Corwin, 1996). Fate mapping of SCs in our models revealed traced Myo7a+ cells; thus, HC loss led SCs towards a HC fate in all turns of the cochlea, albeit predominantly in the apex. By applying mitotic tracers, we detected EdU+ SCs and fate-mapped EdU+/Myo7a+ cells, indicating that the neonatal mouse cochlea can, to a limited extent, proliferate in response to HC loss and that some of these proliferating SCs can acquire a HC fate. Interestingly, we observed the expression of the mitotic markers EdU and pH3 in Myo7a+ and Myo7a+/Sox2+ cells, which likely represent a differentiating SC or an immature HC. We postulate that the processes of cell cycle entry and that of differentiation towards a HC fate might overlap. Future studies using time-lapse imaging would be useful in investigating these steps further.
The other reported mechanism for HC regeneration is direct transdifferentiation, in which a SC directly acquires a HC phenotype without mitotic division (Adler and Raphael, 1996; Baird et al., 1996; Jones and Corwin, 1996). The experiments reported here did not directly address this mechanism because of the limited labeling efficiency of the mitotic tracers used. However, the number of EdU-/Myo7a+/tdTomato+ cells observed in Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ mice was ∼15-fold greater than the number of EdU+/Myo7a+/tdTomato+ cells detected, which suggests that some of these cells were derived from direct transdifferentiation. The avian auditory epithelium regenerates via direct transdifferentiation 1-2 days after ototoxic antibiotic exposure (Cafaro et al., 2007; Roberson et al., 2004), and this mechanism has been reported to be the primary mode of HC regeneration in the adult mouse utricle (Forge et al., 1993; Forge et al., 1998; Golub et al., 2012; Kawamoto et al., 2009). In addition, Lgr5+ SCs can generate HCs via both mechanisms in vitro (Chai et al., 2012). Of note, both methods of fate mapping might underestimate the total number of regenerated HCs resulting from direct transdifferentiation because of the incomplete labeling with the Cre-loxP system or Hes5-nlsLacZ allele and the possibility of other progenitor cell types.
The vast majority of regenerated HCs were observed in the apical turn of the cochlea. Our results are in agreement with previous findings that in vitro HC regeneration after laser ablation of embryonic HCs decreased in a basal-apical gradient (Kelley et al., 1995). Both HCs and SCs continue to mature during the first postnatal weeks, with cytoskeletal, morphological and functional changes detected in cells from the basal turn 2-3 days before cells in the apex (Hallworth et al., 2000; Jensen-Smith et al., 2003; Legendre et al., 2008; Lelli et al., 2009; Szarama et al., 2012). Thus, cells in the apical turn are less mature, which might provide a permissive environment for HC regeneration. Alternatively, the presence of two different HC regeneration mechanisms working in concert in the apex might indicate the presence of undifferentiated progenitor cells. It is worth noting that the apical turn of the cochlea is the first to exit the cell cycle and the last to acquire a HC or SC fate during embryonic development (Lee et al., 2006; Lumpkin et al., 2003; Montcouquiol and Kelley, 2003; Ruben, 1967).
Previous studies characterized an age-dependent decrease in stem/progenitor cells isolated from the cochlea (Oshima et al., 2007; White et al., 2006). This decline correlates well with our finding that the ability of the cochlea to spontaneously regenerate HCs diminished 1 week after birth, suggesting the loss of either progenitor cell competence or a corresponding niche. In support of this theory, there is an age-dependent decline in the ability of the cochlea to respond to Atoh1-mediated conversion of SCs into HCs (Kelly et al., 2012; Liu et al., 2012a) and Sox2/p27Kip1 control of SC proliferation (Liu et al., 2012b). Such an age-dependent decline in the ability to self-repair has been observed in other organ systems: hearts from P1 mice can regenerate after damage and this regenerative capacity is lost in P7 mice (Porrello et al., 2011). Interestingly, heart regeneration correlated with the endogenous proliferative capacity of cardiac tissue, whereas we found regeneration capacity in the cochlea more than a week after the cells in the organ of Corti had become quiescent.
Based on the timing of expression for various HC markers, we conclude that regenerated HCs follow a similar pattern of maturation to normal HC development. Interestingly, the Atoh1DTA model retains conditions favorable for the expression of prestin, a terminal outer HC marker that is not expressed in SC-derived HCs generated from ectopic expression of Atoh1 (Liu et al., 2012a). Unfortunately, regenerated HCs only contributed to a modest and transient degree of repopulation of the organ of Corti, and most regenerated HCs died by P15. The inability of regenerated HCs to survive could be caused by the lack of the survival factor Pou4f3 and/or loss of SCs, which normally provide structural and trophic support. In the described HC damage models, 80-90% of HCs degenerate and more than 100 SCs per cochlea change fate to become HCs, with a small amount of proliferation that is evidently insufficient to compensate for the overall cell loss. As a result, the regenerating organ of Corti is not only disarrayed, but contains an abnormal composition of HCs and SCs, which might affect the survival of regenerated HCs. In addition, the endocochlear potential normally develops between P11 and P17 (Rybak et al., 1992) and might play a role in the death of regenerated HCs by causing excitotoxicity. It is probable that these factors, individually or in combination, contribute to the demise of regenerated HCs. Identification of factors capable of promoting the survival and functional maturation of regenerated HCs could have significant therapeutic benefits.
Finally, regenerated HCs in our models come from unmanipulated SCs; thus, the regeneration process we observed is their natural response to HC death. The Atoh1DTA and Pou4f3DTR/+ models likely induce HC death by reactive oxygen species-induced apoptosis (Abrahamsen et al., 2008; Ivanova et al., 2005), which is similar to the major cell death pathway implicated in noise- or drug-induced HC death (Clerici et al., 1996; Henderson et al., 2006). Thus, our observations are likely to have implications for HC regeneration in response to various insults.
In summary, our data demonstrate that the postnatal mammalian cochlea has the intrinsic capacity to spontaneously regenerate HCs after damage, which was believed to occur only in non-mammalian vertebrates. These models are applicable to the further study of the molecular mechanisms of mammalian HC regeneration, which could lead to the identification of drug targets for the treatment of hearing loss in humans and improve our understanding of factors that promote the survival of regenerated HCs and factors that limit HC regeneration in the maturing cochlea.
MATERIALS AND METHODS
Mouse models and treatments
ROSA26DTA (Ivanova et al., 2005), Lgr5CreER/+ (Barker et al., 2007), ROSA26CAG-tdTomato/+ (Madisen et al., 2010) and ROSA26CAG-ZsGreen/+ (Madisen et al., 2010) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Atoh1-CreER™ (Chow et al., 2006), Pou4f3DTR/+ (Golub et al., 2012; Tong et al., 2011) and Hes5-nlsLacZ (Imayoshi et al., 2010) mice were kind gifts from S. Baker (St. Jude Children’s Research Hospital, Memphis, TN, USA), E. Rubel, L. Tong and R. Palmiter (University of Washington, Seattle, WA, USA) and R. Kageyama at (Kyoto University, Kyoto, Japan). Prestin-CreERT2 mice were generated in our lab (Fang et al., 2012). Genotyping for Lgr5CreER/+, ROSA26CAG-tdTomato/+, ROSA26CAG-ZsGreen/+, Atoh1-CreER™ and Prestin-CreERT2 mice was described previously (Barker et al., 2007; Chow et al., 2006; Fang et al., 2012; Madisen et al., 2010). Genotyping for ROSA26DTA mice, Hes5-nlsLacZ mice and Pou4f3DTR/+ mice is described in supplementary material Table S1. Tamoxifen [3 mg/40 g body weight, intraperitoneal injection (IP); Sigma] was injected once at P0 and P1 for Atoh1-CreER™ mice and once at P6, P7 and P8 for Prestin-CreERT2 mice. Tamoxifen (0.75 mg/g) was given by gavage to Lgr5CreER/+ mice. DT (6.25 ng/g, IP, List Biological Laboratories) was injected at either P1 or P6 into Pou4f3DTR/+ mice. EdU (Click-iT EdU Imaging Kit, Invitrogen) was injected (50 μg/g, IP) once daily at P3-P5 into Pou4f3DTR/+ and Atoh1DTA mice; EdU was injected (10 μg/g, IP) once at the designated ages. Mice of both genders were used in this study. The n value throughout the paper reflects the number of animals analyzed per experiment. All animal work was approved by the Institutional Animal Care and Use Committees at St. Jude Children’s Research Hospital and Stanford University.
Immunostaining
Cochlear samples were fixed in 4% paraformaldehyde overnight and standard immunohistochemistry was performed (Mellado Lagarde et al., 2013). For Sox2 staining, 0.02% sodium azide was added during the blocking and antibody incubation steps. TUNEL staining was performed using the In Situ Cell Death Detection Kit, TMR Red (Roche Applied Science) following the manufacturer’s instructions, except for increasing the sodium citrate concentration in the permeabilization step to 1%. The following primary antibodies were used: anti-β-gal (1:500, AB9361 Abcam), anti-calbindin (1:500, AB1778 Millipore), anti-espin (1:5000, a gift from Dr S. Heller, Stanford University, Palo Alto, CA, USA), anti-Myo6 conjugated to Alexa 647 (1:40, specific request Proteus BioSciences), anti-Myo7a (1:200, 25-6790 Proteus BioSciences), anti-pH3 conjugated to Alexa 555 (1:50, 3475 Cell Signaling), anti-Prox1 (1:500, AB5475 Millipore), anti-parvalbumin (1:1000, P3088 Sigma), anti-VGlut3 (1:500, 135203 Synaptic Systems, Goettingen, Germany), anti-prestin (1:200, sc-22692 Santa Cruz Biotechnology), anti-Sox2 (1:1000, sc-17320 Santa Cruz Biotechnology) and anti-Pou4f3 (1:500, sc-81980 Santa Cruz Biotechnology). Nuclear staining employed Hoechst 33342 (1:2000, Invitrogen). EdU staining was performed using the Click-iT EdU Imaging Kit (Invitrogen) following the manufacturer’s instructions. All secondary antibodies were Alexa conjugated (Invitrogen) and were used at 1:500 or 1:1000. Images were taken using a Zeiss LSM 700 confocal microscope and 3D reconstruction was performed using Imaris 7.1 software (Bitplane).
Cell counts
For counts of traced HCs in the Pou4f3DTR/+; Lgr5CreER/+; ROSA26CAG-tdTomato/+ and Atoh1DTA; Hes5-nlsLacZ samples, we imaged the entire cochlea using a 40× objective and counted Myo7a+ or Sox2+/Myo7a- cells that co-expressed lacZ or tdTomato. The same procedure was used to quantify EdU+/Myo7a+ cells, while EdU+ or traced SC counts were obtained from counting 225 μm regions of each turn. For Pou4f3+ HC counts, two 200 μm regions of the apical turn were analyzed.
Scanning electron microscopy
Samples were prepared as previously described (Steigelman et al., 2011) and were imaged using a JEOL 7000 field emission gun scanning electron microscope (University of Alabama, Tuscaloosa, AL, USA).
Auditory brainstem response
Animals were anesthetized with Avertin (0.6 mg/g, IP) and frequency-specific auditory responses were measured using the Tucker-Davis Technology System III System (Alachua, FL, USA) as previously described (Mellado Lagarde et al., 2013).
Statistical analysis
All data are presented as mean ± s.e.m. Statistical analyses were conducted using GraphPad Prism 5.0 software.
Supplementary Material
Acknowledgments
We thank E. Rubel, L. Tong and R. Palmiter (University of Washington) for Pou4f3DTR/+ mice and discussion; S. Baker (St. Jude) for Atoh1-CreER™ mice and discussion; R. Kageyama (Kyoto University) for Hes5-nlsLacZ mice; P. Chambon (Institut Genetique Biologie Moleculaire Cellulaire) for the CreERT2 construct; S. Heller (Stanford University) for the anti-espin antibody and critical reading, J. Corwin, J. Burns and other members of the Corwin laboratory (University of Virginia) as well as members of our laboratories for discussion and critical comments; S. Connell, V. Frohlich, Y. Ouyang and J. Peters (St. Jude) for expertise in confocal imaging; A. Xue, V. Nookala, N. Pham, A. Vu, G. Huang and W. Liu (Stanford University) for excellent technical support; and L. Boykins (University of Memphis), R. Martens and J. Goodwin (University of Alabama) for assistance and expertise in scanning electron microscopy.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
B.C.C., R.C., A.G.C. and J.Z. developed the concepts or approach; B.C.C., R.C., A.L., Z.L., L.Z., D.-H.N., K.C., K.A.S., J.F., A.G.C. and J.Z. performed experiments or data analysis; B.C.C., R.C., A.G.C. and J.Z. prepared or edited the manuscript prior to submission.
Funding
This work was supported by grants from the National Institutes of Health [DC006471 (J.Z.), DC008800 (J.Z.), DC009393 (K.A.S.), DC010310 (B.C.C.), DC011043 (A.G.C.), CA21765 (St. Jude) and P30DC010363 (Stanford University)], Office of Naval Research [N000140911014, N000141210191 and N000141210775 to J.Z.], Stanford Dean’s Fellowship (R.C.), Hearing Health Foundation (R.C.), National Organization for Hearing Research Foundation [J.F. and A.G.C.], Stanford Children’s Health Research Institute Akiko Yamazaki and Jerry Yang Faculty Scholar Fund (A.G.C.), California Institute for Regenerative Medicine [RN3-06529 to A.G.C.] and American Lebanese Syrian Associated Charities (ALSAC) of St. Jude Children’s Research Hospital. We thank the St. Jude/University of Paris Diderot/Paris 7 graduate student program for support of A.L. J.Z. is a recipient of The Hartwell Individual Biomedical Research Award. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.103036/-/DC1
References
- Abrahamsen B., Zhao J., Asante C. O., Cendan C. M., Marsh S., Martinez-Barbera J. P., Nassar M. A., Dickenson A. H., Wood J. N. (2008). The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321, 702–705 [DOI] [PubMed] [Google Scholar]
- Adler H. J., Raphael Y. (1996). New hair cells arise from supporting cell conversion in the acoustically damaged chick inner ear. Neurosci. Lett. 205, 17–20 [DOI] [PubMed] [Google Scholar]
- Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. (2002). Molecular Biology of the Cell. New York, NJ: Garland Science; [Google Scholar]
- Baird R. A., Steyger P. S., Schuff N. R. (1996). Mitotic and nonmitotic hair cell regeneration in the bullfrog vestibular otolith organs. Ann. N. Y. Acad. Sci. 781, 59–70 [DOI] [PubMed] [Google Scholar]
- Balak K. J., Corwin J. T., Jones J. E. (1990). Regenerated hair cells can originate from supporting cell progeny: evidence from phototoxicity and laser ablation experiments in the lateral line system. J. Neurosci. 10, 2502–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N., van Es J. H., Kuipers J., Kujala P., van den Born M., Cozijnsen M., Haegebarth A., Korving J., Begthel H., Peters P. J., et al. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007 [DOI] [PubMed] [Google Scholar]
- Belyantseva I. A., Adler H. J., Curi R., Frolenkov G. I., Kachar B. (2000). Expression and localization of prestin and the sugar transporter GLUT-5 during development of electromotility in cochlear outer hair cells. J. Neurosci. 20, RC116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohne B. A., Ward P. H., Fernández C. (1976). Irreversible inner ear damage from rock music. Trans. Sect. Otolaryngol. Am. Acad. Ophthalmol. Otolaryngol. 82, ORL50–ORL59 [PubMed] [Google Scholar]
- Burns J. C., Cox B. C., Thiede B. R., Zuo J., Corwin J. T. (2012). In vivo proliferative regeneration of balance hair cells in newborn mice. J. Neurosci. 32, 6570–6577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafaro J., Lee G. S., Stone J. S. (2007). Atoh1 expression defines activated progenitors and differentiating hair cells during avian hair cell regeneration. Dev. Dyn. 236, 156–170 [DOI] [PubMed] [Google Scholar]
- Chai R., Kuo B., Wang T., Liaw E. J., Xia A., Jan T. A., Liu Z., Taketo M. M., Oghalai J. S., Nusse R., et al. (2012). Wnt signaling induces proliferation of sensory precursors in the postnatal mouse cochlea. Proc. Natl. Acad. Sci. USA 109, 8167–8172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow L. M., Tian Y., Weber T., Corbett M., Zuo J., Baker S. J. (2006). Inducible Cre recombinase activity in mouse cerebellar granule cell precursors and inner ear hair cells. Dev. Dyn. 235, 2991–2998 [DOI] [PubMed] [Google Scholar]
- Clerici W. J., Hensley K., DiMartino D. L., Butterfield D. A. (1996). Direct detection of ototoxicant-induced reactive oxygen species generation in cochlear explants. Hear. Res. 98, 116–124 [DOI] [PubMed] [Google Scholar]
- Corwin J. T., Cotanche D. A. (1988). Regeneration of sensory hair cells after acoustic trauma. Science 240, 1772–1774 [DOI] [PubMed] [Google Scholar]
- Dabdoub A., Puligilla C., Jones J. M., Fritzsch B., Cheah K. S., Pevny L. H., Kelley M. W. (2008). Sox2 signaling in prosensory domain specification and subsequent hair cell differentiation in the developing cochlea. Proc. Natl. Acad. Sci. USA 105, 18396–18401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daudet N., Vago P., Ripoll C., Humbert G., Pujol R., Lenoir M. (1998). Characterization of atypical cells in the juvenile rat organ of corti after aminoglycoside ototoxicity. J. Comp. Neurol. 401, 145–162 [PubMed] [Google Scholar]
- Dechesne C. J., Thomasset M. (1988). Calbindin (CaBP 28 kDa) appearance and distribution during development of the mouse inner ear. Brain Res. 468, 233–242 [DOI] [PubMed] [Google Scholar]
- Doetzlhofer A., White P., Lee Y. S., Groves A., Segil N. (2006). Prospective identification and purification of hair cell and supporting cell progenitors from the embryonic cochlea. Brain Res. 1091, 282–288 [DOI] [PubMed] [Google Scholar]
- Doetzlhofer A., Basch M. L., Ohyama T., Gessler M., Groves A. K., Segil N. (2009). Hey2 regulation by FGF provides a Notch-independent mechanism for maintaining pillar cell fate in the organ of Corti. Dev. Cell 16, 58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkman L., McEvilly R. J., Luo L., Ryan A. K., Hooshmand F., O’Connell S. M., Keithley E. M., Rapaport D. H., Ryan A. F., Rosenfeld M. G. (1996). Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature 381, 603–606 [DOI] [PubMed] [Google Scholar]
- Fang J., Zhang W. C., Yamashita T., Gao J., Zhu M. S., Zuo J. (2012). Outer hair cell-specific prestin-CreERT2 knockin mouse lines. Genesis 50, 124–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forge A., Li L., Corwin J. T., Nevill G. (1993). Ultrastructural evidence for hair cell regeneration in the mammalian inner ear. Science 259, 1616–1619 [DOI] [PubMed] [Google Scholar]
- Forge A., Li L., Nevill G. (1998). Hair cell recovery in the vestibular sensory epithelia of mature guinea pigs. J. Comp. Neurol. 397, 69–88 [PubMed] [Google Scholar]
- Golub J. S., Tong L., Ngyuen T. B., Hume C. R., Palmiter R. D., Rubel E. W., Stone J. S. (2012). Hair cell replacement in adult mouse utricles after targeted ablation of hair cells with diphtheria toxin. J. Neurosci. 32, 15093–15105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallworth R., McCoy M., Polan-Curtain J. (2000). Tubulin expression in the developing and adult gerbil organ of Corti. Hear. Res. 139, 31–41 [DOI] [PubMed] [Google Scholar]
- Hartman B. H., Basak O., Nelson B. R., Taylor V., Bermingham-McDonogh O., Reh T. A. (2009). Hes5 expression in the postnatal and adult mouse inner ear and the drug-damaged cochlea. J. Assoc. Res. Otolaryngol. 10, 321–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins J. E., Jr, Johnsson L. G., Stebbins W. C., Moody D. B., Coombs S. L. (1976). Hearing loss and cochlear pathology in monkeys after noise exposure. Acta Otolaryngol. 81, 337–343 [DOI] [PubMed] [Google Scholar]
- Henderson D., Bielefeld E. C., Harris K. C., Hu B. H. (2006). The role of oxidative stress in noise-induced hearing loss. Ear Hear. 27, 1–19 [DOI] [PubMed] [Google Scholar]
- Hume C. R., Bratt D. L., Oesterle E. C. (2007). Expression of LHX3 and SOX2 during mouse inner ear development. Gene Expr. Patterns 7, 798–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I., Sakamoto M., Yamaguchi M., Mori K., Kageyama R. (2010). Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J. Neurosci. 30, 3489–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova A., Signore M., Caro N., Greene N. D., Copp A. J., Martinez-Barbera J. P. (2005). In vivo genetic ablation by Cre-mediated expression of diphtheria toxin fragment A. Genesis 43, 129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen-Smith H. C., Eley J., Steyger P. S., Ludueña R. F., Hallworth R. (2003). Cell type-specific reduction of beta tubulin isotypes synthesized in the developing gerbil organ of Corti. J. Neurocytol. 32, 185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. E., Corwin J. T. (1996). Regeneration of sensory cells after laser ablation in the lateral line system: hair cell lineage and macrophage behavior revealed by time-lapse video microscopy. J. Neurosci. 16, 649–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto K., Izumikawa M., Beyer L. A., Atkin G. M., Raphael Y. (2009). Spontaneous hair cell regeneration in the mouse utricle following gentamicin ototoxicity. Hear. Res. 247, 17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley M. W. (2006). Hair cell development: commitment through differentiation. Brain Res. 1091, 172–185 [DOI] [PubMed] [Google Scholar]
- Kelley M. W., Talreja D. R., Corwin J. T. (1995). Replacement of hair cells after laser microbeam irradiation in cultured organs of corti from embryonic and neonatal mice. J. Neurosci. 15, 3013–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly M. C., Chang Q., Pan A., Lin X., Chen P. (2012). Atoh1 directs the formation of sensory mosaics and induces cell proliferation in the postnatal mammalian cochlea in vivo. J. Neurosci. 32, 6699–6710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan A. E., Pelling A. L., Leung K. K., Tang A. S., Bell D. M., Tease C., Lovell-Badge R., Steel K. P., Cheah K. S. (2005). Sox2 is required for sensory organ development in the mammalian inner ear. Nature 434, 1031–1035 [DOI] [PubMed] [Google Scholar]
- Kuan C. Y., Schloemer A. J., Lu A., Burns K. A., Weng W. L., Williams M. T., Strauss K. I., Vorhees C. V., Flavell R. A., Davis R. J., et al. (2004). Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J. Neurosci. 24, 10763–10772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford P. J., Shailam R., Norton C. R., Gridley T., Kelley M. W. (2000). Expression of Math1 and HES5 in the cochleae of wildtype and Jag2 mutant mice. J. Assoc. Res. Otolaryngol. 1, 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. S., Liu F., Segil N. (2006). A morphogenetic wave of p27Kip1 transcription directs cell cycle exit during organ of Corti development. Development 133, 2817–2826 [DOI] [PubMed] [Google Scholar]
- Legendre K., Safieddine S., Küssel-Andermann P., Petit C., El-Amraoui A. (2008). alphaII-betaV spectrin bridges the plasma membrane and cortical lattice in the lateral wall of the auditory outer hair cells. J. Cell Sci. 121, 3347–3356 [DOI] [PubMed] [Google Scholar]
- Lelli A., Asai Y., Forge A., Holt J. R., Géléoc G. S. (2009). Tonotopic gradient in the developmental acquisition of sensory transduction in outer hair cells of the mouse cochlea. J. Neurophysiol. 101, 2961–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir M., Vago P. (1997). Does the organ of Corti attempt to differentiate new hair cells after antibiotic intoxication in rat pups? Int. J. Dev. Neurosci. 15, 487–495 [DOI] [PubMed] [Google Scholar]
- Li S., Mark S., Radde-Gallwitz K., Schlisner R., Chin M. T., Chen P. (2008). Hey2 functions in parallel with Hes1 and Hes5 for mammalian auditory sensory organ development. BMC Dev. Biol. 8, 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Dearman J. A., Cox B. C., Walters B. J., Zhang L., Ayrault O., Zindy F., Gan L., Roussel M. F., Zuo J. (2012a). Age-dependent in vivo conversion of mouse cochlear pillar and Deiters’ cells to immature hair cells by Atoh1 ectopic expression. J. Neurosci. 32, 6600–6610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Walters B. J., Owen T., Brimble M. A., Steigelman K. A., Zhang L., Mellado Lagarde M. M., Valentine M. B., Yu Y., Cox B. C., et al. (2012b). Regulation of p27Kip1 by Sox2 maintains quiescence of inner pillar cells in the murine auditory sensory epithelium. J. Neurosci. 32, 10530–10540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombarte A., Yan H. Y., Popper A. N., Chang J. S., Platt C. (1993). Damage and regeneration of hair cell ciliary bundles in a fish ear following treatment with gentamicin. Hear. Res. 64, 166–174 [DOI] [PubMed] [Google Scholar]
- Lumpkin E. A., Collisson T., Parab P., Omer-Abdalla A., Haeberle H., Chen P., Doetzlhofer A., White P., Groves A., Segil N., et al. (2003). Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr. Patterns 3, 389–395 [DOI] [PubMed] [Google Scholar]
- Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R., et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahendrasingam S., Beurg M., Fettiplace R., Hackney C. M. (2010). The ultrastructural distribution of prestin in outer hair cells: a post-embedding immunogold investigation of low-frequency and high-frequency regions of the rat cochlea. Eur. J. Neurosci. 31, 1595–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahrt E. J., Perkel D. J., Tong L., Rubel E. W., Portfors C. V. (2013). Engineered deafness reveals that mouse courtship vocalizations do not require auditory experience. J. Neurosci. 33, 5573–5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak A. C., Szeto I. Y., Fritzsch B., Cheah K. S. (2009). Differential and overlapping expression pattern of SOX2 and SOX9 in inner ear development. Gene Expr. Patterns 9, 444–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellado Lagarde M. M., Cox B. C., Fang J., Taylor R., Forge A., Zuo J. (2013). Selective ablation of pillar and deiters’ cells severely affects cochlear postnatal development and hearing in mice. J. Neurosci. 33, 1564–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M., Kelley M. W. (2003). Planar and vertical signals control cellular differentiation and patterning in the mammalian cochlea. J. Neurosci. 23, 9469–9478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesterle E. C., Campbell S., Taylor R. R., Forge A., Hume C. R. (2008). Sox2 and JAGGED1 expression in normal and drug-damaged adult mouse inner ear. J. Assoc. Res. Otolaryngol. 9, 65–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima K., Grimm C. M., Corrales C. E., Senn P., Martinez Monedero R., Géléoc G. S., Edge A., Holt J. R., Heller S. (2007). Differential distribution of stem cells in the auditory and vestibular organs of the inner ear. J. Assoc. Res. Otolaryngol. 8, 18–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parietti C., Vago P., Humbert G., Lenoir M. (1998). Attempt at hair cell neodifferentiation in developing and adult amikacin intoxicated rat cochleae. Brain Res. 813, 57–66 [DOI] [PubMed] [Google Scholar]
- Porrello E. R., Mahmoud A. I., Simpson E., Hill J. A., Richardson J. A., Olson E. N., Sadek H. A. (2011). Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson D. W., Alosi J. A., Cotanche D. A. (2004). Direct transdifferentiation gives rise to the earliest new hair cells in regenerating avian auditory epithelium. J. Neurosci. Res. 78, 461–471 [DOI] [PubMed] [Google Scholar]
- Romand R., Chardin S., Le Calvez S. (1996). The spontaneous appearance of hair cell-like cells in the mammalian cochlea following aminoglycoside ototoxicity. Neuroreport 8, 133–137 [DOI] [PubMed] [Google Scholar]
- Ruben R. J. (1967). Development of the inner ear of the mouse: a radioautographic study of terminal mitoses. Acta Otolaryngol. 220, 1–44 [PubMed] [Google Scholar]
- Ryals B. M., Rubel E. W. (1988). Hair cell regeneration after acoustic trauma in adult Coturnix quail. Science 240, 1774–1776 [DOI] [PubMed] [Google Scholar]
- Rybak L. P., Whitworth C., Scott V. (1992). Development of endocochlear potential and compound action potential in the rat. Hear. Res. 59, 189–194 [DOI] [PubMed] [Google Scholar]
- Savary E., Hugnot J. P., Chassigneux Y., Travo C., Duperray C., Van De Water T., Zine A. (2007). Distinct population of hair cell progenitors can be isolated from the postnatal mouse cochlea using side population analysis. Stem Cells 25, 332–339 [DOI] [PubMed] [Google Scholar]
- Shi F., Kempfle J. S., Edge A. S. (2012). Wnt-responsive Lgr5-expressing stem cells are hair cell progenitors in the cochlea. J. Neurosci. 32, 9639–9648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F., Hu L., Edge A. S. (2013). Generation of hair cells in neonatal mice by β-catenin overexpression in Lgr5-positive cochlear progenitors. Proc. Natl. Acad. Sci. USA 110, 13851–13856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkkonen S. T., Chai R., Jan T. A., Hartman B. H., Laske R. D., Gahlen F., Sinkkonen W., Cheng A. G., Oshima K., Heller S. (2011). Intrinsic regenerative potential of murine cochlear supporting cells. Sci. Rep. 1, 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobkowicz H. M., Slapnick S. M., August B. K. (1995). The kinocilium of auditory hair cells and evidence for its morphogenetic role during the regeneration of stereocilia and cuticular plates. J. Neurocytol. 24, 633–653 [DOI] [PubMed] [Google Scholar]
- Steigelman K. A., Lelli A., Wu X., Gao J., Lin S., Piontek K., Wodarczyk C., Boletta A., Kim H., Qian F., et al. (2011). Polycystin-1 is required for stereocilia structure but not for mechanotransduction in inner ear hair cells. J. Neurosci. 31, 12241–12250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern C. D., Fraser S. E. (2001). Tracing the lineage of tracing cell lineages. Nat. Cell Biol. 3, E216–E218 [DOI] [PubMed] [Google Scholar]
- Szarama K. B., Gavara N., Petralia R. S., Kelley M. W., Chadwick R. S. (2012). Cytoskeletal changes in actin and microtubules underlie the developing surface mechanical properties of sensory and supporting cells in the mouse cochlea. Development 139, 2187–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L., Hume C., Palmiter R., Rubel E. (2011). Ablation of mouse cochlea hair cells by activating the human diphtheria toxin receptor (DTR). Gene targeted to the Pou4f3 locus. In Abstracts of the 34th Annual Midwinter Research Meeting of the Association for Research in Otolaryngology (ed. Santi P. A.), Abs. 836, p. 280 Mount Royal, NJ: ARO; [Google Scholar]
- Warchol M. E., Corwin J. T. (1996). Regenerative proliferation in organ cultures of the avian cochlea: identification of the initial progenitors and determination of the latency of the proliferative response. J. Neurosci. 16, 5466–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warchol M. E., Lambert P. R., Goldstein B. J., Forge A., Corwin J. T. (1993). Regenerative proliferation in inner ear sensory epithelia from adult guinea pigs and humans. Science 259, 1619–1622 [DOI] [PubMed] [Google Scholar]
- Weber T., Corbett M. K., Chow L. M., Valentine M. B., Baker S. J., Zuo J. (2008). Rapid cell-cycle reentry and cell death after acute inactivation of the retinoblastoma gene product in postnatal cochlear hair cells. Proc. Natl. Acad. Sci. USA 105, 781–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White P. M., Doetzlhofer A., Lee Y. S., Groves A. K., Segil N. (2006). Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature 441, 984–987 [DOI] [PubMed] [Google Scholar]
- Xiang M., Gao W. Q., Hasson T., Shin J. J. (1998). Requirement for Brn-3c in maturation and survival, but not in fate determination of inner ear hair cells. Development 125, 3935–3946 [DOI] [PubMed] [Google Scholar]
- Yamamoto N., Tanigaki K., Tsuji M., Yabe D., Ito J., Honjo T. (2006). Inhibition of Notch/RBP-J signaling induces hair cell formation in neonate mouse cochleas. J. Mol. Med. 84, 37–45 [DOI] [PubMed] [Google Scholar]
- Zheng J. L., Gao W. Q. (1997). Analysis of rat vestibular hair cell development and regeneration using calretinin as an early marker. J. Neurosci. 17, 8270–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. L., Gao W. Q. (2000). Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat. Neurosci. 3, 580–586 [DOI] [PubMed] [Google Scholar]
- Zheng J., Shen W., He D. Z., Long K. B., Madison L. D., Dallos P. (2000). Prestin is the motor protein of cochlear outer hair cells. Nature 405, 149–155 [DOI] [PubMed] [Google Scholar]
- Zine A., de Ribaupierre F. (1998). Replacement of mammalian auditory hair cells. Neuroreport 9, 263–268 [DOI] [PubMed] [Google Scholar]
- Zine A., Aubert A., Qiu J., Therianos S., Guillemot F., Kageyama R., de Ribaupierre F. (2001). Hes1 and Hes5 activities are required for the normal development of the hair cells in the mammalian inner ear. J. Neurosci. 21, 4712–4720 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.