

Abstract
The Drosophila heart is composed of two distinct cell types, the contractile cardial cells (CCs) and the surrounding non-muscle pericardial cells (PCs), development of which is regulated by a network of conserved signaling molecules and transcription factors (TFs). Here, we used machine learning with array-based chromatin immunoprecipitation (ChIP) data and TF sequence motifs to computationally classify cell type-specific cardiac enhancers. Extensive testing of predicted enhancers at single-cell resolution revealed the added value of ChIP data for modeling cell type-specific activities. Furthermore, clustering the top-scoring classifier sequence features identified novel cardiac and cell type-specific regulatory motifs. For example, we found that the Myb motif learned by the classifier is crucial for CC activity, and the Myb TF acts in concert with two forkhead domain TFs and Polo kinase to regulate cardiac progenitor cell divisions. In addition, differential motif enrichment and cis-trans genetic studies revealed that the Notch signaling pathway TF Suppressor of Hairless [Su(H)] discriminates PC from CC enhancer activities. Collectively, these studies elucidate molecular pathways used in the regulatory decisions for proliferation and differentiation of cardiac progenitor cells, implicate Su(H) in regulating cell fate decisions of these progenitors, and document the utility of enhancer modeling in uncovering developmental regulatory subnetworks.
Keywords: Machine learning, Gene regulation, Transcription factors, Progenitor specification, Cell division, Organogenesis, Drosophila
INTRODUCTION
A comparison of the molecular mechanisms governing heart development in Drosophila and vertebrates reveals a remarkable conservation of all major regulatory components, including both signals and transcription factors (TFs) (Olson, 2006; Bodmer and Frasch, 2010). Moreover, mutations in many of these conserved regulators of heart development have been shown to cause congenital heart disease in man (Bodmer and Frasch, 2010). Thus, understanding mechanisms of cardiogenesis in Drosophila can inform and guide similar analyses in vertebrate species, including human.
The formation of a complex organ such as the Drosophila heart involves the coordination of a diverse array of developmental processes, such as cell fate specification, differentiation and diversification (Bodmer and Frasch, 2010), by transcriptional regulation through enhancers (Davidson, 2006). Enhancers are stretches of DNA composed of DNA subsequences recognized by sequence-specific DNA-binding TFs that integrate the activity of tissue-specific, cell-specific, ubiquitously expressed and signal-activated TFs to guide gene expression programs at the level of both individual cells and the particular developmental steps that those cells undergo (Davidson, 2006; Busser et al., 2008). Recent genome-scale studies in Drosophila have confirmed the crucial role of transcriptional regulation in orchestrating cardiogenesis (Liu et al., 2009; Ahmad et al., 2012; Junion et al., 2012; Jin et al., 2013).
The Drosophila heart is a linear tube composed of two classes of cells: an inner row of contractile cardial cells (CCs) and an outer layer of non-muscle pericardial cells (PCs). Although CCs and PCs can be distinguished on the basis of morphological differences, unique lineages and the cell-specific expression patterns of distinct TFs (Bodmer and Frasch, 2010), little is known about the molecular mechanisms that underlie these cell-specific differences.
We previously used a machine learning approach to decipher the motifs and enhancers that govern the gene expression patterns of Drosophila muscle founder cells (Busser et al., 2012a), fusion competent myoblasts (Busser et al., 2012c) and cells of the human heart (Narlikar et al., 2010). Here, we utilized a similar multidimensional research strategy involving a combination of machine learning, array-based ChIP data for key mesodermal regulators, and experimental analyses to computationally classify, predict and validate cell type-specific cardiac enhancers and the crucial TF binding sites that are responsible for their activities. This integrative approach also enabled us to identify regulators of cell specification in the heart, and to characterize a molecular pathway involving two forkhead domain TFs, Myb and Polo kinase, which together mediate appropriate progenitor cell divisions in the heart. In addition, our findings allowed us to document a molecular mechanism for how Su(H) acts in the Notch signaling pathway to transcriptionally regulate the cell fates acquired by particular cardiac progenitors.
RESULTS
Rationale and overview
Cardiogenesis involves multiple biological processes acting in concert during development. This coordination is achieved by the regulation of diverse cardiac genes by a finite set of cell-specific, tissue-specific, signal-activated and ubiquitously expressed TFs that drive heart gene expression. Thus, certain combinations of TF binding in vivo, as well as binding motifs for these TFs, constitute sequence features that are expected to be enriched in heart enhancers compared with other regions of DNA. The goal of this study was to take advantage of this distinct distribution of sequence features to build computational models that discriminate known cardiac enhancers from the rest of the genome, and to use this information in turn to predict novel cardiac enhancers, to identify the DNA sequence features (i.e. unique combinations of TF binding motifs) that act as positive predictors of cardiac cell type-specific enhancers, and to functionally validate the roles of these sequence features and their associated TFs in cardiogenic regulatory networks.
The strategy we used to achieve this objective is outlined in Fig. 1. First, we compiled training sets of known enhancers expressed in different cardiac cell subtypes. We next mapped known TF binding motifs from public databases and ChIP signals corresponding to in vivo TF binding to these training sets and control sequences. This sequence annotation data along the DNA sequence of cardiac enhancers was then used by machine learning classification algorithms (classifiers) to separate cardiac enhancer sequences from controls. The reliability and efficacy of these cell type-specific classifiers were examined and compared by multiple approaches including a statistical analysis of enrichment of predicted enhancers in the loci of concordantly expressed genes followed by experimental validation of the functional specificity of enhancer predictions using transgenic reporter assays. Finally, the TF weights estimated by this computational classification were utilized to define and determine the role of novel TF binding motifs and their corresponding TFs in cis and trans assays based on the premise that a positive TF weight indicates a TF that is positively associated with heart activity.
Fig. 1.

Schematic overview of the computational and experimental strategy utilized in this study. Training sets of enhancers expressed in different cardiac cell types (increased in size through phylogenetic profiling) and control sequences were compiled and subsequently scanned to map sequence features corresponding to known binding site motifs collected from public databases and to in vivo TF binding signals obtained from published ChIP data profiles. Classifiers were built to create enhancer models that discriminate cell type-specific enhancers from respective controls. For each cell type, two classifiers were independently constructed: one based solely on motif features (‘motif-alone’) and the other on motif features and ChIP signals (‘motif+ChIP′). The enhancer models were used to scan the Drosophila genome to identify novel cell-specific enhancers similar to the training sets, and the reliability and efficacy of the motif-alone and motif+ChIP classifiers for different cardiac cell types were examined and compared. Sequence features positively associated with computational classification were examined further using cis and trans in vivo experimental assays to identify and determine the functional roles of both the binding motifs and their associated regulatory TFs.
Training sets of enhancers active in the Drosophila heart and individual cardiac cell subtypes
The PCs and CCs of the heart develop from a population of progenitor cells derived from the dorsal-most mesodermal cells termed the cardiac mesoderm (CM) (Bodmer and Frasch, 2010). In order to build classifiers, i.e. computational models that can discriminate cardiac cell type-specific enhancers from other DNA sequences, we first compiled training sets of enhancers with activity in the different cells of the Drosophila heart (Fig. 1). These included regions curated from the literature as well as a small set of unpublished enhancer sequences with activity in the heart (supplementary material Fig. S1), which we had previously identified and empirically verified (supplementary material Table S1). We sought to classify all cardiac enhancers by first compiling a training set of sequences with activity in any differentiated heart cells or their progenitors, including those unique to or active in any combination of PCs, CCs and CM (referred to hereafter as ‘heart’; Fig. 1; supplementary material Table S1). We then attempted to refine this generic heart classification by categorizing expression in the individual subsets of the heart by training on sets of enhancers with activity in either PCs or CCs (supplementary material Table S1). Owing to the small size of the training sequences (23 heart enhancers, ten PC enhancers and 16 CC enhancers), and in order to avoid over-fitting decision rules learned by the classifier, we expanded these training sets using phylogenetic profiling (Busser et al., 2012a). Enhancer orthologs were extracted from the other 11 fully sequenced Drosophila species, mosquito, honeybee and red flour beetle by searching for regions with at least 50% but less than 80% sequence identity and similar length, GC content and repeat density as their D. melanogaster counterparts (supplementary material Table S1) (Busser et al., 2012a). Of note, we previously demonstrated the value of phylogenetic profiling as such orthologous sequences are active in the appropriate cells and their inclusion improves enhancer classification performance (Busser et al., 2012a). This approach led to a training set of 47 heart, 33 CC and 25 PC enhancer sequences (supplementary material Table S1).
Controls were generated by randomly sampling sequences from D. melanogaster non-coding regions with similar length, GC content and repeat content to the training enhancers (Fig. 1). Ten control sequences were retrieved for each training enhancer.
Accurate classification of cell-specific cardiac enhancers
Prior studies in Drosophila documented the difficulty in using machine learning for identifying cell- or tissue-specific enhancer activities simply by relying primarily on known TF binding motifs from available databases (Kantorovitz et al., 2009; Busser et al., 2012a). We hypothesized that this poor performance might be improved with the inclusion of relevant ChIP data as an additional classifier feature (Busser et al., 2012a). Furlong and colleagues have previously determined the genome-wide binding profiles of a set of key Drosophila mesodermal regulatory TFs, including Tin, Twi and Mef2 (Zinzen et al., 2009). Because all three of these TFs are known to be required for cardiogenesis (Bodmer and Frasch, 2010), we included array-based ChIP data for these TFs as additional sequence features to build enhancer classifiers (Fig. 1). To investigate the contribution of their inclusion, we constructed separate classifiers for each training set that either added ChIP data (‘motif+ChIP′) or omitted this information (‘motif-alone’). In this study, we used linear support vector machines (SVMs) as enhancer classifiers. SVMs are designed to identify combinations of sequence motifs that optimally separate enhancers from controls.
The ability of the classifiers to accurately predict regulatory activity was assessed by a tenfold cross-validation strategy (in which the original training set data was partitioned into complementary subsets, with one subset utilized to build the cell type-specific classifier, and the other subset used to validate the constructed classifier), with performance gauged by the area under the receiver operating characteristic curve (AUC). The AUC values for the motif-alone classifiers for heart, CC and PC enhancers are 0.85, 0.83 and 0.91, respectively, which suggests an accurate ability to discriminate appropriate enhancers from background sequences (Fig. 2A). The inclusion of ChIP data significantly improved the performance of each classifier, with AUC values increasing to 0.96 for heart enhancers, 0.96 for CCs and 0.97 for PCs (Fig. 2A). These results confirm that we are able to generate reliable classifiers, with significant improvements obtained with the inclusion of relevant ChIP data.
Fig. 2.

Cell-specific cardiac enhancer classifiers perform with high specificity and sensitivity. (A) Average receiver operating characteristic curves and standard deviations for tenfold cross-validation performed for both motif-alone and motif+ChIP classifiers in 20 independent runs. (B) Enrichment in validated heart, CC and PC genes in the neighborhood of putative cardiac enhancers at different ranks for the motif-alone and motif+ChIP classifiers. Each of these genes is generally associated with only one prediction. Double asterisks indicate significant differences (P<0.005).
We next applied the developed classifiers to predict the presence of novel cardiac and cell type-specific enhancers throughout the Drosophila genome (supplementary material Table S2). These models predicted ∼2000 enhancers at a false positive rate (FPR) of 0.01 for each cardiac cell type (supplementary material Fig. S2). Putative enhancers are strongly associated with genes expressed in the corresponding cardiac cell type (Fig. 2B; supplementary material Tables S1, S2). Furthermore, the addition of the ChIP data significantly improved the enhancer model, as such predictions are more often associated with genes having known heart expression (Fig. 2B). Finally, all (100%) of the 28 cardiac enhancers reported in a recent study (Jin et al., 2013) that were not included in the training set were predicted by our motif+ChIP classifier for the heart at an FPR of 0.05, whereas 15 (53.6%) were identified by motif-alone (supplementary material Fig. S3; Table S2). Collectively, these results document the generation of classifiers that can reliably detect cell type-specific cardiac enhancers, and further show that the inclusion of ChIP data significantly improves the enhancer model.
Large-scale validation of cell-specific cardiac enhancers
To evaluate the in vivo functions of classifier-predicted enhancers, we used genomic site-specific transgenic reporter assays (Busser et al., 2012b; Busser et al., 2012a) to test 80 enhancer predictions with varying scores in the classifier rankings for the different enhancer models (supplementary material Table S2). Such analyses revealed that the top-scoring predictions for all classifications were often functional enhancers: 21 out of 41 (51.2%) top-scoring (i.e. <0.01 FPR) cardiac predictions were active in the heart, whereas 15 out of 37 (40.5%) top-scoring CC predictions were active in CCs, and 15 out of 31 (48.4%) top-scoring PC predictions were active in PCs (supplementary material Fig. S4A; Table S2). Not surprisingly, given that CCs and PCs define subsets of heart cells, many of these predictions were highly ranked in more than one classifier. For comparison, low scoring (>0.05 FPR) successful predictions were one out of 13 (7.7%), nine out of 38 (23.7%), and 11 out of 38 (28.9%) for heart, CC and PC classifiers, respectively.
These analyses also revealed that the classifiers that included ChIP data performed better in predicting functional enhancer activity on a genome-wide scale (supplementary material Fig. S4B). This result is most pronounced for the cell-specific classifiers, as many successful enhancer predictions for classifiers that relied solely on motifs were not included in the set identified with the relatively stringent 0.05 FPR cutoff. For example, whereas only 11 out of 20 successful motif-alone CC predictions (55.0%) and 13 out of 24 motif-alone successful PC predictions (54.1%) fell within the 0.05 FPR cutoff, 19 out of 20 successful CC predictions (95.0%) and 23 out of 24 successful PC predictions (95.8%) that utilized ChIP data also scored within this same cutoff (supplementary material Table S2; Fig. S4B). However, this difference was not seen for general heart predictions, as 25 out of 26 (96.2%) motif-alone and motif+ChIP predictions fell within the 0.05 FPR cutoff. These results suggest that the inclusion of ChIP data as an additional feature generates more reliable cell type-specific enhancer models, but is less necessary for predicting general cardiac activity.
Representative examples of transgenic embryos for top-scoring cardiac enhancer predictions are shown in Fig. 3, including those predicted regulatory elements associated with senseless-2 (sens-2), sprouty (sty), Trim9, CG9650 and dally-like (dlp) genes. Each of these predictions was highly ranked in both the heart and cell type-specific classifiers, and all were appropriately active in both CCs and PCs. Thus, both the cardiac and cell type-specific enhancer models are able to accurately predict appropriate in vivo reporter activity.
Fig. 3.

Candidate enhancers predicted by the cell-specific cardiac classifiers are active in the appropriate cardiac cell types. (A-J′′) lacZ reporter gene activity (β-galactosidase, green) driven by classifier-predicted enhancers. All PCs are marked by Zfh1 expression (blue) whereas the posterior-most four CCs in each hemisegment, the Tin-PCs and the Eve-PCs are marked by Tin expression (red).
Interestingly, the predictions associated with the rolling pebbles (rols) and Dorsocross 3 (Doc3) genes ranked much higher in the CC than in the PC classifier and indeed were found to be only active in CCs (Fig. 3). Conversely, the predictions associated with the CG13822, Stromalin-2 (SA-2) and CG14207 genes ranked much higher in the PC than in the CC classifier and were only active in PCs (Fig. 3). Thus, the distribution of enhancer ranks in these cell-specific classifications can be used to uncover cell-specific enhancer predictions.
Motif features associated with cardiac and cell type-specific enhancers
The analyses described above suggest that the sequence features learned by the classifier are sufficiently robust to predict cell type-specific cardiac enhancer activities. To understand these decision rules, we examined the sequence features positively associated with the classification of cardiac cell-type enhancers. For linear SVMs, features associated with the training set are given positive weights, those associated with the controls are given negative weights, and irrelevant features receive zero weight. The motifs most relevant to the heart and cell type-specific motif+ChIP classifications were then grouped according to the protein families of their respective TFs (Fig. 4). Similar results were seen with the motif-alone classification (supplementary material Fig. S5).
Fig. 4.
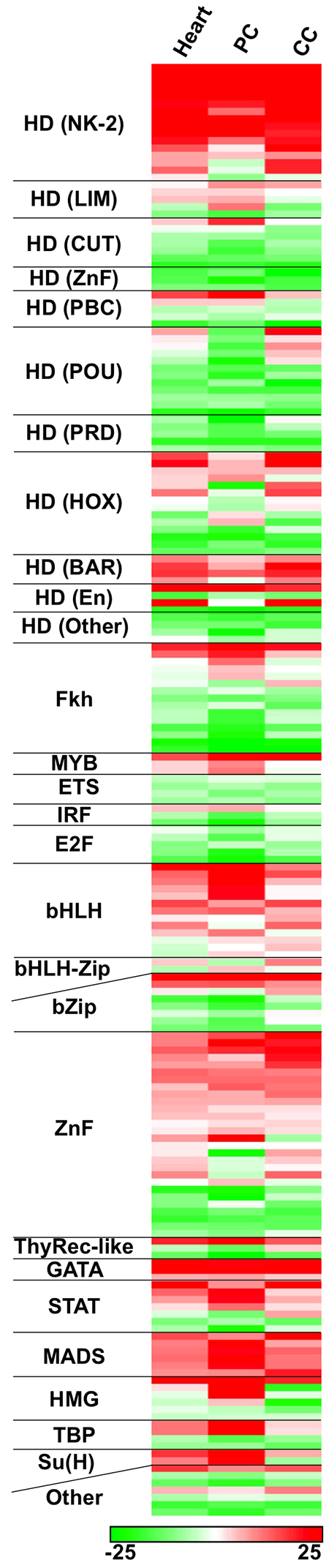
Machine learning modeling of cardiac enhancers reveals sequence motif features that are relevant to their cell type-specific functional classification. A linear SVM was trained for each of three cardiac training sets (heart, PC and CC) using motif features and relevant ChIP data. TF binding motifs are ranked according to their linear SVM weights, with positive weights reflecting enrichment and negative weights reflecting depletion from the training set compared with background. TF binding motifs were grouped according to DNA-binding domain class of their respective TFs.
The presence of numerous known cardiac regulatory motifs, including those for NK-2 homeodomains (which include Tin), MADS box (which include Mef2) and basic helix-loop-helix (bHLH) TFs (which include Twi and Hand), as some of the most discriminating features of heart enhancers was an encouraging finding (Bodmer and Frasch, 2010). These motifs are likely to be directly occupied by the relevant regulatory TFs because the features representing Tin, Mef2 and Twi ChIP data are also highly weighted in the corresponding classifiers (supplementary material Table S3). Furthermore, additional enriched sequence features include previously characterized cardiac regulatory TFs, such as members of the GATA family, zinc finger family, LIM homeodomain family, Signal transducer and activator of transcription (Stat) and forkhead family of TFs (Bodmer and Frasch, 2010; Johnson et al., 2011; Ahmad et al., 2012; Zhu et al., 2012).
Interestingly, certain sequence features previously not known to play a role in heart development are enriched among all classifications. Such motifs include those recognized by the family of Myb domain TFs, which in Drosophila consists of a single ubiquitously expressed DNA-binding protein (Tomancak et al., 2007) that controls the regulation of proliferation or differentiation of progenitor cells (Ramsay, 2005), suggesting that Myb might be an unrecognized regulator of cardiogenesis. Finally, visualizing the separate classifications permits the identification of motifs that may discriminate PC and CC enhancer activities. For example, motifs for the Notch signaling pathway TF Su(H) are enriched in the PC classification and depleted in or irrelevant to the CC classification. This result suggests a role for Su(H) in governing the cell fate decision between PCs and CCs. In total, these results provide a comprehensive analysis of the motif preferences directing heart and cardiac cell type-specific gene expression programs.
Myb is an activator of the Ndg heart enhancer
Neither the CCs nor the PCs constitute a uniform population, as revealed both by their distinct cell lineages and by the complexity of their individual gene expression programs. From anterior to posterior, and named for the TFs they express, there are two Seven-up-CCs (Svp-CCs, unstained in Fig. 5A-C) and four Tinman-CCs (Tin-CCs, red in Fig. 5A-C) in each repeating hemisegment. In order to assess whether the Myb motif is functionally relevant to driving the activity of heart enhancers, we examined its potential role in the Nidogen (Ndg) cardiac enhancer (Philippakis et al., 2006). We previously showed that the wild-type Ndg enhancer is active in only the two anteriormost Tin-CCs in a hemisegment (Fig. 5A,D; supplementary material Table S4) (Zhu et al., 2012) and contains three Myb motifs required for restricting somatic mesodermal enhancer activity (Busser et al., 2012a). Here we show that mutagenesis of these same Myb binding sites results in significant partial inactivation of the enhancer (NdgMyb) in CCs (Fig. 5B,D; supplementary material Table S4), thereby demonstrating the functional importance of these sites to cardiac activity.
Fig. 5.
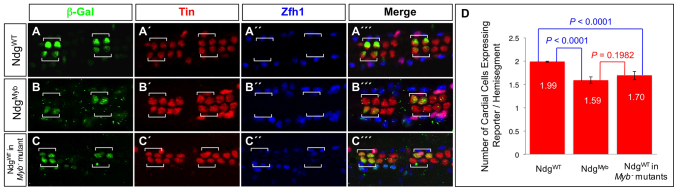
Myb is an activator of the Ndg enhancer in the heart. (A-C′′) The posterior-most four CCs are marked by Tin expression (red), and the PCs are marked by Zfh1 expression (blue). (A-A′′) A β-galactosidase reporter (green) driven by the wild-type Ndg enhancer is expressed in only two Tin-expressing CCs per hemisegment (square brackets). (B-B′′) Mutations in the Myb binding sites result in partial but significant inactivation of reporter expression in these two CCs. (C-C′′) Reporter expression from the wild-type Ndg enhancer is also partially but significantly inactivated in these two CCs in embryos hemizygous for the MybMH30 null mutation. (D) Histogram showing the mean number of CCs with 95% confidence intervals expressing the reporter and the significance of partial inactivation as a result of either the Myb binding site mutations in the Ndg enhancer or the MybMH30 null mutation.
We next determined whether a similar phenotype could be achieved by eliminating Myb function in trans. Embryos hemizygous for MybMH30 (Manak et al., 2002), a chromosomal deficiency that completely deletes the Myb gene, do indeed exhibit significant partial inactivation of the wild-type enhancer (NdgWT) reporter (Fig. 5C,D; supplementary material Table S4).
The convergence of results for these cis and trans experiments suggests that Myb protein functionally activates the cardiac enhancer of at least one heart gene, thus providing a plausible rationale for the identification of Myb motifs as positive discriminators in the classification of CC, PC and heart enhancers. In turn, this finding raises the question of in which cardiogenic processes this TF might be involved.
Myb is required for two distinct categories of cardiac progenitor cell divisions
We undertook a more detailed analysis of the cardiogenic roles of Myb by examining embryos in which Myb activity was knocked down by CM and heart-targeted RNA interference (RNAi) directed by the TinD-GAL4 (Yin et al., 1997) and Hand-GAL4 (Han and Olson, 2005) drivers. Staining with appropriate antibodies showed hemisegments with localized increases and decreases in both Svp-CC and Tin-CC numbers, compared with control embryos (supplementary material Fig. S6). Similar cardiac phenotypes were also observed in embryos hemizygous for the MybMH30 deficiency (Fig. 6A-D).
Fig. 6.

Cardiac progenitor cell division defects associated with Myb loss of function. (A) A heart from a wild-type embryo bearing the svp-lacZ enhancer trap showing hemisegments consisting of four Tin-CCs (green), two Svp-CCs (yellow) and two Svp-PCs (red). (B-D) Hearts from embryos that are hemizygous for the MybMH30 null mutation demonstrating mutant hemisegments with either excess or too few CCs (dashed ovals) and illustrating the two distinct types of progenitor cell division defects that underlie these cardiac phenotypes. (E) Schematic showing cell lineage relationships in a wild-type heart, and the three previously characterized cardiac progenitor cell divisions defects known to be responsible for localized changes in heart cell number (Ahmad et al., 2012). Note that only two types of developmental errors, those involved in symmetric cell divisions and those involved in an earlier step to determine the number of Svp progenitors, are primarily responsible for the Myb cardiac phenotypes. (F) Fraction of hemisegments exhibiting each type of cardiac progenitor cell division defect in embryos that are wild type or hemizygous for the MybMH30 mutation. The significance of each type of cell division defect in the Myb mutants compared with wild-type embryos is shown.
The localized changes in CC number in embryos lacking Myb function are reminiscent of the effects of cardiac progenitor cell division defects observed in embryos mutant for either of the two forkhead TF-encoding genes jumu or CHES-1-like, or for the downstream kinase-encoding gene polo (Ahmad et al., 2012). The two Svp-CCs in wild-type cardiac hemisegments are generated by two asymmetric progenitor cell divisions, with each division from an Svp progenitor cell producing one Svp-CC and one Svp-PC (Fig. 6E, yellow and red cells, respectively), whereas two symmetric cell divisions give rise to the four Tin-CCs (Fig. 6E, green cells) (Gajewski et al., 2000; Ward and Skeath, 2000; Han and Olson, 2005; Bodmer and Frasch, 2010). Eliminating jumu, CHES-1-like or polo function results in characteristic asymmetric and symmetric cell division defects affecting the number of Svp heart cell types and Tin-CCs, respectively (Fig. 6E) (Ahmad et al., 2012). All three genes have also been implicated in cell divisions at an earlier developmental stage that generate the Svp progenitor cells, with mutations in these genes producing either one or three Svp progenitors per hemisegment in place of the expected two, thus subsequently resulting in one or three pairs of Svp-CCs and Svp-PCs, respectively (Ahmad et al., 2012). Note that each of these distinct types of cell division defects (schematically summarized in Fig. 6E) can be uniquely identified by assessing the number of Svp-CCs, Svp-PCs and Tin-CCs in a hemisegment by staining with appropriate markers.
We attempted to ascertain whether one or more of these cell division defects were also responsible for the localized changes in CC number detected in Myb mutants. Examination of embryos hemizygous for MybMH30 revealed a significant increase over wild type both in the fraction of hemisegments with either excess or fewer Tin-CCs, corresponding to symmetric cell division defects, and in the fraction of hemisegments with either one or three pairs of Svp-CCs and Svp-PCs, corresponding to earlier cell division defects affecting the number of Svp progenitors (Fig. 6A-D,F; supplementary material Table S4). An alternative explanation for the latter phenotype is defective Myb-mediated selection of Svp progenitors from cardiac mesoderm equivalence groups. However, given the well-characterized role of Myb in governing mitosis and cytokinesis (Katzen et al., 1998; DeBruhl et al., 2013), we consider defects in earlier rounds of cell division to be the more likely cause. By contrast, the fraction of hemisegments exhibiting asymmetric cell division defects in MybMH30 hemizygotes was both minuscule and not significantly different from that in wild-type embryos (Fig. 6F).
Collectively, these results indicate that Myb regulates two distinct categories of cardiac progenitor cell divisions to specify correct cardiac cell numbers: symmetric cell divisions of the Tin-CC progenitors, and an earlier round of cell divisions that gives rise to the Svp progenitors. The observations that these two classes of cell division are also mediated by a pathway involving jumu and CHES-1-like regulating polo (Ahmad et al., 2012), and that Myb itself also regulates polo in other systems (Wen et al., 2008), suggested that Myb too might be acting through the same pathway, a hypothesis that we examined in the following experiments.
Synergistic genetic interactions between jumu, CHES-1-like, polo and Myb
If Myb functions through the same pathway as jumu, CHES-1-like and polo, then strong pairwise genetic interactions are likely to occur between Myb and each of these genes. In order to assess this possibility, the cardiac phenotypes of single heterozygotes for mutations in these four genes were quantified and compared with those of embryos that were doubly heterozygous for mutations in both Myb and jumu, for mutations in both Myb and CHES-1-like, or for mutations in both Myb and polo (supplementary material Figs S7, S8; Table S4). Double heterozygotes for both Myb and jumu mutations exhibit symmetric cell division defects and earlier Svp cell division defects that are significantly more severe than the additive effects of each of the two single heterozygotes. Similar synergistic genetic interactions occur between Myb and CHES-1-like for both symmetric cell divisions and earlier cell divisions generating Svp progenitors, as well as between Myb and polo. Collectively, these results suggest that Myb, jumu, CHES-1-like and polo act through the same pathway to mediate these two classes of cardiac progenitor cell divisions.
The Notch signaling pathway TF Su(H) discriminates between PC and CC enhancer activities
The observation that Su(H) motifs were positively weighted among PC sequences and were either depleted or irrelevant for the classification of CC sequences (Fig. 4; supplementary material Fig. S5) raised the possibility that Su(H) may act as a discriminator of these two cell types, which is supported by the expression of Su(H) in the CM (Tomancak et al., 2007). Furthermore, Su(H) is the terminal TF in the Notch signaling pathway, and inactivating mutations in other genes in the pathway, such as Notch, Delta or sanpodo, result in PC to CC transformations (Park et al., 1998; Gajewski et al., 2000; Ward and Skeath, 2000; Mandal et al., 2004; Grigorian et al., 2011), providing additional support for this hypothesis.
To test this model experimentally, we used site-directed mutagenesis to abolish the ability of Su(H) to recognize its well-characterized binding site (Rebeiz et al., 2011) in the PC enhancer of Holes in muscle (Him), a gene previously shown to be a direct target of Su(H) in vivo (Krejcí et al., 2009; Bernard et al., 2010), and assessed the effects of this mutation on enhancer function. The wild-type version of this enhancer (HimWT) is active exclusively in all PCs of the differentiated heart (Fig. 7A), whereas mutagenesis of the Su(H) binding site in the Him enhancer [HimSu(H)] induced additional ectopic reporter activity in all of the CCs without altering expression in PCs (Fig. 7B; supplementary material Fig. S9). Thus, Su(H) binding is necessary for repressing the activity of the Him enhancer in CCs, consistent with its well-characterized role in mediating transcriptional repression in other contexts (Bray and Furriols, 2001; Bray and Bernard, 2010).
Fig. 7.

Su(H) discriminates between PC and CC enhancer activities. (A-C′′) lacZ reporter gene activity (β-galactosidase, green) driven by relevant Him enhancers in indicated genotypes. All CCs express Mef2 (red) whereas PCs are marked by Zfh1 (blue). (A-A′′) The wild-type Him enhancer (HimWT) is active only in the Zfh1-expressing PCs. (B-B′′) When the Su(H) binding site is mutated in the Him enhancer [HimSu(H)], the reporter is still active in Zfh1-expressing PCs but is de-repressed in Mef2-positive CCs (arrows). (C-C′′) Knockdown of Su(H) with dorsal mesoderm-targeted RNAi driven by the TinD-GAL4 driver induces ectopic HimWT enhancer-driven β-galactosidase reporter activity in CCs (arrows). (D-E′′) Fluorescent in situ hybridization analysis of stage 16 embryos for Him mRNA (D,E) and antibody analysis for Mef2 (D′,E′) and Zfh1 (D′,E′) of the indicated genotypes. (D-D′′) Him mRNA is restricted to the Zfh1-expressing PCs of the wild-type heart. (E-E′′) Overexpression of Nicd in all cells of the heart driven by the Hand-GAL4 driver leads to ectopic Him mRNA being expressed in the weakly Mef2-expressing CCs (arrows).
In order to examine further the mechanism by which Su(H) normally represses the Him enhancer in CCs, and to avoid the pleiotropic effects of the Notch signaling pathway on additional tissues, we targeted gain and loss of function of Su(H) to cardiac progenitors. We reasoned that loss of Su(H) function by RNAi directed to the CM should lead to ectopic Him reporter activity in CCs, mimicking the loss of Su(H) binding sites in the Him enhancer. We confirmed this prediction by showing that the wild-type Him enhancer is indeed ectopically active in a subset of CCs in embryos in which Su(H) levels are depleted by RNAi knockdown (Fig. 7C). The fact that not all CCs express ectopic reporter activity probably reflects an incomplete RNAi knockdown of Su(H) levels in all progenitor cells.
In addition, we reasoned that the overexpression of the processed intracellular domain of Notch (Nicd), which has been shown to convert the Su(H) complex from a repressor into an activator for target genes (Bray and Furriols, 2001; Bray and Bernard, 2010), should induce ectopic activity of the Him gene in CCs. In agreement with this prediction, overexpression of Nicd in all cardiac progenitors by the Hand-Gal4 driver (Han and Olson, 2005) induced ectopic expression of Him, which is normally restricted to PCs, in Mef2-expressing CCs (Fig. 7D,E). The disorganized heart cells and the very weak expression of Mef2 in CCs suggest that CC cell fate specification is highly abnormal in Hand>Nicd embryos, with the CCs in the process of being partly transformed to PCs. Nevertheless, this partial transformation of CCs to PCs also entails the ectopic expression of PC markers, and it is this latter process that is illustrated by the detection of Him transcript in CCs as a consequence of the overexpression of Nicd (Fig. 7E). Collectively, these results suggest that the Notch signaling pathway TF Su(H) is required for differentially regulating appropriate enhancer activities in PCs, and is crucial for mediating the lineage decision between PCs and CCs.
DISCUSSION
In this study, we used machine learning with DNA motifs and relevant array-based ChIP data to computationally classify cell type-specific Drosophila cardiac enhancers. We show that 48.7% of tested low FPR predictions with a classifier built solely on the presence of DNA motifs are active in the heart. This result is much better than one we had previously obtained for a similar classifier built to uncover muscle founder cell (FC) enhancers (Busser et al., 2012a), and may reflect a larger training set with activity restricted to the heart, a more limited number of distinct cell states for the heart compared with FCs, and/or the sequence features discriminating cardiac cell types being more unique than those for FCs (Busser et al., 2012a). In support of the latter possibility, the accuracy of Drosophila cardiac predictions in the present study is more consistent with our prior classification of human heart enhancers (Narlikar et al., 2010), which are likely to be subject to similar transcriptional regulatory mechanisms (Bodmer and Frasch, 2010).
Our large-scale testing of predicted enhancers also reveals the importance of ChIP data in improving enhancer modeling for cell type-specific predictions. Of note, the ChIP data for Twi, Tin and Mef2 used in this study are not specific solely for cardiac cell types; these TFs are broadly expressed throughout the mesoderm, are involved in the development of numerous types of muscles, and thus bind to a wide range of mesodermal/muscle enhancers. The observation that these non-cell type-specific ChIP data are nevertheless able to significantly improve enhancer predictions specific to particular cardiac cell types suggests that our machine learning procedure is readily applicable to other systems for which ChIP data are available primarily for TFs expressed in multiple cell types.
As both Tin and Mef2 are known to confer both general and subtype properties on mesodermal cells, this latter function of these two TFs may be the crucial determinant for discovering cell type-specific predictions (Bodmer and Frasch, 2010). For example, Mef2 expression is restricted to the CCs in the differentiated heart and Tin expression is restricted to subsets of CCs and PCs. Thus, defining the in vivo binding profile for additional cell-specific regulators of cardiogenesis should vastly improve cell type-specific predictions, as we observed. Moreover, further refinements of our research strategy could lead to the discovery of additional features of the transcriptional codes that regulate the diversity of PC and CC subtypes (Bodmer and Frasch, 2010).
Finally, the inclusion of ChIP data significantly improved the quality of some successful enhancer predictions, as certain predictions that relied solely on motifs as discriminating features did not fall within low FPR cutoffs. This finding suggests that a unique set of sequence features are probably discriminating such enhancers, which supports the conclusion that the currently available training set of PC and CC enhancer sequences may not completely reflect the diversity of transcriptional codes that direct such enhancer activities (Busser et al., 2012a). Therefore, an iterative application of cell-specific enhancer modeling with both the training set and the newly discovered enhancer sequences is likely to improve the reliability of such predictions.
A subnetwork of two forkhead proteins, Polo kinase and Myb9 regulates cardiac progenitor cell divisions
Our previous work revealed that two forkhead TF-encoding genes, jumu and CHES-1-like, ensure the correct number of both CCs and PCs in the heart by regulating Polo activity to mediate three distinct classes of cardiac progenitor cell divisions: asymmetric cell divisions, symmetric cell divisions, and cell divisions occurring at an earlier developmental stage that generate Svp progenitor cells (Ahmad et al., 2012). In the present study, we identify another TF-encoding gene, Myb, which works in concert with jumu, CHES-1-like and polo to mediate the two latter classes of cardiac progenitor cell divisions. As Drosophila Myb was previously shown to regulate the expression of polo in a different context (Wen et al., 2008), a plausible model that both illustrates and explains our observations is presented in Fig. 8A. Consistent with this model’s interpretation of the interconnected roles of Myb, jumu, CHES-1-like and polo in governing specific subsets of cardiac progenitor cell divisions, the loss of function of any one of these genes also results in abnormal spindles (Moutinho-Santos et al., 1999; Goshima et al., 2007; Wen et al., 2008), which could account for both the observed karyokinesis defects and the increase in CCs.
Fig. 8.

Distinct developmental subnetworks regulate cardiac progenitor cell division and specification. (A) Schematic of Jumu, CHES-1-like and Myb regulation of cardiac progenitor cell divisions by Polo kinase. The forkhead TFs Jumu and CHES-1-like regulate Polo kinase activity to mediate three distinct classes of cardiac progenitor cell divisions: asymmetric cell divisions, symmetric cell divisions, and an earlier round of cell division that determines the number of Svp progenitors (Ahmad et al., 2012). Mutations in Myb result in defects in only the latter two categories of cell divisions, which also exhibit synergistic genetic interactions among Myb, jumu, CHES-1-like and polo. As Myb transcriptionally regulates polo (Wen et al., 2008), Myb and the forkhead TFs act in concert to control Polo activity and thus govern both the symmetric and earlier class of cell divisions. (B) Schematic of the involvement of the Notch signaling pathway in the lineage decision between PCs and CCs for PC enhancers like that of Him. Modes of regulation activating and repressing target genes are shown as green and red arrows, respectively. In CCs, the enhancers of PC genes are repressed by the Su(H)-co-repressor complex. The Delta ligand expressed by CCs activates Notch receptor in neighboring PCs, with the resulting cleaved Nicd fragment associating with Su(H) and displacing the co-repressor. The consequent elimination of repressor complex binding in PCs is sufficient to initiate transcription due to the presence of other local TF activators, and is enhanced further by the Nicd-Su(H) complex.
Given that the regulation of polo by the forkhead genes is also crucial for the asymmetric cell division of cardiac progenitors, an intriguing question is why Myb, a known regulator of polo, does not appear to be involved in this process. One possible explanation might be a requirement for the regulation of other components of asymmetric cell division in addition to polo. In this context, it is worth noting that the forkhead genes do indeed regulate additional genes involved in asymmetric cell division; for example, ectopic overexpression of jumu in the mesoderm results in the transcriptional upregulation of abnormal spindle and Inner centromere protein (Ahmad et al., 2012).
The Notch-dependent signaling pathway TF Su(H) discriminates between PC and CC enhancer activities
Another notable TF identified by our machine learning classifier study of Drosophila heart enhancers is Su(H), an integral component of the Notch signaling pathway the presence of which we found unexpectedly discriminates between PC and CC regulatory elements. Previous work had shown that in the absence of Notch signaling, co-repressors such as Groucho or C-terminal binding protein associate with Su(H) to form a repressor complex that binds to the enhancers of target genes to prevent their transcription (Bray and Furriols, 2001; Barolo and Posakony, 2002; Bray and Bernard, 2010). The activation of Notch receptors by ligand binding results in a proteolytic cleavage that releases Nicd from the cell membrane, allowing it to enter the nucleus where it associates with Su(H), displaces the co-repressor and converts the Su(H) complex from a transcriptional repressor into an activator.
The motif preferences associated with PC and CC enhancer classifications in the present study revealed an enrichment of Su(H) binding sites among PC enhancers, and either depletion or nondiscrimination among CC enhancers. We also showed that either mutating Su(H) binding motifs in the Him PC enhancer or reducing Su(H) levels by RNAi knockdown causes this enhancer to drive expression ectopically in CCs, implying that Su(H) normally represses Him in CCs. Finally, we showed that ectopically expressing Nicd in both CCs and PCs results in de-repression of the Him transcript in CCs, demonstrating that Notch signaling activates this PC gene. As we describe below, these studies confirm a requirement of Su(H) binding sites for restricting enhancer activity to PCs.
These results are in agreement with previous investigations of the role of Notch function in cardiogenesis. For example, inactivating mutations of Notch or Delta, the relevant ligand for Notch in cardiogenesis, lead to an overproduction of CCs at the expense of PCs (Mandal et al., 2004; Grigorian et al., 2011), thus indicating a role for Notch signaling in activating key PC genes, whereas relieving the antagonism of Notch signaling through numb loss-of-function mutations results in additional PCs at the expense of CCs (Gajewski et al., 2000; Ward and Skeath, 2000; Han and Olson, 2005). Interestingly, a recent study has shown that Delta expression is restricted to CCs (Grigorian et al., 2011), suggesting that cell-cell signaling between CC and PC progenitors is crucial for the proper restriction of cell fates in the CM, a result consistent with the trans-activating role of Notch (Niessen and Karsan, 2008). Furthermore, the cardiogenic functions of Notch signaling are conserved in vertebrates, as this pathway is involved in the differentiation and proliferation of cardiomyocytes as well as in the formation of the atrioventricular canal and the outflow tract (Niessen and Karsan, 2008). Collectively, these results suggest that Notch signaling mediated by progenitors fated to become CCs instructs nearby progenitors to assume the PC fate, a process that is in part mediated by Su(H) directly regulating key PC target genes. A model that synthesizes and explains the entirety of our observations on Notch-Su(H) regulation of cardiogenesis is presented in Fig. 8B and supplementary material Fig. S10A-D.
Conclusions
Here, we combined sequence features with ChIP data for key cardiac regulators to computationally classify cell type-specific Drosophila cardiac enhancers, thereby identifying heart regulatory elements on a genome-wide scale, their shared and unique sequence motifs, and novel TFs that direct cell type-specific genetic programs during cardiogenesis. Illustrative examples of these computational predictions were validated by appropriate in vivo experiments. The combination of computational and experimental approaches that we employed are generalizable and should readily be applicable to augment an understanding of the developmental gene regulatory networks that operate in other cell types and species.
MATERIALS AND METHODS
Fly stocks and analysis of transgenic reporter constructs
The analysis of site-specific transgenic reporter constructs, antibodies and fly stocks used in this study were previously described (Manak et al., 2002; Ahmad et al., 2012; Busser et al., 2012c; Busser et al., 2012b; Busser et al., 2012a).
Identification and mutagenesis of TF binding sites
Su(H) binding sites were identified by searching sequences for matches to YGTGDGAA (while omitting matches to TGTGTGAA), and single-base mutations were engineered to abrogate Su(H) binding in otherwise wild-type enhancers, as documented in a prior study (Rebeiz et al., 2011). Mutagenesis of the Myb binding sites in the Ndg enhancer was previously described (Busser et al., 2012a).
Classifier training
The methodology behind the isolation of orthologous and control sequences, development of sequence-based classifiers, genome-wide prediction of enhancers, and association of cell-specific genes with predicted enhancers was previously described (Busser et al., 2012a). The full dataset has been deposited at Figshare in the form of an extended version of supplementary material Table S2 (http://dx.doi.org/10.6084/m9.figshare.867687).
Supplementary Material
Acknowledgments
We thank B. Patterson, M. Frasch, R. Bodmer, J. Skeath, W. Grueber and J. Lipsick for providing fly strains and antibodies; M. Bloom, T. Tansey and C. Sonnenbrot for technical assistance; and the TRiP at Harvard Medical School [NIH/NIGMS RO1-GM084947] for providing transgenic RNAi fly stocks.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
B.W.B., D.H., I.O. and A.M.M. designed the overall research project. S.M.A. and B.W.B. designed individual experiments and interpreted the results. S.M.A., B.W.B., D.H., I.O. and A.M.M. wrote the manuscript. B.W.B., E.J.C. and X.Z. performed transgenic reporter assays. D.H. and I.O. constructed classifiers and performed related statistical analyses. S.M.A., X.Z., S.M. and B.W.B. performed cis tests of Ndg and Him enhancer function. S.M.A. performed trans tests of Ndg and Him enhancer function and genetic interaction studies. A.A. and M.L.B. contributed data/materials/analysis tools. N.J. performed Myb-related statistical analyses.
Funding
This work was supported by the National Heart, Lung, and Blood Institute (NHLBI) Division of Intramural Research (A.M.M.); the Intramural Research Program of the NIH; the National Library of Medicine (I.O.); and an American Heart Association Postdoctoral Fellowship (S.M.A.). Deposited in PMC for immediate release.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.101709/-/DC1
References
- Ahmad S. M., Tansey T. R., Busser B. W., Nolte M. T., Jeffries N., Gisselbrecht S. S., Rusan N. M., Michelson A. M. (2012). Two forkhead transcription factors regulate the division of cardiac progenitor cells by a Polo-dependent pathway. Dev. Cell 23, 97–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T. L., Gribskov M. (1998). Combining evidence using p-values: application to sequence homology searches. Bioinformatics 14, 48–54 [DOI] [PubMed] [Google Scholar]
- Barolo S., Posakony J. W. (2002). Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev. 16, 1167–1181 [DOI] [PubMed] [Google Scholar]
- Berger M. F., Philippakis A. A., Qureshi A. M., He F. S., Estep P. W., III, Bulyk M. L. (2006). Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 24, 1429–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard F., Krejci A., Housden B., Adryan B., Bray S. J. (2010). Specificity of Notch pathway activation: twist controls the transcriptional output in adult muscle progenitors. Development 137, 2633–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer R., Frasch M. (2010). Development and aging of the Drosophila heart. In Heart Development and Regeneration (ed. Rosenthal N., Harvey R. P.). London: Academic Press; [Google Scholar]
- Bray S., Bernard F. (2010). Notch targets and their regulation. Curr. Top. Dev.Biol. 92, 253–275 [DOI] [PubMed] [Google Scholar]
- Bray S., Furriols M. (2001). Notch pathway: making sense of suppressor of hairless. Curr. Biol. 11, R217–R221 [DOI] [PubMed] [Google Scholar]
- Busser B. W., Bulyk M. L., Michelson A. M. (2008). Toward a systems-level understanding of developmental regulatory networks. Curr. Opin. Genet. Dev. 18, 521–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busser B. W., Taher L., Kim Y., Tansey T., Bloom M. J., Ovcharenko I., Michelson A. M. (2012a). A machine learning approach for identifying novel cell type-specific transcriptional regulators of myogenesis. PLoS Genet. 8, e1002531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busser B. W., Shokri L., Jaeger S. A., Gisselbrecht S. S., Singhania A., Berger M. F., Zhou B., Bulyk M. L., Michelson A. M. (2012b). Molecular mechanism underlying the regulatory specificity of a Drosophila homeodomain protein that specifies myoblast identity. Development 139, 1164–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busser B. W., Huang D., Rogacki K. R., Lane E. A., Shokri L., Ni T., Gamble C. E., Gisselbrecht S. S., Zhu J., Bulyk M. L., et al. (2012c). Integrative analysis of the zinc finger transcription factor Lame duck in the Drosophila myogenic gene regulatory network. Proc. Natl. Acad. Sci. USA 109, 20768–20773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. C., Lin C. J. (2011). LIBSVM: A library for support vector machines. ACM Transactions on Intelligent Systems and Technology 2, 27 [Google Scholar]
- Cortes C., Vapnik V. (1995). Support-vector networks. Mach. Learn. 20, 273–297 [Google Scholar]
- Davidson E. (2006). The Regulatory Genome: Gene Regulatory Networks in Development And Evolution. London: Academic Press; [Google Scholar]
- Davison A., Hinkley D. (1997). Bootstrap Methods and Their Application. Cambridge: Cambridge University Press; [Google Scholar]
- DeBruhl H., Wen H., Lipsick J. S. (2013). The complex containing Drosophila Myb and RB/E2F2 regulates cytokinesis in a histone H2Av-dependent manner. Mol. Cell. Biol. 33, 1809–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski K., Choi C. Y., Kim Y., Schulz R. A. (2000). Genetically distinct cardial cells within the Drosophila heart. Genesis 28, 36–43 [DOI] [PubMed] [Google Scholar]
- Good P. (1994). Permutation Tests: a Practical Guide to Resampling Methods for Testing Hypotheses. New York, NY: Springer-Verlag; [Google Scholar]
- Goshima G., Wollman R., Goodwin S. S., Zhang N., Scholey J. M., Vale R. D., Stuurman N. (2007). Genes required for mitotic spindle assembly in Drosophila S2 cells. Science 316, 417–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorian M., Mandal L., Hakimi M., Ortiz I., Hartenstein V. (2011). The convergence of Notch and MAPK signaling specifies the blood progenitor fate in the Drosophila mesoderm. Dev. Biol. 353, 105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z., Olson E. N. (2005). Hand is a direct target of Tinman and GATA factors during Drosophila cardiogenesis and hematopoiesis. Development 132, 3525–3536 [DOI] [PubMed] [Google Scholar]
- Jin H., Stojnic R., Adryan B., Ozdemir A., Stathopoulos A., Frasch M. (2013). Genome-wide screens for in vivo Tinman binding sites identify cardiac enhancers with diverse functional architectures. PLoS Genet. 9, e1003195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A. N., Mokalled M. H., Haden T. N., Olson E. N. (2011). JAK/Stat signaling regulates heart precursor diversification in Drosophila. Development 138, 4627–4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junion G., Spivakov M., Girardot C., Braun M., Gustafson E. H., Birney E., Furlong E. E. (2012). A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell 148, 473–486 [DOI] [PubMed] [Google Scholar]
- Kantorovitz M. R., Kazemian M., Kinston S., Miranda-Saavedra D., Zhu Q., Robinson G. E., Göttgens B., Halfon M. S., Sinha S. (2009). Motif-blind, genome-wide discovery of cis-regulatory modules in Drosophila and mouse. Dev. Cell 17, 568–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzen A. L., Jackson J., Harmon B. P., Fung S. M., Ramsay G., Bishop J. M. (1998). Drosophila myb is required for the G2/M transition and maintenance of diploidy. Genes Dev. 12, 831–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejcí A., Bernard F., Housden B. E., Collins S., Bray S. J. (2009). Direct response to Notch activation: signaling crosstalk and incoherent logic. Sci. Signal. 2, ra1 [DOI] [PubMed] [Google Scholar]
- Liu Y. H., Jakobsen J. S., Valentin G., Amarantos I., Gilmour D. T., Furlong E. E. (2009). A systematic analysis of Tinman function reveals Eya and JAK-STAT signaling as essential regulators of muscle development. Dev. Cell 16, 280–291 [DOI] [PubMed] [Google Scholar]
- Manak J. R., Mitiku N., Lipsick J. S. (2002). Mutation of the Drosophila homologue of the Myb protooncogene causes genomic instability. Proc. Natl. Acad. Sci. USA 99, 7438–7443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal L., Banerjee U., Hartenstein V. (2004). Evidence for a fruit fly hemangioblast and similarities between lymph-gland hematopoiesis in fruit fly and mammal aorta-gonadal-mesonephros mesoderm. Nat. Genet. 36, 1019–1023 [DOI] [PubMed] [Google Scholar]
- Moutinho-Santos T., Sampaio P., Amorim I., Costa M., Sunkel C. E. (1999). In vivo localisation of the mitotic POLO kinase shows a highly dynamic association with the mitotic apparatus during early embryogenesis in Drosophila. Biol. Cell 91, 585–596 [PubMed] [Google Scholar]
- Narlikar L., Sakabe N. J., Blanski A. A., Arimura F. E., Westlund J. M., Nobrega M. A., Ovcharenko I. (2010). Genome-wide discovery of human heart enhancers. Genome Res. 20, 381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen K., Karsan A. (2008). Notch signaling in cardiac development. Circ. Res. 102, 1169–1181 [DOI] [PubMed] [Google Scholar]
- Olson E. N. (2006). Gene regulatory networks in the evolution and development of the heart. Science 313, 1922–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M., Yaich L. E., Bodmer R. (1998). Mesodermal cell fate decisions in Drosophila are under the control of the lineage genes numb, Notch, and sanpodo. Mech. Dev. 75, 117–126 [DOI] [PubMed] [Google Scholar]
- Philippakis A. A., Busser B. W., Gisselbrecht S. S., He F. S., Estrada B., Michelson A. M., Bulyk M. L. (2006). Expression-guided in silico evaluation of candidate cis regulatory codes for Drosophila muscle founder cells. PLOS Comput. Biol. 2, e53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay R. G. (2005). c-Myb a stem-progenitor cell regulator in multiple tissue compartments. Growth Factors 23, 253–261 [DOI] [PubMed] [Google Scholar]
- Rebeiz M., Miller S. W., Posakony J. W. (2011). Notch regulates numb: integration of conditional and autonomous cell fate specification. Development 138, 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A., Alkema W., Engström P., Wasserman W. W., Lenhard B. (2004). JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 32, D91–D94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomancak P., Berman B. P., Beaton A., Weiszmann R., Kwan E., Hartenstein V., Celniker S. E., Rubin G. M. (2007). Global analysis of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 8, R145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward E. J., Skeath J. B. (2000). Characterization of a novel subset of cardiac cells and their progenitors in the Drosophila embryo. Development 127, 4959–4969 [DOI] [PubMed] [Google Scholar]
- Warner J. B., Philippakis A. A., Jaeger S. A., He F. S., Lin J., Bulyk M. L. (2008). Systematic identification of mammalian regulatory motifs’ target genes and functions. Nat. Methods 5, 347–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H., Andrejka L., Ashton J., Karess R., Lipsick J. S. (2008). Epigenetic regulation of gene expression by Drosophila Myb and E2F2-RBF via the Myb-MuvB/dREAM complex. Genes Dev. 22, 601–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E., Chen X., Fricke E., Geffers R., Hehl R., Liebich I., Krull M., Matys V., Michael H., Ohnhäuser R., et al. (2001). The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 29, 281–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z., Xu X. L., Frasch M. (1997). Regulation of the twist target gene tinman by modular cis-regulatory elements during early mesoderm development. Development 124, 4971–4982 [DOI] [PubMed] [Google Scholar]
- Zhu X., Ahmad S. M., Aboukhalil A., Busser B. W., Kim Y., Tansey T. R., Haimovich A., Jeffries N., Bulyk M. L., Michelson A. M. (2012). Differential regulation of mesodermal gene expression by Drosophila cell type-specific Forkhead transcription factors. Development 139, 1457–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzen R. P., Girardot C., Gagneur J., Braun M., Furlong E. E. (2009). Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature 462, 65–70 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.