

Abstract
Background
Mitochondrial DNA (mtDNA) encodes key proteins of the electron transfer chain (ETC), which produces ATP through oxidative phosphorylation (OXPHOS) and is essential for cells to perform specialised functions. Tumor-initiating cells use aerobic glycolysis, a combination of glycolysis and low levels of OXPHOS, to promote rapid cell proliferation and tumor growth. Glioblastoma multiforme (GBM) is an aggressively malignant brain tumor and mitochondria have been proposed to play a vital role in GBM tumorigenesis.
Results
Using next generation sequencing and high resolution melt analysis, we identified a large number of mtDNA variants within coding and non-coding regions of GBM cell lines and predicted their disease-causing potential through in silico modeling. The frequency of variants was greatest in the D-loop and origin of light strand replication in non-coding regions. ND6 was the most susceptible coding gene to mutation whilst ND4 had the highest frequency of mutation. Both genes encode subunits of complex I of the ETC. These variants were not detected in unaffected brain samples and many have not been previously reported. Depletion of HSR-GBM1 cells to varying degrees of their mtDNA followed by transplantation into immunedeficient mice resulted in the repopulation of the same variants during tumorigenesis. Likewise, de novo variants identified in other GBM cell lines were also incorporated. Nevertheless, ND4 and ND6 were still the most affected genes. We confirmed the presence of these variants in high grade gliomas.
Conclusions
These novel variants contribute to GBM by rendering the ETC. partially dysfunctional. This restricts metabolism to anaerobic glycolysis and promotes cell proliferation.
Keywords: Mitochondrial DNA, Tumorigenesis, Genetic variants, Aerobic glycolysis, Depletion, Oxidative phosphorylation
Background
Mitochondrial DNA (mtDNA) resides exclusively within the matrix of the mitochondrion [1]. MtDNA is ~16.6 kb in size and encodes 13 proteins that are essential to the electron transfer chain, which produces the vast majority of cellular ATP through the biochemical process of oxidative phosphorylation (OXPHOS) [2]. It also consists of 22 tRNAs and 2 rRNAs and the D-Loop, which is the site of interaction for the nuclear-encoded factors that regulate mtDNA transcription and replication. The D-loop also contains two hypervariable regions, which are indicative of maternal ancestry [3].
The absence of intronic regions increases the likelihood of a variant residing within the coding regions of mtDNA [4], especially since the non-coding region accounts for only ~38% of the entire mitochondrial genome [5]. In the disease state, mutations are commonly observed throughout the mitochondrial genome [6,7]. Due to the presence of multiple copies of mtDNA in the matrix of each mitochondrion and cells having large numbers of mitochondria, mutant and wild-type mtDNA can co-exist.
Primarily, aerobic glycolysis is central to tumor cell metabolism, as these cells exhibit three-times higher glycolytic activity than their normal counterparts [8]. This reflects the Warburg effect, whereby cancer cells undergo a metabolic transition, regardless of the oxygen concentration available [9]. This causes the cell to favour glycolysis rather than OXPHOS, as glycolysis generates ATP at a higher rate than OXPHOS, which produces ATP at a higher yield [10], and is thus more effective for cellular proliferation and growth. It is therefore likely that the presence of multiple mtDNA variants in a single cell or cohort of cells would promote tumorigenesis as there would be a decreased ability to generate ATP through OXPHOS and an increased reliance on glycolysis.
Generally, there is an accumulation in the number of mtDNA variants before a cell experiences detrimental or phenotypic effects [11] and this is likely to be the case for tumor cells. Nevertheless, it remains to be determined whether mtDNA variants present in tumors arise from the clonal expansion of existing variants or if they occur de novo. Normally, variants must be present at significant levels to allow for their detection [12]. However, next generation sequencing enables whole mitochondrial genomes to be sequenced at a high coverage of over 1000-fold, which provides sufficient depth to identify sequence variants at even low levels [13,14].
Although the benefits of using such technology are clear, variants can be called that are interpreted as being true variants but are erroneous [15]. High resolution melt (HRM) analysis is an appropriate validation method to confirm next generation sequencing results by eliminating any errors that may have arisen from the sequencing reactions [16] or from long PCR amplification [17]. Confirmed variants can then be analysed by bioinformatics programs, such as SNPs & GO and MutPred that predict the impact of the SNPs on protein function [18].
Gliomas account for 80% of all primary malignant brain tumors and about 50% of gliomas give rise to Glioblastoma Multiforme (GBM) [19]. GBM has been rated most severe (level 4) on the World Health Organisation’s (WHO) scale of severity, due to its malignant nature [20]. For patients diagnosed with GBM, prognosis is poor with median survival estimated at 12–14 months [21]. Indeed, only about 3-5% of patients survive beyond 3 years [21].
A number of cell lines are representative of GBM. These include the frequently investigated lines, such as U87MG, SF-767 [22] and HSR-GBM1 [23]. Within patient GBM tissues, amplification of epidermal growth factor receptor (EGFR) genes is often observed [24]. This is evident in the GBM 6 and HK301 neurosphere cell lines, both of which express EGFRvIII. Others have been established from expanded cultures from high-grade human GBM tumors.
We have analysed a series of GBM cell lines using a combination of next generation sequencing and HRM analysis to identify variants within the mitochondrial genome that are indicative of tumorigenesis in GBM. We have then depleted one of the cell lines to varying degrees to determine whether the same variants reoccur following mtDNA replenishment as tumorigenesis progresses. We have applied several bioinformatic approaches to predict the severity of each mutation and to model the effects on protein structure. We have confirmed that these variants are indicative of GBM through their identification in GBM tumors. The frequency of variants is greatest within the D-loop and the origin of light strand replication in the non-coding regions whilst NADH dehydrogenase 6 (ND6) is the most susceptible gene for the development of mutations in the coding region.
Methods
Cell culture
The GBM cell lines GBM L1, GBM L2, GBM4, GBM6, CSC020, CSC014, BAH-1 and HK301 were established in-house directly from patient tumors using neural stem cell conditions, as recently described by us [25]. None of these cell lines was exposed to fetal calf serum (FCS). HSR-GBM1, GBM L1, GBM L2 and hNSCs derived from the NIH-approved H9 (WA09) human ESC line (Invitrogen, Carlsbad, CA, USA) were cultured as neurospheres on ultra-low attachment plates (Sigma, St. Louis, MO) in DMEM/F12 Media containing 2% StemPro neural supplement (both Gibco, Carlsbad, CA, USA), 20 ng/ml basic fibroblast growth factor (bFGF) and 20 ng/ml epidermal growth factor (EGF; both Millipore, Billerica, MA). They were passaged every 3 days to maintain their undifferentiated states. The GBM4, GBM6, CSC020, CSC014, BAH-1 and HK301 cell lines were cultured in the same manner but with knockout DMEM/F12 media and with the addition of GlutaMAX (2 mM) only for the GBM 4, CSC-020 and CSC-014 cell lines. The NO7-152 cell line is an in-house cell line and is an early passage line grown in the traditional manner using DMEM high glucose, 4% non-essential amino acids (NEAA), 20% FCS and 2 mM GlutaMAX (all from Gibco, Carlsbad, CA, USA). Both U87MG (sourced directly from ATCC) and SF-767 (sourced directly from Brain Tumor Research Center Tissue Core, UCSF) adherent cell lines were cultured in DMEM/F12 Media supplemented with 5% FCS. Cells underwent enzymatic passaging every 3 days using TrypLE™ Express (Gibco) to dissociate cells into a single cell suspension.
MtDNA depletion
HSR-GBM1 cells were cultured as described above but in the presence of the mtDNA depletion agent, 2′3′-dideoxycytidine (ddC), which was added daily during half media changes at a concentration of 10 μg/ml, alongside 50 μg/ml of uridine (both: Sigma, St Louis, MO). Cells were passaged every 3 days. They were allowed to recover in vitro by withdrawal of ddC from the culture media.
Normal brain and tumor tissue samples
Normal human brain and tumor tissue samples were obtained from the Victorian Brain Bank Network under the human research ethics number 09023B, Southern Health, HREC. The normal human tissues were obtained from individuals with no known history of neoplastic or neurological pathology. The brain tumor samples were from patients diagnosed with high grade glioma.
Xenograft tumor models
Mouse experiments were approved by the Animal Ethics Committee, Monash University, Approval Number: MMCA/2011/76. HSR-GBM1 cells were depleted of their mtDNA over 50 days in culture. They were harvested after depletion to 50%, 20%, 3% and 0.2% of their original mtDNA content. Following depletion, 5 × 105 cells were resuspended in 100μl of Complete DMEM/StemPro Media and inoculated subcutaneously into both leg regions of immunedeficient female BALB/c nude mice (Animal Research Centre, Perth, Australia) aged between 4 to 6 weeks old. Tumors were extracted and stored at -80°C.
DNA extraction
DNA was extracted from cell pellets and tumors using the DNeasy Blood & Tissue kit in the presence of RNase A (Qiagen, West Sussex, UK), following the manufacturer’s recommendations.
Long PCR
Templates for next generation sequencing were generated by long PCR amplification of two overlapping fragments that each spans half of the mitochondrial genome. 50 ng DNA template was used per reaction, together with 1× High Fidelity PCR buffer, 100 mM MgSO4, 1 mM dNTPs (Bioline, London, UK), 1U of Platinum Taq High Fidelity (Invitrogen, Carlsbad, CA, USA) in the presence of 10 μM each of the forward and reverse primers (long1 F: 5′-GACGGGCTCACATCACCCCATAA-3′; long1 R: 5′-GCGTACGGCCAGGGCTATTGGT-3′; long2 F: 5′-GCCACAACTAACCTCCTCGGACTCCT-3′; long2 R: 5′-GGTGGCTGGCACGAAATTGACC-3′). All reactions were performed in a total volume of 50μl. Conditions were 94°C for 2 minutes, 35 cycles of 94°C for 15 seconds, 63°C for 30 seconds and 68°C for 8 minutes 45 seconds. Products were purified using the QIAquick PCR purification kit (Qiagen).
Next generation sequencing using the Ion Torrent Personal Genome Machine (PGM)™
Purified amplicon pairs generated from long PCR were combined at equal concentrations, prior to generation of the amplicon libraries. Amplicon libraries were generated using the recommended workflow procedures from the Ion Fragment Library Kit and Ion Xpress™ Template kit (Life Technologies). MtDNA was sheared using the Covaris Adaptive Focused Acoustics™ system. Fragments of approximately 200 bp were selected following electrophoretic separation with the E-gel system (Life Technologies). Confirmation of product and the quality of the mtDNA was assessed by the Agilent Bioanalyzer using the Agilent High Sensitivity DNA Kit (Agilent, Santa Clara, CA). For multiplexing of the samples, each DNA library was barcoded using different ligation adaptors. Libraries were then pooled at equal concentrations and loaded onto 316 chips for sequencing. Sequence alignment to the reference genome was performed using the Ion Torrent Suite (v.2.2).
Variant selection was performed using CLC Genomics Workbench (v5.5.1). For quality control, reads were filtered to exclude those of a nucleotide length of <15 bp, with one nucleotide being trimmed from both ends of each read. All reads accepted into analysis surpassed a Phred quality score of 15. The following parameters were applied to score reads during the selection process for inclusion into the final alignment: setting a mismatch cost of 2 and an insertion/deletion cost of 3; acceptance of reads that had a minimum of 80% identity to the reference sequence; and exclusion of all duplicate reads. For single nucleotide polymorphism (SNP) analysis, a minimum mutation threshold of 3% was applied.
HRM analysis
Genomic DNA samples for PCR were loaded on 96-well plates (Bio-Rad Laboratories, Hercules, CA) with each sample analysed in triplicate. The starting template for all reactions was 10 ng in total, amplified in the presence of 1× HRM master mix containing LCGreen® Plus + (TrendBio), and 2.5 μM of forward and reverse primers (Additional file 1: Table S1). Reactions were performed in a total volume of 10 μl, with an overlay of 20 μl mineral oil (Sigma). Amplification conditions were: 95°C for 2 minutes, 45 cycles of 94°C for 30 seconds and 55°C for 30 seconds, followed by 1 cycle at 94°C for 30 seconds and cooling to 25°C for heteroduplex formation.
Products underwent melt analysis on the LightScanner (Idaho Technologies, Salt Lake City, Utah) with analysis performed using the LightScanner Instrument & Analysis software with Call-IT 2.0 (V.2.0.0.1331). Data acquisition began at 70°C and increased incrementally by 0.1°C until the reaction terminated at 96°C [26].
Validation of next generation sequencing by HRM is described in: Additional file 2: Supplementary Methods.
In silico predictive analysis on protein function
Web application tools SNPs & GO (University of Bologna) and MutPred (v.1.2.) (Buck Institute, Indiana University) were used to predict the potential effects of amino acid changes to protein function. SNPs & GO uses the known protein sequence, protein function and evolutionary data based on the Swiss-Prot protein database to determine whether the variant is ‘neutral’ or capable of causing ‘disease’, and provides a reliability value in association with its prediction [27]. MutPred predicts the top five features within the protein that are likely to occur as a result of the amino acid substitution, and provides a probability value for each prediction given [28].
Molecular protein modeling
Protein sequences were obtained from the National Center for Biotechnology Institute database. The Clustal Omega tool (EMBL-EBI) was used for sequence alignment. If a protein structure shared a sequence identity of over 70% to a protein of interest, the location of the mutation was identified and its effect predicted using Swiss-PDB viewer (v.4.1.0). Protein structures were obtained from the RCSB Protein Data Bank (PDB) database (PDB ID for COX I: 1OCC_A, COX II: 1OCC_B, COX III: 1V54_C, CYT B: 1BGY_C, CYT C1: 1BGY_D, Rieske protein: 1BGY_E).
Results
Validation of the Ion Torrent PGM™ to detect a range of sequence variants
PCR products containing a known mtDNA variant and its wild-type counterpart were sub-cloned into DNA plasmids. They were mixed at varying percentages to generate a series of standard dilutions to test the sensitivity of the Ion Torrent and were validated by HRM (Additional file 1: Table S1; Additional file 3: Figure S1A-C).
Identification of variants in the GBM cell lines
The full mitochondrial genomes of 12 GBM cell lines were then analysed by Ion Torrent to determine whether the pattern of variants present are shared amongst all cell lines. Altogether, 13 variants were detected in the non-coding region (Table 1) and 19 in the coding region of the mitochondrial genome (Table 2). Within the non-coding region, the majority of variants occurred within the triple-stranded D-loop region. The D-loop harbored the highest frequency of mutation (77.9%) at position 16224 bp in the U87MG cells. For GBM L1, GBM L2 and NO7-152 cell lines, the occurrence of a variant at 310 bp of the D-loop region was shared between the three lines at similar frequencies whilst it was three-fold higher in the CSC014 cells. Variants at positions 2130 bp and 5752 bp were also common, with 6 lines sharing the presence of both variants at similar levels (GBM L1, GBM L2, GBM 4, CSC014, CSC020 and NO7-152). Likewise, the GBM 6, SF-767, U87MG, HK301 and BAH1 lines shared the presence of the 16519 bp variant at high levels (range 51.2 to 58.2%), whilst the GBM 6 and U87MG cell lines shared the presence of the 3168 bp variant (16 s rRNA) at a frequency of 3.7%. For some shared variants, the frequency differed considerably between lines. For example, the D-loop variant at 302 bp was present in the GBM L1 cells at 16.4%, approximately two-fold lower than for NO7-152 (at 39.9%) and CSC014 (at 41.5%) cells.
Table 1.
MtDNA variants identified from sequencing the non-coding region of mtDNA using the Ion Torrent PGM on 12 cell lines representative of GBM
| |
|
|
Percentage change in variant (%) |
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference position | Reference | Variant | HSR-GBM1 | GBM L1 | GBM L2 | GBM 4 | GBM 6 | CSC 014 | CSC 020 | NO7 152 | SF-767 | U87MG | HK301 | BAH1 | Gene region |
| 16186 |
C |
C/T |
4.5 |
|
|
|
|
|
|
|
|
|
|
|
D loop - Hypervariable segment 1, 7S DNA, membrane attachment site |
| 16218 |
C |
C/T |
16.9 |
|
|
|
|
|
|
|
|
|
|
|
|
| 16224 |
T |
T/C |
|
|
|
|
|
|
|
|
|
77.9 |
|
|
|
| 16519 |
T |
T/C |
|
|
|
|
53.7 |
|
|
|
55.8 |
58.2 |
51.2 |
52.9 |
D loop - 7S DNA, membrane attachment site |
| 194 |
C |
T/C |
|
|
|
4.1 |
|
|
|
|
|
|
|
|
D loop - Hypervariable segment 2, membrane attachment site |
| 302 |
A |
A/C |
|
16.4 |
|
|
|
41.5 |
|
39.9 |
|
|
|
|
|
| 310 |
T |
T/C |
|
5.6 |
5.0 |
|
|
17.5 |
|
6.0 |
|
|
|
|
|
| 1386 |
T |
T/C |
9.9 |
|
|
|
|
|
|
|
|
|
|
|
12s rRNA |
|
2002 |
G |
G/A |
|
|
|
|
11.1 |
|
|
|
|
|
|
|
16s rRNA |
|
2130 |
A |
A/G |
|
3.9 |
5.5 |
4.0 |
|
3.6 |
6.3 |
7.4 |
|
|
|
|
|
|
2817 |
G |
G/A |
|
|
|
|
|
|
|
|
|
|
|
11.7 |
|
|
3168 |
C |
C/T |
|
|
|
|
3.7 |
|
|
|
|
3.7 |
|
|
|
| 5752 | A | A/G | 11.3 | 13.1 | 11.4 | 10.5 | 10.9 | 9.9 | L strand replication origin | ||||||
Variants represent those that arise at a percentage frequency of or greater than 3%, after strict parameters were applied to exclude low quality reads from the final mapping of each full mitochondrial genome sequence. Variants that have not been previously reported are in bold.
Table 2.
MtDNA variants identified within the coding region of 12 GBM cell lines
| |
|
Percentage change in variant (%) |
|
|
|
MutPred |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference position | Variant change | HSR-GBM1 | GBM L1 | GBM L2 | GBM 4 | GBM 6 | CSC 014 | CSC 020 | NO7 152 | SF-767 | U87MG | HK301 | BAH1 | Gene region | Amino acid change | SNPs & GO | Probability of deleterious mutation | Top 5 predicted features caused by the amino acid mutation |
| 6422 |
C→T |
3.8 |
|
|
|
|
|
|
|
|
|
|
|
COX I |
Syn (P) |
- |
- |
|
|
6999 |
G→A |
6.5 |
|
|
|
|
|
|
|
|
|
|
|
V366M |
Neutral, RI 4, uniprot P00395 |
0.454 |
Loss of stability (P = 0.0688) |
|
| Loss of sheet (P = 0.0817) | ||||||||||||||||||
| Loss of catalytic residue at V366 (P = 0.1011) | ||||||||||||||||||
| Loss of glycosylation at S362 (P = 0.2022) | ||||||||||||||||||
| Gain of loop (P = 0.4661) | ||||||||||||||||||
| 8251 |
G→A |
|
43.5 |
|
50.8 |
|
|
|
|
|
|
|
|
COX II |
Syn (G) |
- |
- |
|
|
8252 |
C→A |
|
55.5 |
|
46.8 |
|
|
|
|
|
|
|
|
P223T |
Neutral, RI 9, uniprot P00403 |
0.327 |
Gain of glycosylation at P223 (P = 0.1135) |
|
| Loss of disorder (P = 0.1694) | ||||||||||||||||||
| Loss of catalytic residue at G222 (P = 0.2169) | ||||||||||||||||||
| Loss of phosphorylation at T226 (P = 0.3735) | ||||||||||||||||||
| Loss of helix (P = 0.3949) | ||||||||||||||||||
|
10473 |
C→G |
|
|
|
|
|
4.3 |
|
|
|
|
|
|
ND4L |
P2A |
Neutral, RI 9, uniprot P03901 |
0.344 |
Loss of disorder (P = 0.0496) |
| Loss of catalytic residue at L3 (P = 0.1395) | ||||||||||||||||||
| Gain of helix (P = 0.2684) | ||||||||||||||||||
| Loss of loop (P = 0.3664) | ||||||||||||||||||
| Loss of phosphorylation at Y5 (P = 0.4053) | ||||||||||||||||||
|
10814 |
A→C |
|
6.0 |
|
5.3 |
|
|
5.0 |
5.2 |
|
|
|
|
ND4 |
K19Q |
Neutral, RI 8, uniprot P03905 |
0.551 |
Loss of methylation at K19 (P = 0.0012) |
|
Loss of ubiquitination at K19 (P = 0.0283) | ||||||||||||||||||
| Loss of MoRF binding (P = 0.134) | ||||||||||||||||||
| Gain of helix (P = 0.2684) | ||||||||||||||||||
| Loss of catalytic residue at K19 (P = 0.2966) | ||||||||||||||||||
| 11361 |
T→C |
|
|
|
|
|
6.0 |
|
|
|
|
|
|
M201T |
Neutral, RI 5, uniprot P03905 |
0.706 |
Loss of stability (P = 0.0853) |
|
| Gain of ubiquitination at K206 (P = 0.1204) | ||||||||||||||||||
| Gain of catalytic residue at M201 (P = 0.1253) | ||||||||||||||||||
| Gain of methylation at K206 (P = 0.1903) | ||||||||||||||||||
| Loss of MoRF binding (P = 0.2081) | ||||||||||||||||||
| 11512 |
C→A |
|
6.0 |
|
9.6 |
|
11.0 |
6.4 |
15.2 |
|
|
|
|
N251K |
Neutral, RI 4, uniprot P03905 |
0.495 |
Gain of methylation at N251 (P = 0.0194) |
|
| Gain of MoRF binding (P = 0.0632) | ||||||||||||||||||
| Loss of stability (P = 0.0709) | ||||||||||||||||||
| Loss of ubiquitination at K255 (P = 0.0768) | ||||||||||||||||||
| Gain of solvent accessibility (P = 0.0837) | ||||||||||||||||||
| 11674 |
C→T |
|
|
|
3.5 |
|
|
|
|
|
|
|
|
Syn (T) |
- |
- |
|
|
|
12101 |
T→C |
|
|
|
3.8 |
|
3.1 |
|
4.2 |
|
|
|
|
S448P |
Neutral, RI 7, uniprot P03905 |
0.484 |
Loss of helix (P = 0.0093) |
|
| Gain of loop (P = 0.0321) | ||||||||||||||||||
| Gain of relative solvent accessibility (P = 0.09) | ||||||||||||||||||
| Gain of sheet (P = 0.1451) | ||||||||||||||||||
| Gain of catalytic residue at L447 (P = 0.1502) | ||||||||||||||||||
| 12102 |
C→T |
|
|
|
3.7 |
|
|
|
3.2 |
|
|
|
|
S448F |
Neutral, RI 3, uniprot P03905 |
0.472 |
Loss of disorder (P = 0.0619) |
|
| Gain of helix (P = 0.2059) | ||||||||||||||||||
| Loss of loop (P = 0.2897) | ||||||||||||||||||
| Loss of phosphorylation at S448 (P = 0.5302) | ||||||||||||||||||
| Gain of catalytic residue at S448 (P = 0.5425) | ||||||||||||||||||
|
12877 |
G→C |
|
|
|
|
|
|
|
|
27.7 |
|
|
|
ND5 |
G181R |
Disease, RI 7, Uniprot P03915 |
0.795 |
Loss of catalytic residue at I183 (P = 0.1945) |
| Gain of MoRF binding (P = 0.2553) | ||||||||||||||||||
| Gain of methylation at G181 (P = 0.3559) | ||||||||||||||||||
| Loss of helix (P = 0.4763) | ||||||||||||||||||
| Loss of stability (P = 0.5598) | ||||||||||||||||||
| 13043 |
C→T |
|
|
|
|
|
|
|
|
|
3.3 |
|
|
A236V |
Neutral, RI 3, Uniprot P03915 |
0.786 |
Loss of glycosylation at P234 (P = 0.0757) |
|
| Loss of disorder (P = 0.0789) | ||||||||||||||||||
| Gain of helix (P = 0.132) | ||||||||||||||||||
| Loss of phosphorylation at T241 (P = 0.2504) | ||||||||||||||||||
| Loss of loop (P = 0.2897) | ||||||||||||||||||
|
13061 |
C→A |
|
|
|
|
|
|
4.0 |
|
|
|
|
|
P242Q |
Disease, RI 0, uniprot P03915 |
0.776 |
Loss of glycosylation at P242 (P = 0.035) |
|
| Loss of phosphorylation at T241 (P = 0.1079) | ||||||||||||||||||
| Loss of disorder (P = 0.1807) | ||||||||||||||||||
| Loss of catalytic residue at E238 (P = 0.1978) | ||||||||||||||||||
| Loss of helix (P = 0.2271) | ||||||||||||||||||
|
14159 |
C→G |
|
|
4.1 |
|
|
|
|
|
|
|
|
|
ND6 |
R172P |
Disease, RI 3, uniprot P03923 |
0.423 |
Loss of methylation at R172 (P = 0.0305) |
| Gain of catalytic residue at R172 (P = 0.0632) | ||||||||||||||||||
| Loss of sheet (P = 0.0817) | ||||||||||||||||||
| Loss of stability (P = 0.126) | ||||||||||||||||||
| Gain of disorder (P = 0.1619) | ||||||||||||||||||
|
14160 |
G→C |
|
|
4.0 |
3.5 |
3.3 |
7.5 |
|
4.3 |
3.2 |
|
8.1 |
5.0 |
R172G |
Neutral, RI 2, uniprot P03923 |
0.442 |
Loss of methylation at R172 (P = 0.0305) |
|
| Loss of stability (P = 0.0532) | ||||||||||||||||||
| Loss of sheet (P = 0.0817) | ||||||||||||||||||
| Gain of disorder (P = 0.1578) | ||||||||||||||||||
| Gain of loop (P = 0.2045) | ||||||||||||||||||
|
14426 |
C→T |
|
|
|
|
|
8.9 |
|
|
|
|
|
|
G85E |
Neutral, RI 7, uniprot P03923 |
0.364 |
Loss of glycosylation at S84 (P = 0.0357) |
|
|
Gain of solvent accessibility (P = 0.0456) | ||||||||||||||||||
| Loss of catalytic residue at V86 (P = 0.1017) | ||||||||||||||||||
| Gain of disorder (P = 0.1294) | ||||||||||||||||||
| Gain of loop (P = 0.2045) | ||||||||||||||||||
|
15264 |
C→T |
14.1 |
|
|
|
|
|
|
|
|
|
|
|
CYTB |
P173L |
Disease, RI 4, uniprot P00156 |
0.361 |
Loss of relative solvent accessibility (P = 0.0793) |
| Loss of solvent accessibility (P = 0.089) | ||||||||||||||||||
| Gain of methylation at R177 (P = 0.1226) | ||||||||||||||||||
| Loss of glycosylation at S172 (P = 0.1763) | ||||||||||||||||||
| Loss of disorder (P = 0.2084) | ||||||||||||||||||
|
15267 |
C→G |
20.5 |
|
|
|
|
|
|
|
|
|
|
|
T174S |
Disease, RI 3, uniprot P00156 |
0.866 |
Gain of glycosylation at T174 (P = 0.0587) |
|
| Gain of disorder (P = 0.0665) | ||||||||||||||||||
| Loss of catalytic residue at T174 (P = 0.1513) | ||||||||||||||||||
| Loss of methylation at R177 (P = 0.2045) | ||||||||||||||||||
| Loss of sheet (P = 0.3635) | ||||||||||||||||||
In silico analysis was performed using the online tools SNPs & GO and MutPred to predict the impact these variants exert on the corresponding protein of interest. Variants that have not been previously reported are in bold.
For the 19 variants identified within the coding region of the mitochondrial genome (Table 2), the levels ranged from 3.1% at position 12101 bp in the NADH dehydrogenase 4 (ND4) gene in the CSC-014 cell line to 55.5% at position 8252 bp (Cytochrome C Oxidase II; COX II) in the GBM L1 cell line. The most frequently observed variants were at positions 11512 bp (ND4) and 14160 bp (ND6), which were present in 5/12 and 8/12 lines, respectively. 16 of the variants resulted in non-synonymous amino acid changes whilst 3 were synonymous. Of the 16 non-synonymous substitutions, 5 were predicted by the in silico web-based tool, SNPs & GO, to be disease causing (Table 2). The 13061 bp variant was predicted by MutPred to cause impaired protein function due to the substitution of a glutamine for a proline at position 242 in the NADH dehydrogenase 5 (ND5) polypeptide chain (P = 0.035) resulting in the loss of glycosylation. The variant at position 14159 bp substituted a proline for a methylated arginine at position 172, again disrupting protein function (P = 0.03). The cytochrome B (CYT B) disease-causing variants at positions 15264 bp and 15267 bp were predicted to result in the loss of relative solvent accessibility and gain of glycosylation, respectively.
Overall, the majority of variants in the coding regions for all 12 cell lines arose in the ND4 region (n = 6 variants). Both the ND5 and ND6 genes, the latter of which is encoded on the light strand of mtDNA, possessed the second highest number of variants (n = 3). The ND6 variant at position 14160 bp was the most common mutational event with 8/12 lines harboring this variant. All variants were confirmed using HRM analysis, for example see Additional file 4: Figure S2.
The susceptibility of the different mtDNA gene regions to the development of variants
To determine the susceptibility of various mtDNA regions to mutation, we normalized the number of variants identified in each mtDNA region to the size of the gene region (bp) on which the variant was located. The overall probability of deriving a mutation in the non-coding region (2.08 × 10-3) was greater than in the coding region (1.84 × 10-3) (Additional file 5: Table S2). The 12S rRNA region was found to be the least susceptible to mutation of all the non-coding regions (n = 1 variant within 953 bp: Additional file 5: Table S2) whilst the origin of light strand replication (1 variant within 77 bp) was the region most likely to develop mutation (12.38X greater than for the 12S rRNA region) (Additional file 5: Table S3). Although the D-loop harboured 7 variants, due to its size, it was the second most susceptible non-coding region to mutation. In the coding region, Cytochrome C Oxidase I (COX I) was the least susceptible to acquiring mutation (1.298 × 10-3), whilst ND6 was the most susceptible (5.725 × 10-3), representing a 4.41X fold difference between the two genes (Additional file 5: Table S3).
Protein modeling of non-synonymous amino acid substitutions
To understand the impact of non-synonymous amino acid changes induced by the introduction of variants into the mitochondrial genome, we performed protein modeling on the bovine protein structures for COX I, COX II and CYT B equivalents due to the unavailability of the human structures. These are 91%, 72% and 78.4% homologous to their respective human proteins. The ND subunits do not have protein structures available that closely resemble the corresponding human protein sequence.
The CYT B variants at mtDNA nucleotide positions 15264 bp and 15267 bp induce a transition from proline to leucine at position 173 (P173L) for the variant at 15264 bp, and a change from threonine to serine at position 174 (T174S) for the 15267 bp variant. These variants are located at the terminal end of one of the transmembrane helical domains on CYT B (Figure 1). However, their relatively central location in relation to the catalytic centre of CYT B is predicted to impact on function. The mtDNA variant identified at position 6999 bp in the COX I region of the HSR-GBM1 cells induces a change from valine to methionine at position 366 of the polypeptide chain. However, the mutational change resides within a transmembrane helix region that faces towards the mitochondrial intermembrane space (Additional file 6: Figure S3) and is predicted to have a neutral effect (Table 2). The variant at position 8252 bp in COX II of the HSR-GBM1 cells results in a proline to a threonine transition at position 223 (P223T) of the protein sequence. It maps to a helix region located away from the catalytic core of complex IV in the mitochondrial intermembrane space (Additional file 7: Figure S4) and is thus predicted to be neutral (Table 2).
Figure 1.
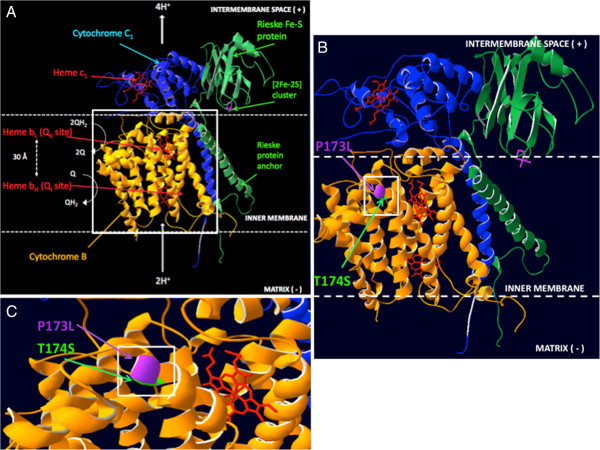
Protein structure of the complex III catalytic centre (the cytochrome bc1 complex) with annotated locations for the variants in cytochrome B. (A) The catalytic centre of complex III is shown, represented by three interacting subunits, cytochrome B (yellow), cytochrome C1 (blue) and the Rieske iron-sulfur protein (green). The key redox cofactors are Heme bL and bH, which reside within cytochrome B, and heme c1 in cytochrome C1. Oxidation of ubihydroquinone (QH2) occurs at the Qo site, and reduction of ubiquinone to ubiquinol at the Qi site. Following these events, a membrane potential is generated across the inner mitochondrial membrane. The Rieske iron-sulfur protein interconnects the cytochrome B and C1 subunits. (B) Location of the proline to leucine (P173L) and threonine to serine (T174S) mutations are highlighted on cytochrome B. Both of these reside on a transmembrane helical region of the subunit. (C) Magnified view of the adjacent P173L and T174S mutations.
Variants identified in the brain tissue of normal brain biopsy patients
In order to determine the type and frequency of mtDNA variants in brain samples from unaffected individuals, we analysed brain tissue samples from 13 individuals with no known brain disease. The majority of variants (14/21) were present in hypervariable regions I and II of the D-loop (Table 3). Sample 05–470 possessed the highest number of non-coding variants (n = 12) whilst the remaining 12 samples possessed 5 or fewer. Apart from the variant at position 16519 bp, the other variants were present at frequencies <10% except for the A→G substitution at position 16293 bp, which was present at 16.1% in 04–273, and the G→C substitution at 12.1% at position 2647 bp in the 16 s rRNA region for sample 05–470.
Table 3.
Variants detected within the non-coding region of mtDNA in normal brain tissue (n = 13)
| |
|
|
Percentage change in variant (%) |
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference position | Reference | Variant | 03-965 | 04-273 | 05-470 | 07-104 | 07-445 | 07-635 | 08-012 | 08-026 | 09-005 | 09-152 | 16-1 | 22-1 | 20-1 | Gene region |
| 16069 |
C |
T |
|
|
7.2 |
|
|
|
|
|
|
|
|
|
|
D loop - Membrane attachment site, Hypervariable segment 1 |
| 16126 |
T |
C |
|
|
5.2 |
|
|
|
|
|
|
|
|
|
|
D loop - Membrane attachment site, Hypervariable segment 1, 7S DNA |
| 16145 |
G |
A |
|
|
4.8 |
|
|
|
|
|
|
|
|
|
|
D loop - Membrane attachment site, Hypervariable segment 1, 7S DNA, termination-associated sequence |
| 16179 |
C |
T |
|
|
|
|
|
|
|
|
4.3 |
|
|
|
|
D loop - Membrane attachment site, Hypervariable segment 1, 7S DNA |
| 16222 |
C |
T |
|
|
5.1 |
|
|
|
|
|
|
|
|
|
|
|
| 16261 |
C |
T |
|
|
9.4 |
|
|
|
|
|
|
|
|
|
|
|
| 16293 |
A |
G |
|
16.1 |
|
|
|
|
|
|
|
|
|
|
|
|
| 16356 |
T |
C |
|
|
|
|
|
|
|
|
3.0 |
|
|
|
|
|
| 16519 |
T |
C |
57.5 |
|
54.5 |
|
|
56.0 |
|
57.6 |
58.3 |
59.4 |
|
|
|
D loop - Membrane attachment site, 7S DNA |
| 9 |
G |
T |
|
|
5.8 |
|
|
|
|
|
|
|
|
|
|
|
| 10 |
T |
C |
|
|
5.8 |
|
|
|
|
|
|
|
|
|
|
|
| 66 |
G |
T |
|
|
|
|
|
|
|
|
|
|
|
|
3.2 |
D-loop - Hypervariable segment 2, membrane attachment site, 7S DNA |
| 73 |
A |
G |
|
|
6.2 |
|
|
|
|
|
|
|
|
|
|
|
| 150 |
C |
T |
|
|
|
|
|
|
|
|
4.4 |
|
|
|
|
D loop - Hypervariable segment 2, H-strand origin, Membrane attachment site, 7S DNA |
| 189 |
A |
G |
|
|
|
|
|
|
3.0 |
|
|
|
|
|
|
|
| 195 |
T |
C |
|
|
|
|
|
|
|
|
4.5 |
|
|
|
|
D loop - Hypervariable segment 2, H-strand origin, Membrane attachment site |
| 242 |
C |
T |
|
|
7.7 |
|
|
|
|
|
|
|
|
|
|
D loop - Hypervariable segment 2, H-strand origin, mtTF1 binding site, Membrane attachment site |
| 751 |
A |
T |
|
|
|
|
|
|
3.9 |
|
|
|
|
|
|
12s ribosomal RNA (rRNA) |
|
2232 |
A |
T |
|
|
|
|
|
3.0 |
|
|
|
|
|
|
|
16s ribosomal RNA (rRNA) |
|
2647 |
G |
C |
|
|
12.1 |
|
|
|
|
|
|
|
|
|
|
|
| 3010 | G | A | 3.5 | |||||||||||||
Variants represented those that were present at or above the 3% mutation threshold selection criteria. Variants that have not been previously reported are in bold.
In the coding region of the normal samples, 39 variants were identified (Table 4). CYT B harboured the largest proportion of variants (9/39) whilst ND4 possessed the second highest proportion of variants (6/39). The majority of variants (21/39) led to non-synonymous amino acid changes, whilst 1 variant generated a stop codon at position 8630 bp.
Table 4.
Variants identified in the coding region of the mitochondrial genome in normal brain tissue samples
| |
|
|
Percentage change in variant (%) |
|
|
|
MutPred |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference position | Reference | Variant | 03-965 | 04-273 | 05-470 | 07-104 | 07-445 | 07-635 | 08-012 | 08-026 | 09-005 | 09-152 | 16-1 | 22-1 | 20-1 | Gene region | Amino acid change | SNPs & GO | Probability of deleterious mutation | Top 5 predicted features caused by amino acid mutation |
| 4646 |
T |
C |
|
|
|
|
|
|
|
|
3.4 |
|
|
|
|
ND2 |
Syn (Y) |
- |
|
|
|
4701 |
A |
T |
|
|
|
4.8 |
|
|
|
|
|
|
|
|
|
N78Y |
Neutral, RI 7, Uniprot P03891 |
0.329 |
|
|
| 4703 |
T |
C |
|
|
|
4.8 |
|
|
|
|
|
|
|
|
|
Syn (N) |
- |
|
|
|
|
4878 |
G |
A |
|
|
|
|
|
|
|
|
4.6 |
|
|
|
|
A137T |
Neutral, RI 5 |
0.786 |
|
|
|
4879 |
C |
G |
|
|
|
|
|
|
|
|
5.0 |
|
|
|
|
A137G |
Neutral, RI 5 |
0.78 |
Loss of stability (P = 0.0465) |
|
| 6146 |
A |
G |
|
|
|
|
|
|
|
|
3.1 |
|
|
|
|
COX I |
Syn (W) |
- |
|
|
|
8497 |
A |
G |
|
12.5 |
|
|
|
|
|
|
4.8 |
|
|
|
|
ATP8 |
Syn (M) |
- |
|
|
|
8630 |
A |
G |
|
|
|
|
|
|
|
|
|
|
3.7 |
|
|
ATP6 |
K35 Stop |
Uniprot P00846 |
|
|
| 8756 |
T |
C |
|
|
|
|
|
|
|
6.5 |
|
|
|
|
|
I77T |
Neutral, RI 5 |
0.684 |
Loss of stability (P = 0.0062) |
|
| 8790 |
G |
A |
|
|
|
|
18.4 |
|
|
|
|
|
|
|
|
Syn (L) |
- |
|
|
|
| 8994 |
G |
A |
|
|
|
|
|
|
|
4.7 |
|
|
|
|
|
Syn (L) |
- |
|
|
|
| 9070 |
T |
G |
|
|
|
|
|
|
|
|
6.2 |
|
|
|
|
S182A |
Neutral RI 5 |
0.162 |
Loss of glycosylation at S182 (P = 0.0094) |
|
| 9258 |
C |
T |
|
|
|
|
3.2 |
|
|
|
|
|
|
|
|
COX III |
Syn (L) |
- |
|
|
|
9526 |
C |
G |
|
5.0 |
|
|
|
3.8 |
|
|
|
|
|
|
|
A107G |
Neutral, RI 5, Uniprot P00414 |
0.667 |
Loss of stability (P = 0.0332) |
|
|
9528 |
C |
A |
|
9.9 |
6.4 |
|
|
|
4.8 |
|
|
|
|
|
|
P108T |
Neutral, RI 2 |
0.731 |
|
|
|
9558 |
C |
A |
|
|
|
|
5.8 |
|
|
3.6 |
|
|
|
|
|
P118T |
Disease, RI 3 |
0.579 |
Loss of catalytic residue at P117 (P = 0.0195) |
|
|
Gain of glycosylation at P118 (P = 0.0283) | ||||||||||||||||||||
| 10398 |
A |
G |
|
|
5.4 |
|
|
|
|
|
|
|
|
|
|
ND3 |
T114A |
Neutral, RI 10, Uniprot P03897 |
0.071 |
|
| 10993 |
G |
A |
|
|
4.6 |
|
|
|
|
|
|
|
|
|
|
ND4 |
Syn (M) |
- |
|
|
| 11332 |
C |
T |
|
|
|
|
|
|
|
|
3.4 |
|
|
|
|
Syn (A) |
- |
|
|
|
| 11467 |
A |
G |
|
|
|
|
|
|
|
|
3.1 |
|
|
|
|
Syn (L) |
- |
|
|
|
|
11516 |
C |
A |
|
|
|
|
|
|
4.9 |
|
|
|
|
|
|
L253M |
Neutral, RI 7, Uniprot P03905 |
0.369 |
|
|
|
11725 |
A |
T |
|
|
|
|
|
|
3.0 |
|
|
|
|
|
|
Syn (T) |
- |
|
|
|
|
11791 |
C |
T |
|
|
|
|
|
|
|
|
|
|
11.8 |
|
|
Syn (L) |
|
|
|
|
|
12719 |
T |
C |
3.2 |
|
|
|
|
|
|
|
|
|
|
|
|
ND5 |
M128T |
Disease, RI 2, Uniprot P03915 |
0.795 |
Loss of stability (P = 0.0126) |
| 12774 |
C |
T |
|
|
|
|
|
|
|
|
|
|
9.3 |
|
|
Syn (G) |
|
|
|
|
| 13984 |
C |
T |
|
|
|
|
|
|
|
|
|
3.7 |
|
|
|
Syn (L) |
- |
|
|
|
|
13985 |
T |
C |
|
|
3.1 |
|
|
|
|
|
3.1 |
3.7 |
|
|
|
L550P |
Neutral, RI 1 |
0.457 |
Gain of loop (P = 0.0013) |
|
|
Loss of helix (P = 0.0041) | ||||||||||||||||||||
|
Gain of catalytic residue at P549 (P = 0.0244) | ||||||||||||||||||||
|
Gain of glycosylation at L550 (P = 0.0364) | ||||||||||||||||||||
|
Gain of relative solvent accessibility (P = 0.0479) | ||||||||||||||||||||
| 14155 |
C |
T |
|
|
|
|
|
|
|
|
|
3.8 |
|
|
|
ND6 |
Syn (G) |
- |
|
|
|
14159 |
C |
G |
|
|
|
|
|
|
12.5 |
|
|
|
|
|
|
R172P |
Disease, RI 3, Uniprot P03923 |
0.423 |
Loss of methylation at R172 (P = 0.0305) |
|
|
14160 |
G |
C |
|
|
|
|
8.1 |
|
12.5 |
|
|
|
|
|
|
R172G |
Neutral, RI 2 |
0.442 |
Loss of methylation at R172 (P = 0.0305) |
|
| 14770 |
C |
A |
|
|
|
|
|
|
|
|
|
|
|
8.0 |
|
CYT B |
N8K |
Neutral RI 1, Uniprot P00156 |
0.446 |
Gain of methylation at N8 (P = 0.0039) |
|
Gain of MoRF binding (P = 0.0159) | ||||||||||||||||||||
|
Gain of ubiquitination at N8 (P = 0.0452) | ||||||||||||||||||||
|
14823 |
A |
C |
|
|
|
|
4.0 |
|
|
|
|
|
|
|
|
N26T |
Disease, RI 6 |
0.752 |
|
|
| 14857 |
T |
C |
|
|
|
|
|
|
3.8 |
|
|
|
|
|
|
Syn (L) |
- |
|
|
|
|
14861 |
G |
T |
|
|
|
|
|
|
3.6 |
|
|
|
|
|
|
A39S |
Neutral, RI 5 |
0.33 |
|
|
| 14866 |
C |
T |
|
|
|
|
|
|
|
|
5.4 |
|
|
|
|
Syn (C ) |
- |
|
|
|
| 15287 |
T |
C |
|
|
|
|
26.9 |
|
|
|
|
|
|
|
|
F181L |
Neutral, RI 3 |
0.617 |
|
|
| 15452 |
C |
A |
|
|
3.2 |
|
|
|
|
|
|
|
|
|
|
L236I |
Neutral, RI 7 |
0.307 |
|
|
|
15579 |
A |
T |
|
|
|
|
|
3.7 |
|
|
|
|
|
|
|
Y278F |
Disease, RI 6 |
0.796 |
|
|
| 15693 | T | C | 4.9 | M316T | Neutral, RI 8 | 0.312 | ||||||||||||||
Variants represented those that were present at or above the 3% mutation threshold selection criteria. Variants that have not been previously reported are in bold. Predictions on the effects of amino acid substitutions were performed using MutPred and SNPs & GO online tools.
The D-loop region was the most susceptible to mutation (1.517 × 10-2) amongst the coding and non-coding regions of mtDNA with COX I being the least susceptible (6.4893 × 10-4) (Additional file 5: Table S4) representing a 23.37-fold difference (Additional file 5: Table S5). Within the coding region, CYT B was the most susceptible amongst all other coding genes with a score 12.17X higher than that for COX I (Additional file 5: Table S5).
Identification of variants through cell culture
In order to determine whether the culture environment would induce mtDNA variants over time, we sequenced the mitochondrial genome of a human neural stem cell (hNSC) line that had been subjected to multiple passaging (x20). We identified 2 variants in the non-coding region, which were restricted to the D-loop region. The variant at position 16519 bp had the highest percentage change at 51.3% whilst the variant at position 496 was present at 3.3%. In the coding region, there were 4 variants, namely three located in ND5 at positions 13762 bp (3.2%), 13984 (3.6%) (D-loop) and 13985 (3.6%) and 1 residing in CYT B at position 15153 bp (9.4%). The low level of percentage variant present reflects the low levels identified in the normal samples. Furthermore, comparison of the variants present in the traditional immortalised lines (NO7 152, SF-767 and U87MG) and the primary glioma lines (GBM L1; GBM L2; GBM 4; GBM 6; CSC014; CSC020, HK301 and BAH1) did not reveal a greater distribution of mtDNA variants for one set of lines.
Phylogenetic analysis of the GBM cell lines and normal brain samples
In order to determine that the normal samples and the GBM cell lines were not indicative of tight cohorts of mtDNA clusters but were from comparable lineages, we subjected them to phylogenetic analysis. As can be seen from Additional file 8: Figure S5, there is no bias due to the equal distribution of samples across a wide mtDNA genetic distribution and that the variants observed appear to be unique to the tumor samples.
Identification of mtDNA variants essential to tumorigenesis
In order to determine which mtDNA variants identified in the GBM cell lines are essential to tumorigenesis, we depleted HSR-GBM1 cells to varying degrees, namely to 50% (mtDNA50), 20% (mtDNA20), 3% (mtDNA3) and 0.2% (mtDNA0.2) of their original mtDNA content. We transplanted these and non-depleted HSR-GBM1 (mtDNA100) cells into BALB/c nude mice and assessed whether the same or other mtDNA variants were selected for as cells replenished their mtDNA content during tumorigenesis.
Through HRM, we determined that 10/22 of the tumors harboured all of the variants present in the parental cells whilst 9 (2 mtDNA100, 5 mtDNA50, 1 mtDNA20 and 1 mtDNA0.2) shared 15/16 variants. Another 2 tumors (1 mtDNA100 and 1 mtDNA3) shared 14/16 variants and 1 (mtDNA20) shared 13/16 variants (Figure 2). In the non-coding region, the variants at 16186 bp, 16218 bp, 302 bp, 310 bp and 1386 bp (12 s rRNA) persisted in all tumors, whereas the variants at 6999 bp (COX I), 11361 bp and 11674 bp (ND4), 14159 bp and 14160 bp (ND6), and 15264 bp and 15267 bp (CYT B) persisted in the coding region of all tumors.
Figure 2.
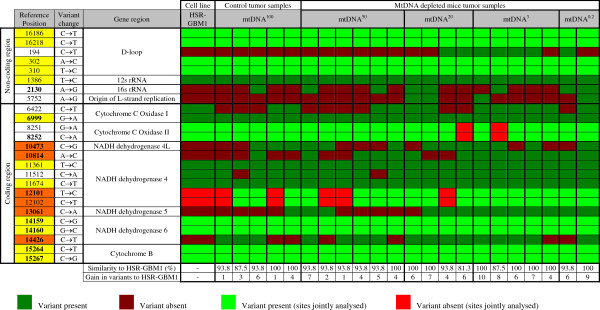
HRM analysis of HSR-GBM1 cells that have undergone mtDNA depletion for up to 50 days, followed by the replenishment of mtDNA in vivo in immunedeficient nude mice, based on the panel of variants identified across the different GBM cell lines. Tumors mtDNA50, mtDNA20, mtDNA3 and mtDNA0.2 were generated with HSR-GBM1 cells depleted of their mtDNA copy number to 50%, 20%, 3% and 0.2% of their original copy number relative to non-depleted HSR-GBM1 cells (mtDNA100). Multiple samples for each tumor type represent experimental replicates. The variants highlighted in yellow indicate variants shared between all samples analysed. Variants highlighted in orange represent those that have been acquired de novo during tumor formation and were identified within the other GBM cell lines.
Nevertheless, there was an overall gain in the number of variants (Figure 3) as a function of cells having undergone increased levels of mtDNA depletion (P = 0.0027). Specifically, there were significant differences in the incremental gain of variants between the parental cells and the mtDNA20 (P < 0.05), mtDNA3 (P < 0.01) and mtDNA0.2-derived (P < 0.01) tumors (Figure 3). These variants were identified in the other GBM cell lines. The variants that were persistently selected were at 10473 bp (NADH dehydrogenase 4L; ND4L), 10814 bp, 12101 bp and 12102 bp (ND4), 13061 bp (ND5) and 14426 bp (ND6). Interestingly, the variants associated with the ND4 and ND5 were more frequently present in the mtDNA20-derived, mtDNA3-derived and mtDNA0.2-derived tumors, as was the ND6 tumor-specific variant at 14426 bp (Figure 2). We then assessed the presence of variants in cells depleted to 50%, 20% and 3% of their original mtDNA content and then allowed to recover in culture over 14 days. As can be seen from Additional file 5: Table S6, the vast majority of the variants are present in recovered cells, although those variants at 10473 bp (ND4L), 10814 bp, 12101 bp and 12102 bp (ND4) and 13061 bp (ND5) were less frequently present. This suggests that a specific set of mtDNA variants are required to initiate GBM tumorigenesis and that they are selected for whilst those not essential to tumorigenesis appear to be selected against.
Figure 3.
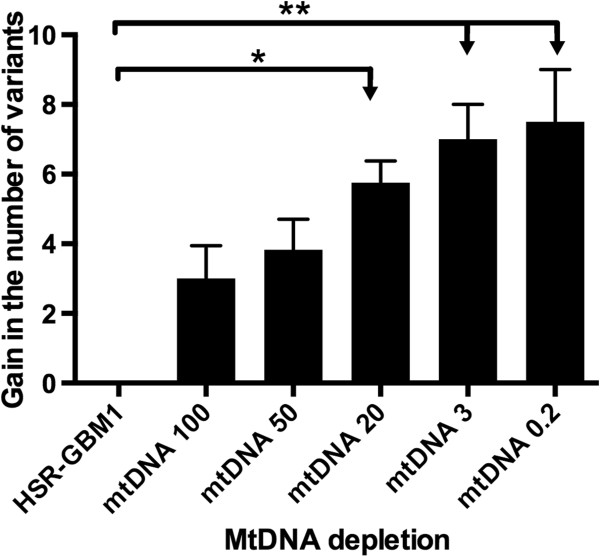
HSR-GBM1 cells and HSR-GBM1 cells depleted of their mtDNA to levels of 50%, 20%, 3% and 0.2% of their original mtDNA copy number were transplanted into immunedeficient nude mice and developed as tumors with mtDNA copy number being replenished in vivo. The number of variants gained relative to the non-depleted HSR-GBM1 cells was assessed using HRM. *P = <0.05, **P = <0.01.
Identification of the tumor-specifying variants in patient samples
In order to determine whether the variants identified in our in vivo tumorigenesis model are present in GBM patient samples, we screened high grade tumors obtained from 22 patients diagnosed with glioma (Additional file 5: Table S7). Each of the variants was present in the majority of patient tumor samples with tumor sample T16 sharing all the variants. Furthermore, the variants at 6999 bp (COX I), 8251 bp, 8252 bp (both COX II) and 11361 bp (ND4) were present in all patient tumors whilst the variants at 6999 bp and 11361 bp were also common to all tumors derived from our in vivo model.
Discussion
This is the first study using next generation sequencing to identify the presence of mtDNA variants in GBM cell lines, which are representative of the relapse and/or refractory stage of the disease. We used a two-fold strategy to ensure that the variants identified were present within the cells and were not artifacts. Firstly, we amplified the two overlapping regions of the mitochondrial genome by long PCR followed by next generation sequencing, an approach used by others to, for example, identify the degree of heteroplasmy amongst different populations [29]. Importantly, this strategy ensured that we were not screening and, thus identifying variants, in nuclear pseudo-genes but rather targeting the mitochondrial genome itself [30]. Indeed, isolation of mitochondria and subsequent extraction of mtDNA can result in DNA samples possessing contaminating nuclear DNA. Secondly, we validated the presence of the variants using HRM on genomic DNA, which enabled us to confirm that the variants were ‘true’ calls, and not introduced during long PCR.
Overall, the GBM cell lines harbored proportionally more previously reported variants (16/29) than the normal, unaffected brain samples (20/60) following searches on the MitoMap variant database. Specifically, for the GBM cell lines, 4/13 variants that have not been previously reported resided within the non-coding region and 12/19 within the coding region. For the normal samples, there were two previously unidentified variants in the non-coding region and 18 in the coding region. Indeed, it was most apparent that there were only three variants common to the GBM cell lines and the normal samples, one in the D-loop at position 16519 bp (D-loop) and two in the coding genes at positions 14159 bp and 14160 bp (ND6). The 16519 bp variant has been identified in multiple studies [31] and was present in both cohorts at similar levels. However, the 14159 bp and 14160 bp variants have not been previously reported. Consequently, it appears that two distinct populations of variants define the two cohorts.
Although the variants identified in the GBM and normal cohorts appear to be randomly distributed throughout the mitochondrial genome, certain regions were not affected. No variants were identified in the NADH dehydrogenase 1 (ND1) gene or the 22 tRNAs suggesting that the incorporation of variants was highly selective to the other 12 coding genes and the D-loop region. Moreover, different regions of the mitochondrial genome were more susceptible to mutation in the normal samples than the GBM cell lines. For the GBM cells, ND6 and ND4 were the most susceptible genes in the coding region whilst the origin of light strand replication was the most susceptible non-coding gene. The ND6, ND4 and D-loop variants were also present in the majority of patient samples. For the normal samples, the D-loop was the most susceptible non-coding region whilst CYT B was the most susceptible gene in the coding region. Interestingly, for both cohorts, COX I was the least susceptible. COX I is one of the subunits of complex IV of the electron transfer chain protein and is amongst the most conserved of the proteins encoded by mtDNA [32]. Nevertheless, the COX II and Cytochrome C Oxidase III (COX III) genes, which also encode complex IV subunits, were more frequently susceptible to mutation than COX I in the GBM cell lines (2.26 fold) and the normal samples (7.87 fold), respectively. CYT B is also highly conserved [32] and the presence of variants in this gene would explain why they were predicted to be disease causing.
Although there was overlap between the various regions of the mitochondrial genome that were affected, there were regions that were specific to each cohort. The variants detected in the origin of light strand replication, COX II and ND4L were specific to the GBM cell lines, whereas variants specific to the normal samples were detected in the NADH dehydrogenase 2 (ND2), NADH dehydrogenase 3 (ND3), ATPase 6 (ATP6), ATPase 8 (ATP8) and COX III genes. This suggests that tumorigenesis targets specific regions of the mitochondrial genome for mutagenesis. To ascertain whether our outcomes were not biased by the cell lines and samples used, we confirmed through PHYLIP analysis that the cell lines and normal samples were from similar lineages and that their respective variants were not indicative of clustered cohorts. Furthermore, as many of the variants have not been previously reported, it appears that we have identified a series of mtDNA variants that are indicative of GBM, which can be validated in a large cohort.
Mitochondrial genetics is becoming increasingly important to our understanding of aging, metabolic disease, demographic patterns of human migration and cancer [33]. However, there is no association between mitochondrial dysfunction and the development of tumors in patients diagnosed with mitochondrial disease, where a single variant or deletion is present at high levels [34]. Moreover, no association has been effectively described for the existence of specific variants with a potential hotspot region for mutation [33]. Furthermore, others have argued that the nuclear genome is primarily responsible for the onset of tumorigenesis [35,36]. However, multiple mtDNA variants scattered throughout the mitochondrial genome [37,38] and in specific genes, such as ND5, are associated with the progression of tumorigenesis [39]. Nevertheless, studies using mouse transmitochondrial cybrids, where specific mtDNA variants have been introduced into cells with identical nuclear backgrounds, have shown that G→A at position 13997 bp and a frame-shift mutation at position 13885 bp caused by the insertion of a C residue, in the ND6 gene, were involved in metastasis and can affect tumor phenotype [40]. Other cybrid studies have also shown that mtDNA mutations act to positively induce tumorigenesis [41-45]. However, others hypothesise that there is active selection against unfavourable mtDNA mutations in tumor cells, due to the monoclonal origin of tumors whilst normal tissues are highly polyclonal in nature, thus promoting the selection of non-mutated mtDNA [46].
There are a number of studies that do not support the presence of mitochondrial mutations as a cause for tumorigenesis. In 2001, Kirches et al. analysed the D-loop of 17 paired GBM samples for mtDNA mutations, only to observe that 7 of the 17 tumors possessed variants [47]. The same authors later published a short report describing the absence of any sequence variant in 10 paired astrocytoma samples [48]. Moreover, in a study that focused on mtDNA mutations in mouse brain tumors, no pathogenic variants were detected across the mitochondrial genome in both induced and spontaneously developed tumors [49]. Seoane et al. also argued that the maintenance of mtDNA integrity is essential in order for tumor cells to survive increased reactive oxygen species production, which is associated with tumorigenesis [50,51]. Altogether, it remains controversial whether mtDNA mutations arise as a cause or consequence of tumorigenesis. However, our data support the role of mtDNA as an initiator of tumorigenesis.
From our results, not only were the vast majority of HSR-GBM1 specific variants reestablished in cells depleted to varying degrees, but new variants were introduced as a function of mtDNA depletion. This is highlighted by those tumors generated from cells depleted to 20%, 3% and 0.2% of their original copy number. Interestingly, those variants that had been identified in the MitoMap database were present in all or nearly all of the newly formed tumors suggesting that there is a common core of mtDNA variants in tumors. Furthermore, 5/6 variants that had been established as de novo variants in the tumors have not been previously reported. To confirm that the newly derived variants were not indicative of clonal expansion from very few residual variants present in the cells, we analysed the raw data from the next generation sequencing runs for the HSR-GBM1 cell line and were able to confirm that these variants had not been called. Consequently, the induction of de novo mtDNA variants appears to be essential to the onset of tumorigenesis. Furthermore, our results reaffirm that ND4 and ND6 are the most susceptible genes to mutation in GBM and that GBM is likely to be a complex I disorder, which is supported by the one previous finding, prior to next generation sequencing, that complex I along with the D-loop is implicated in GBM [47]. Consequently, complex I would be a pharmaceutical target for the development of potential therapies.
Overall, the repopulation of depleted cells with mitochondria appears to be essential during tumorigenesis, as recently demonstrated [52]. The accumulation of mtDNA variants would result in a dysfunctional electron transfer chain that promotes reduced OXPHOS output, as is the case for GBM [53] and forces cells to adopt glycolysis, supported by low levels of OXPHOS. As a result, mtDNA copy number remains suppressed, which promotes the metabolic state of aerobic glycolysis, first proposed by Otto Warburg in 1956 [9]. This form of metabolism promotes self-renewal and cellular proliferation, which is also favoured by adult and embryonic stem cells [54-57]. However, embryonic and adult stem cells have the potential to differentiate into mature cell types, increase their mtDNA copy number and, consequently, switch from anaerobic metabolism to OXPHOS as their primary method of metabolism [52,56,58,59].
Conclusion
We have identified a series of novel mtDNA variants by screening GBM cell lines. Mutations at positions 2130 bp within the 16 s rRNA and 5752 bp within the origin of light strand replication were sites of frequent mutation across the cell lines. Most of the GBM variants identified in the non-coding regions of mtDNA were within the D-loop region, whereas most variants in the coding portion resided within the ND4 region. However, ND6 was determined to be the most susceptible mtDNA region for the development of mutation in the GBM cells, given that it contained the most variants following normalization for the different sizes of the mtDNA regions. These outcomes were further endorsed by mtDNA depletion and in vivo replenishment during tumorigenesis and analysis of patient tumor samples, which support the hypothesis that mtDNA variants in complex I act as initiators of GBM.
Competing interests
The authors declare no competing financial interests.
Author contribution
KYY carried out the collection, assembly, analysis and interpretation of the data, and drafting of the manuscript; AD carried out the collection of data and analysis; JFD carried out the collection and analysis of all the in vivo data; GP designed and assisted in the analysis and interpretation of the protein modeling work; SJW carried out the collection of data; DKG carried out data analysis and provided financial support for the study; MM carried out the analysis and interpretation of the data; TGJ designed the in vivo work, carried out the analysis and interpretation of the data and provided the GBM cell lines and mice used in this study; JCSJ carried out the analysis and interpretation of the data, designed the experiments, wrote the manuscript and provided financial support. All authors read and approved the final manuscript.
Supplementary Material
HRM primers used to screen for mtDNA variants.
Supplementary Methods.
Determining the threshold sensitivity of the Ion Torrent PGM with HRM. (A) Mutant and wild-type recombinant plasmids containing a 223 bp amplicon carrying the MELAS A3243G mutation were sequenced using capillary sequencing. (B) Sensitivity levels of the Ion Torrent to detect specific percentage of mutation generated by mixing wild-type and mutant plasmid DNA. Confirmation of variant detection is also indicated for HRM. (C) The mutant/wild-type specific dilutions at various known percentages were examined by HRM and the results from the difference curves obtained are presented. The 100% mutant plasmid DNA is set as the baseline for comparison for this experiment.
Example of the HRM analysis conducted to confirm the presence of mtDNA variants on the GBM cell lines HK301, SF-767 and BAH1.
Normalisation of the identified variants to the size of the mtDNA region on which they reside to determine susceptibility of each region to the acquisition of mutations, based on the variant screen across all GBM cell lines. Table S3. Comparison of the different mtDNA regions to their susceptibility to develop mutations based on normalised data against the size of the different mtDNA regions. Table S4. Susceptibility table for each mtDNA region, based on the next generation sequencing results obtained from normal brain samples, to the development of variants. The size of each mtDNA region was normalised to allow for comparative analyses to identify the regions most susceptible and least susceptible to mutation. Table S5. Fold change comparison of the different susceptibility scores calculated for each mtDNA region of normal brain samples based on the information from Additional file 5: Table S4. Table S6. HRM analysis of HSR-GBM1 cells depleted to 50% (mtDNA50), 20% (mtDNA20) and 3% (mtDNA3) of their original mtDNA content. They were then allowed to recover in culture over 14 days and screened by HRM using the panel of primers for the variants identified from the GBM cell lines in Tables 1 and 2, and Figure 2 to determine whether they harbored the same mtDNA variants. Table S7. HRM analysis on 22 GBM patient tumor samples screened using the panel of primers for the variants identified from the GBM cell lines in Tables 1, 2 and Figure 2 to identify whether these tumor samples harbor the same mtDNA variants.
Protein modeling of bovine complex IV of the mitochondrial respiratory chain, with identification of the COX I V366M mutation. (A) The catalytic centre of complex IV is represented in its entirety with subunits COX I (purple), II (blue) and III (green). Several of the key components are shown; heme a and a 3 reside within COX I. Heme a 3 and the copper B (CuB) centre together form a bimetallic site. Through these redox cofactors an electron transfer process occurs, which is initiated from ferrocytochrome c transferring electrons to the copper A (CuA) site. The cascade of electron flow occurs as follows from the CuA centre: heme a to the heme a 3 - CuB site, which binds to and reduces molecular oxygen to water. The magnesium (Mg2+) centre functions to maintain activity of complex IV. (B) The valine to methionine substitution (yellow) is shown at position 366 of the COX I protein subunit, residing within a transmembrane helical domain close to COX II and the intermembrane space. (C) Magnified view of the V366M mutation.
Location of the proline to threonine (P223T) mutation in the bovine COX II protein subunit. (A) The P223T mutation resides within a helical segment of COX II in the mitochondrial intermembrane space. (B) Magnified observation of the same P223T mutation.
Phylogenetic analysis using the PHYLIP software package to determine the evolutionary relationships between the GBM cell lines and normal brain samples.
Contributor Information
Ka Yu Yeung, Email: vanessa.yeung@monash.edu.
Adam Dickinson, Email: adamdickinson8686@gmail.com.
Jacqueline F Donoghue, Email: Jacqueline.Donoghue@monash.edu.
Galina Polekhina, Email: Galina.Polekhina@monash.edu.
Stefan J White, Email: Stefan.White@monash.edu.
Dimitris K Grammatopoulos, Email: D.Grammatopoulos@warwick.ac.uk.
Matthew McKenzie, Email: matthew.mckenzie@monash.edu.
Terrance G Johns, Email: Terry.Johns@monash.edu.
Justin C St John, Email: justin.stjohn@monash.edu.
Acknowledgements
This work was supported by the Victorian Government’s Operational Infrastructure Support Program, Monash Institute of Medical Research start up funds to JCSJ; an NHMRC CDA Fellowship and the James and Vera Lawson Trust to MMcK; NHMRC Project Grant 1012020 to TGJ; the Marian and E.H. Flack Trust to SJW and JCSJ; KYY is funded through a Medical Research Council, UK, PhD scholarship. We are also grateful to the MHTP Medical Genomics Facility - ACRF Centre for Cancer Genomic Medicine for use of next generation sequencing.
References
- Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;2(2):103–123. doi: 10.1016/S0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;2(5806):457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Gill PIP, Kimpton C. et al. Identification of the remains of the Romanov family by DNA analysis. Nat Genet. 1994;2:130–135. doi: 10.1038/ng0294-130. [DOI] [PubMed] [Google Scholar]
- Penta JS, Johnson FM, Wachsman JT, Copeland WC. Mitochondrial DNA in human malignancy. Mutat Res. 2001;2(2):119–133. doi: 10.1016/S1383-5742(01)00053-9. [DOI] [PubMed] [Google Scholar]
- Jakupciak JP, Maragh S, Markowitz ME, Greenberg AK, Hoque MO, Maitra A, Barker PE, Wagner PD, Rom WN, Srivastava S, Sidransky D, O’Connell CD. Performance of mitochondrial DNA mutations detecting early stage cancer. BMC Cancer. 2008;2:285. doi: 10.1186/1471-2407-8-285. doi:10.1186/1471-2407-8-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizi GM, Cardaioli E, Grieco GS, Cavallaro T, Malandrini A, Manneschi L, Dotti MT, Federico A, Guazzi G. The A to G transition at nt 3243 of the mitochondrial tRNALeu(UUR) may cause an MERRF syndrome. J Neurol Neurosurg Psychiatry. 1996;2(1):47–51. doi: 10.1136/jnnp.61.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BO, Hwang JH, Kim J, Cho EM, Cho SY, Hwang SJ, Lee HW, Kim SJ, Chung KW. A MELAS syndrome family harboring two mutations in mitochondrial genome. Exp Mol Med. 2008;2(3):354–360. doi: 10.3858/emm.2008.40.3.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudard S, Arvelo F, Miccoli L, Apiou F, Dutrillaux AM, Poisson M, Dutrillaux B, Poupon MF. High glycolysis in gliomas despite low hexokinase transcription and activity correlated to chromosome 10 loss. Br J Cancer. 1996;2(6):839–845. doi: 10.1038/bjc.1996.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;2(3215):269–270. [PubMed] [Google Scholar]
- Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;2(5516):504–507. doi: 10.1126/science.1058079. doi:10.1126/science.1058079. [DOI] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;2(2):254–260. doi: 10.1016/j.ajhg.2008.07.004. doi:10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, Loeb LA. Quantification of random mutations in the mitochondrial genome. Methods. 2008;2(4):263–268. doi: 10.1016/j.ymeth.2008.10.008. doi:10.1016/j.ymeth.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Dickins B, Afgan E, Paul IM, Taylor J, Makova KD, Nekrutenko A. Dynamics of mitochondrial heteroplasmy in three families investigated via a repeatable re-sequencing study. Genome Biol. 2011;2(6):R59. doi: 10.1186/gb-2011-12-6-r59. doi:10.1186/gb-2011-12-6-r59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza MV, Fass J, Diegoli M, Lin D, Arbustini E. Mitochondrial DNA variant discovery and evaluation in human Cardiomyopathies through next-generation sequencing. PloS one. 2010;2(8):e12295. doi: 10.1371/journal.pone.0012295. doi:10.1371/journal.pone.0012295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;2(5):434–439. doi: 10.1038/nbt.2198. doi:10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- Elliott AM, Radecki J, Moghis B, Li X, Kammesheidt A. Rapid detection of the ACMG/ACOG-recommended 23 CFTR disease-causing mutations using ion torrent semiconductor sequencing. J Biomol Tech. 2012;2(1):24–30. doi: 10.7171/jbt.12-2301-003. doi:10.7171/jbt.12-2301-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RE, Foster PG, Guille M, Littlewood DT. Next generation sequencing and comparative analyses of Xenopus mitogenomes. BMC Genomics. 2012;2(1):496. doi: 10.1186/1471-2164-13-496. doi:10.1186/1471-2164-13-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg J, Olatubosun A, Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat. 2011;2(4)):358–368. doi: 10.1002/humu.21445. [DOI] [PubMed] [Google Scholar]
- Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2(9):494–503. doi: 10.1038/ncpneuro0289. quiz 491 p following 516. doi:10.1038/ncpneuro0289. [DOI] [PubMed] [Google Scholar]
- Kunkle B, Yoo C, Roy D. Discovering gene-environment interactions in Glioblastoma through a comprehensive data integration bioinformatics method. Neurotoxicology. 2012. doi:10.1016/j.neuro.2012.11.001. [DOI] [PubMed]
- Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, Sabel M, Steinbach JP, Heese O, Reifenberger G, Weller M, Schackert G. Long-term survival with glioblastoma multiforme. Brain. 2007;2(Pt 10):2596–2606. doi: 10.1093/brain/awm204. doi:10.1093/brain/awm204. [DOI] [PubMed] [Google Scholar]
- Maletinska L, Blakely EA, Bjornstad KA, Deen DF, Knoff LJ, Forte TM. Human glioblastoma cell lines: levels of low-density lipoprotein receptor and low-density lipoprotein receptor-related protein. Cancer Res. 2000;2(8):2300–2303. [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;2(19):7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. doi:10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- Pandita A, Aldape KD, Zadeh G, Guha A, James CD. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer. 2004;2(1):29–36. doi: 10.1002/gcc.10300. doi:10.1002/gcc.10300. [DOI] [PubMed] [Google Scholar]
- Day B, Stringer B, Wilson J, Jeffree R, Jamieson P, Ensbey K, Bruce Z, Inglis P, Allan S, Winter C, Tollesson G, Campbell S, Lucas P, Findlay W, Kadrian D, Johnson D, Robertson T, Johns T, Bartlett P, Osborne G, Boyd A. Glioma surgical aspirate: a viable source of tumor tissue for experimental research. Cancers. 2013;2(2):357–371. doi: 10.3390/cancers5020357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer CM, Eini R, Gillis AM, Stoop H, Looijenga LH, White SJ. DICER1 RNase IIIb domain mutations are infrequent in testicular germ cell tumours. BMC research notes. 2012;2:569. doi: 10.1186/1756-0500-5-569. doi:10.1186/1756-0500-5-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat. 2009;2(8):1237–1244. doi: 10.1002/humu.21047. doi:10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;2(21):2744–2750. doi: 10.1093/bioinformatics/btp528. doi:10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Schonberg A, Schaefer M, Schroeder R, Nasidze I, Stoneking M. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am J Hum Genet. 2010;2(2):237–249. doi: 10.1016/j.ajhg.2010.07.014. doi:10.1016/j.ajhg.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr RL, Maki J, Reguly B, Dakubo GD, Aguirre A, Wittock R, Robinson K, Jakupciak JP, Thayer RE. The pseudo-mitochondrial genome influences mistakes in heteroplasmy interpretation. BMC Genomics. 2006;2:185. doi: 10.1186/1471-2164-7-185. doi:10.1186/1471-2164-7-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilleri M, Carlson P, Zinsmeister AR, McKinzie S, Busciglio I, Burton D, Zaki EA, Boles RG. Mitochondrial DNA and gastrointestinal motor and sensory functions in health and functional gastrointestinal disorders. Am J Physiol Gastrointest Liver Physiol. 2009;2(3):G510–516. doi: 10.1152/ajpgi.90650.2008. doi:10.1152/ajpgi.90650.2008. [DOI] [PubMed] [Google Scholar]
- Wong LJ. Pathogenic mitochondrial DNA mutations in protein-coding genes. Muscle Nerve. 2007;2(3):279–293. doi: 10.1002/mus.20807. doi:10.1002/mus.20807. [DOI] [PubMed] [Google Scholar]
- Jakupciak JP, Dakubo GD, Maragh S, Parr RL. Analysis of potential cancer biomarkers in mitochondrial DNA. Curr Opin Mol Ther. 2006;2(6):500–506. [PubMed] [Google Scholar]
- Ishikawa K, Hayashi J. A novel function of mtDNA: its involvement in metastasis. Ann N Y Acad Sci. 2010;2:40–43. doi: 10.1111/j.1749-6632.2010.05616.x. doi:10.1111/j.1749-6632.2010.05616.x. [DOI] [PubMed] [Google Scholar]
- Hayashi JI, Yonekawa H, Tagashira Y. Nuclear but not mitochondrial genome involvement in 3-methylcholanthrene-induced expression of tumorigenicity in mouse somatic cells. Cancer Res. 1989;2(17):4715–4720. [PubMed] [Google Scholar]
- Akimoto M, Niikura M, Ichikawa M, Yonekawa H, Nakada K, Honma Y, Hayashi J. Nuclear DNA but not mtDNA controls tumor phenotypes in mouse cells. Biochem Biophys Res Commun. 2005;2(4):1028–1035. doi: 10.1016/j.bbrc.2004.12.105. doi:10.1016/j.bbrc.2004.12.105. [DOI] [PubMed] [Google Scholar]
- He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;2(7288):610–614. doi: 10.1038/nature08802. doi:10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;2(3):291–293. doi: 10.1038/3108. doi:10.1038/3108. [DOI] [PubMed] [Google Scholar]
- Shen L, Fang H, Chen T, He J, Zhang M, Wei X, Xin Y, Jiang Y, Ding Z, Ji J, Lu J, Bai Y. Evaluating mitochondrial DNA in cancer occurrence and development. Ann N Y Acad Sci. 2010;2:26–33. doi: 10.1111/j.1749-6632.2010.05635.x. doi:10.1111/j.1749-6632.2010.05635.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;2(5876):661–664. doi: 10.1126/science.1156906. doi:10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- Ohta S. Contribution of somatic mutations in the mitochondrial genome to the development of cancer and tolerance against anticancer drugs. Oncogene. 2006;2(34):4768–4776. doi: 10.1038/sj.onc.1209602. doi:10.1038/sj.onc.1209602. [DOI] [PubMed] [Google Scholar]
- Mito T, Kikkawa Y, Shimizu A, Hashizume O, Katada S, Imanishi H, Ota A, Kato Y, Nakada K, Hayashi J. Mitochondrial DNA mutations in mutator mice confer respiration defects and B-cell lymphoma development. PloS one. 2013;2(2):e55789. doi: 10.1371/journal.pone.0055789. doi:10.1371/journal.pone.0055789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takibuchi G, Imanishi H, Morimoto M, Ishikawa K, Nakada K, Toyama-Sorimachi N, Kikkawa Y, Takenaga K, Hayashi JI. Polymorphic mutations in mouse mitochondrial DNA regulate a tumor phenotype. Mitochondrion. 2013. doi:10.1016/j.mito.2013.07.117. [DOI] [PubMed]
- Imanishi H, Hattori K, Wada R, Ishikawa K, Fukuda S, Takenaga K, Nakada K, Hayashi J. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PloS one. 2011;2(8):e23401. doi: 10.1371/journal.pone.0023401. doi:10.1371/journal.pone.0023401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume O, Shimizu A, Yokota M, Sugiyama A, Nakada K, Miyoshi H, Itami M, Ohira M, Nagase H, Takenaga K, Hayashi J. Specific mitochondrial DNA mutation in mice regulates diabetes and lymphoma development. Proc Natl Acad Sci U S A. 2012;2(26):10528–10533. doi: 10.1073/pnas.1202367109. doi:10.1073/pnas.1202367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaidakov M, Shmookler Reis RJ. Possibility of selection against mtDNA mutations in tumors. Mol Cancer. 2005;2:36. doi: 10.1186/1476-4598-4-36. doi:10.1186/1476-4598-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirches E, Krause G, Warich-Kirches M, Weis S, Schneider T, Meyer-Puttlitz B, Mawrin C, Dietzmann K. High frequency of mitochondrial DNA mutations in glioblastoma multiforme identified by direct sequence comparison to blood samples. Int J Cancer. 2001;2(4):534–538. doi: 10.1002/ijc.1375. doi:10.1002/ijc. [DOI] [PubMed] [Google Scholar]
- Kirches E, Krause G, Weis S, Mawrin C, Dietzmann K. Comparison between mitochondrial DNA sequences in low grade astrocytomas and corresponding blood samples. Mol Pathol. 2002;2(3):204–206. doi: 10.1136/mp.55.3.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebish MA, Seyfried TN. Absence of pathogenic mitochondrial DNA mutations in mouse brain tumors. BMC Cancer. 2005;2:102. doi: 10.1186/1471-2407-5-102. doi:10.1186/1471-2407-5-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane M, Mosquera-Miguel A, Gonzalez T, Fraga M, Salas A, Costoya JA. The mitochondrial genome is a “genetic sanctuary” during the oncogenic process. PloS one. 2011;2(8):e23327. doi: 10.1371/journal.pone.0023327. doi:10.1371/journal.pone.0023327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias P, Salas A, Costoya JA. The maintenance of mitochondrial genetic stability is crucial during the oncogenic process. Communicative & integrative biology. 2012;2(1):34–38. doi: 10.4161/cib.18160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson A, Yeung KY, Donoghue J, Baker MJ, Kelly RD, McKenzie M, Johns TG, St John JC. The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell Death Differ. 2013;2(12):1644–1653. doi: 10.1038/cdd.2013.115. doi:10.1038/cdd.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Shingu T, Feng L, Chen Z, Ogasawara M, Keating MJ, Kondo S, Huang P. Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. J Biol Chem. 2011;2(37):32843–32853. doi: 10.1074/jbc.M111.260935. doi:10.1074/jbc.M111.260935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casson RJ, Chidlow G, Han G, Wood JP. An explanation for the Warburg effect in the adult mammalian retina. Clin Experiment Ophthalmol. 2012. doi:10.1111/ceo.12050. [DOI] [PubMed]
- Abu Dawud R, Schreiber K, Schomburg D, Adjaye J. Human embryonic stem cells and embryonal carcinoma cells have overlapping and distinct metabolic signatures. PloS one. 2012;2(6):e39896. doi: 10.1371/journal.pone.0039896. doi:10.1371/journal.pone.0039896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facucho-Oliveira JM, Alderson J, Spikings EC, Egginton S, St John JC. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J Cell Sci. 2007;2(Pt 22):4025–4034. doi: 10.1242/jcs.016972. doi:10.1242/jcs.016972. [DOI] [PubMed] [Google Scholar]
- Kelly RD, Mahmud A, McKenzie M, Trounce IA, St John JC. Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res. 2012;2(20):10124–10138. doi: 10.1093/nar/gks770. doi:10.1093/nar/gks770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facucho-Oliveira JM, St John JC. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev. 2009;2(2):140–158. doi: 10.1007/s12015-009-9058-0. doi:10.1007/s12015-009-9058-0. [DOI] [PubMed] [Google Scholar]
- Kelly RD, Sumer H, McKenzie M, Facucho-Oliveira J, Trounce IA, Verma PJ, St John JC. The effects of nuclear reprogramming on mitochondrial DNA replication. Stem Cell Rev. 2013;2(1):1–15. doi: 10.1007/s12015-011-9318-7. doi:10.1007/s12015-011-9318-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HRM primers used to screen for mtDNA variants.
Supplementary Methods.
Determining the threshold sensitivity of the Ion Torrent PGM with HRM. (A) Mutant and wild-type recombinant plasmids containing a 223 bp amplicon carrying the MELAS A3243G mutation were sequenced using capillary sequencing. (B) Sensitivity levels of the Ion Torrent to detect specific percentage of mutation generated by mixing wild-type and mutant plasmid DNA. Confirmation of variant detection is also indicated for HRM. (C) The mutant/wild-type specific dilutions at various known percentages were examined by HRM and the results from the difference curves obtained are presented. The 100% mutant plasmid DNA is set as the baseline for comparison for this experiment.
Example of the HRM analysis conducted to confirm the presence of mtDNA variants on the GBM cell lines HK301, SF-767 and BAH1.
Normalisation of the identified variants to the size of the mtDNA region on which they reside to determine susceptibility of each region to the acquisition of mutations, based on the variant screen across all GBM cell lines. Table S3. Comparison of the different mtDNA regions to their susceptibility to develop mutations based on normalised data against the size of the different mtDNA regions. Table S4. Susceptibility table for each mtDNA region, based on the next generation sequencing results obtained from normal brain samples, to the development of variants. The size of each mtDNA region was normalised to allow for comparative analyses to identify the regions most susceptible and least susceptible to mutation. Table S5. Fold change comparison of the different susceptibility scores calculated for each mtDNA region of normal brain samples based on the information from Additional file 5: Table S4. Table S6. HRM analysis of HSR-GBM1 cells depleted to 50% (mtDNA50), 20% (mtDNA20) and 3% (mtDNA3) of their original mtDNA content. They were then allowed to recover in culture over 14 days and screened by HRM using the panel of primers for the variants identified from the GBM cell lines in Tables 1 and 2, and Figure 2 to determine whether they harbored the same mtDNA variants. Table S7. HRM analysis on 22 GBM patient tumor samples screened using the panel of primers for the variants identified from the GBM cell lines in Tables 1, 2 and Figure 2 to identify whether these tumor samples harbor the same mtDNA variants.
Protein modeling of bovine complex IV of the mitochondrial respiratory chain, with identification of the COX I V366M mutation. (A) The catalytic centre of complex IV is represented in its entirety with subunits COX I (purple), II (blue) and III (green). Several of the key components are shown; heme a and a 3 reside within COX I. Heme a 3 and the copper B (CuB) centre together form a bimetallic site. Through these redox cofactors an electron transfer process occurs, which is initiated from ferrocytochrome c transferring electrons to the copper A (CuA) site. The cascade of electron flow occurs as follows from the CuA centre: heme a to the heme a 3 - CuB site, which binds to and reduces molecular oxygen to water. The magnesium (Mg2+) centre functions to maintain activity of complex IV. (B) The valine to methionine substitution (yellow) is shown at position 366 of the COX I protein subunit, residing within a transmembrane helical domain close to COX II and the intermembrane space. (C) Magnified view of the V366M mutation.
Location of the proline to threonine (P223T) mutation in the bovine COX II protein subunit. (A) The P223T mutation resides within a helical segment of COX II in the mitochondrial intermembrane space. (B) Magnified observation of the same P223T mutation.
Phylogenetic analysis using the PHYLIP software package to determine the evolutionary relationships between the GBM cell lines and normal brain samples.