

Abstract
Erythroid-specific 5-aminolevulinate synthase (ALAS2) is the rate-limiting enzyme for heme biosynthesis in erythroid cells, and a missense mutation of the ALAS2 gene is associated with congenital sideroblastic anemia. However, the gene responsible for this form of anemia remains unclear in about 40% of patients. Here, we identify a novel erythroid-specific enhancer of 130 base pairs in the first intron of the ALAS2 gene. The newly identified enhancer contains a cis-acting element that is bound by the erythroid-specific transcription factor GATA1, as confirmed by chromatin immunoprecipitation analysis in vivo and by electrophoretic mobility shift assay in vitro. A promoter activity assay in K562 human erythroleukemia cells revealed that the presence of this 130-base pair region increased the promoter activity of the ALAS2 gene by 10–15-fold. Importantly, two mutations, each of which disrupts the GATA-binding site in the enhancer, were identified in unrelated male patients with congenital sideroblastic anemia, and the lower expression level of ALAS2 mRNA in bone marrow erythroblasts was confirmed in one of these patients. Moreover, GATA1 failed to bind to each mutant sequence at the GATA-binding site, and each mutation abolished the enhancer function on ALAS2 promoter activity in K562 cells. Thus, a mutation at the GATA-binding site in this enhancer may cause congenital sideroblastic anemia. These results suggest that the newly identified intronic enhancer is essential for the expression of the ALAS2 gene in erythroid cells. We propose that the 130-base pair enhancer region located in the first intron of the ALAS2 gene should be examined in patients with congenital sideroblastic anemia in whom the gene responsible is unknown.
Introduction
The ALAS2 gene encodes for erythroid-specific 5-aminolevulinate synthase (ALAS-E, EC 2.3.1.37), which is the rate-limiting enzyme of the heme biosynthetic pathway in erythroid cells.1 It has been reported that the human ALAS2 gene is mapped on the X chromosome,2 and that a loss-of-function mutation of this gene causes X-linked sideroblastic anemia (XLSA),3,4 which is the most common genetic form of congenital sideroblastic anemia (CSA). Moreover, a missense mutation of ALAS2 was identified in a patient with non-familial CSA (nfCSA),5 in which no family history of sideroblastic anemia was identified. In addition to ALAS2, several other genes were recently identified as causative genes for CSA, including SLC25A38,6 GLRX5,7 ABCB7,8 PUS1,9 and SLC19A2,10 but the cause of sideroblastic anemia still remains undefined in more than 40% of patients with CSA.11
GATA1 transcription factor regulates the expression of several erythroid–specific genes, such as erythropoietin receptor gene,12,13 α- and β-globin genes,14,15 ALAS216 and the GATA1 gene itself,17 during erythroid differentiation.18,19 Ablation of the Gata1 gene in mice resulted in embryonic death because of anemia,20 suggesting that GATA1 is essential for erythroid differentiation in vivo. It has been reported that GATA1 regulates transcription of human ALAS2 through the proximal promoter region16 and the erythroid-specific enhancer located in the eighth intron of ALAS2.21 However, Fujiwara et al. demonstrated that the GATA1 protein binds to the ALAS2 gene only in the middle of its first intron, where no regulatory region had so far been identified, by genome-wide analysis of K562 human erythroleukemia cells using chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq).22
In the present study, we have identified a novel erythroid-specific enhancer region in the first intron of the ALAS2 gene. Moreover, we describe two mutations in the newly identified enhancer of ALAS2: a T-to-C transition, which changes GATA to GGTA at the GATA element in the antisense strand, in a pedigree with XLSA and one proband with nfCSA, and a 35-base pair (bp) deletion including the above-mentioned GATA element in a proband with nfCSA.
Methods
Polymerase chain reaction
DNA polymerases used for polymerase chain reaction (PCR) analysis were purchased from TAKARA BIO Inc. (Shiga, Japan). The sequence of primers and probes used in this study are listed in the Online Supplementary Tables.
Polymerase chain reaction-based quantitative chromatin immunoprecipitation
Real-time PCR-based quantitative chromatin immunoprecipitation (ChIP-qPCR) analysis was conducted essentially as previously described.22
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed using “DIG Gel Shift Kit, 2nd Generation” (Roche Diagnostics GmbH, Mannheim, Germany), according to the manufacturer’s protocol. Sequences of oligonucleotides for probes are indicated by the horizontal bar in the relevant figures. Nuclear extracts were prepared, as described previously,23 from K562 cells or HEK293 human embryonic kidney cells that were transfected with a GATA1-FLAG fusion protein expression vector or its backbone vector.
Promoter/enhancer activity assays
Each target DNA fragment was prepared from genomic DNA from normal volunteers (WT) or patients with CSA (referred to as “GGTA” or “delGATA” in each reporter construct) and was cloned into pGL3basic plasmid (Promega Corporation, Madison, WI, USA). The human ALAS2 proximal promoter region (g.4820_5115, between −267 and +29 from the transcription start site)16,24 was cloned into the multiple-cloning site of pGL3basic [referred to as pGL3-AEpro(−267)]. A single DNA fragment (5.2 kbp), carrying the ALAS2 proximal promoter, first exon, first intron and the untranslated region of the second exon, was sub-cloned into the multiple cloning site of pGL3basic [referred to as pGL3-AEpro(−267)+intron1]. A DNA fragment containing the GATA1-binding region in the first intron of the ALAS2 gene (corresponding to g.7488_7960), which was defined by ChIP-seq analysis,22 is referred to as the ChIP-peak. The length of the WT ChIP-peak is 473 bp. In addition, a 130-bp fragment containing ALAS2int1GATA, the consensus sequence for the GATA1-binding site in the ChIP-peak, is referred to as ChIPmini. Several deletion mutants of ChIPmini were prepared using pGL3-AEpro(−267)+ChIPmini(WT) as a template. The pGL3-TKpro plasmid was constructed by cloning herpes simplex virus thymidine kinase promoter into the multiple cloning site of pGL3basic plasmid. Each reporter vector and pEF-RL25 were introduced into K562 cells or HEK293 cells. Luciferase activity was determined using a dual-luciferase reporter system (Promega).
Identification of mutations of the ALAS2 gene
All exons including exon-intron boundaries, the proximal promoter region, and intron 1 and intron 8 of the ALAS2 gene (GeneBank: NG_8983.1) were directly sequenced according to previously reported methods.26
Measurement of ALAS2 mRNA in purified erythroblasts
Total RNA was extracted from glycophorin A-positive bone marrow mononuclear cells, and was used for cDNA synthesis. ALAS2 expression was measured by real-time PCR, and was normalized to that of GAPDH mRNA.
Statistical analysis
Multiple comparisons between groups were made using the Tukey-Kramer test.
Patients
Eleven probands (eight pedigrees) with CSA of unknown cause were selected to determine the nucleotide sequence of the first intron of ALAS2 gene. In these patients no disease-causative mutation was identified in the coding regions or reported regulatory regions in ALAS2, SLC25A38, GLRX5, ABCB7, PUS1 and SLC19A2, which have been reported to be genes causing CSA11 (see the Online Supplementary Methods for full details of the methods).
The genetic analyses performed in this project were approved by the ethical committee of Tohoku University School of Medicine. Blood samples were withdrawn from the probands and the family members after informed consent.
Results
Polymerase chain reaction-based quantitative chromatin immunoprecipitation analysis of the first intron of the ALAS2 gene
To identify the novel regulatory region for ALAS2 transcription, we first performed ChIP-qPCR analysis in K562 cells to localize the GATA1-binding region of the ALAS2 gene in vivo, which was determined by genome-wide ChIP-seq analysis.22 In fact, ChIP-qPCR enabled us to examine the GATA1-binding activity of an individual GATA element or two adjacent GATA elements in the first intron of the ALAS2 gene. Based on a search of NCBI Reference Sequence (NG_8983.1) using SeqBuilder software (DNASTAR Inc., Madison, WI, USA), we identified 17 GATA elements (16 out of 17 GATA elements are present in the antisense orientation) in the first intron of human ALAS2 (Figure 1A), which is compatible with the previous report.21 We also included the proximal promoter region that contains a functional GATA-binding site (g.4961_4966).16 Overall 13 primer sets were designed to amplify the GATA elements located in the proximal promoter region and the first intron of ALAS2 (Figure 1A and Online Supplementary Table S1). Among the 12 primer sets targeting the first intron, using primer set 10, we could amplify genomic DNA that was precipitated with anti-GATA1 antibody at a similar level to that of the positive control, but not with other primer sets. We refer to this region amplified with primer set 10 as ChIPmini (g.7795_7924), the sequence of which is shown in Figure 1B. In silico analysis identified only one GATA element (g.7860_7865, boxed in Figure 1B) in ChIPmini, termed ALAS2int1GATA. In addition, primer set 1 which targets the proximal promoter region yielded notable amounts of amplified genome DNA. These results indicate that GATA1 protein bound to the regions amplified with primer sets 1 and 10 in K562 cells; that is, GATA1 protein could bind to the proximal promoter region as well as to ALAS2int1GATA in the first intron of the ALAS2 gene in vivo. Since the GATA element located in the proximal promoter has been well examined in vitro,16 we further determined the functional features of ALAS2int1GATA.
Figure 1.
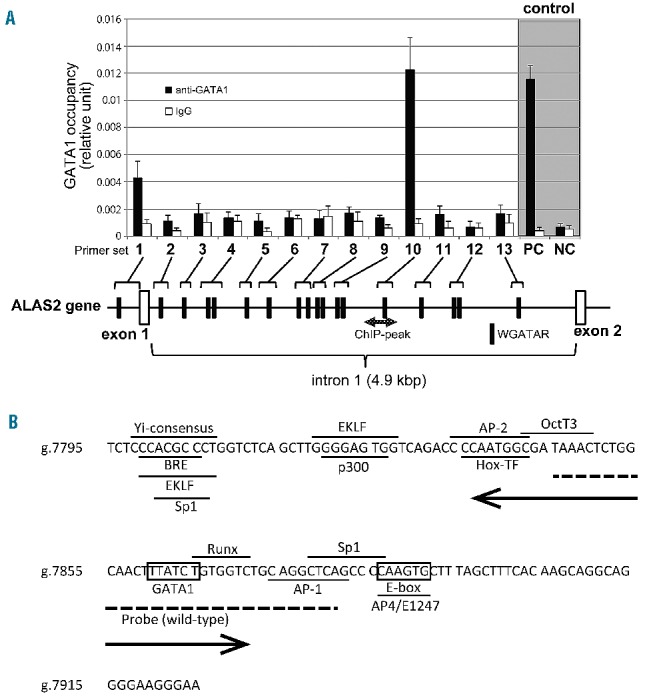
Identification of a functional GATA1 element in the first intron of the ALAS2 gene. (A) Chromatin immunoprecipitation assay. Fragmented genomic DNA segments were immunoprecipitated with anti- GATA1 antibody or control IgG, and then precipitated fragments were quantified using real-time PCR as described in the Online Supplementary Methods. PC or NC indicates positive control or negative control, respectively, for the ChIP assay using anti-GATA1 in K562 cells.22 One GATA element is present in the proximal promoter region and 17 GATA elements in the first intron (black symbols). The shaded double arrow indicates the region corresponding to ChIP-peak. (B) Nucleotide sequence of ChIPmini. The GATA binding site, ALAS2int1GATA, is located in the center of ChIPmini (boxed). A box also indicates the consensus for E-box that is bound by Scl/TAL1.22 The sequence of ChIPmini was further analyzed for putative transcription factor binding sites using GeneQuest software (DNASTAR Inc., Madison, WI, USA), and the results are indicated by the horizontal bar. Yi-consensus, Yi transcription factor consensus site;33 BRE, transcription factor IIB binding site;34 EKLF, erythroid/Kruppel-like factor consensus site;35 Sp1, stimulatory protein 1 binding site;36 P300, P300 transcriptional coactivator consensus site;37 AP-2, AP-2 beta consensus site;38 Hox-TF, C1 element binding factor binding site;39 OctT3, OctT3 binding site;40 Runx, Runx proteins binding site;41 AP-1, activator protein 1 binding site;42 and AP4/E1247, AP4/E1247 binding site.43 The sequence for the wild-type probe used in the EMSA is indicated by a dashed line. A double arrow indicates the deleted region of the delGATA mutation.
GATA1 protein binds to ALAS2int1GATA located in ChIPmini
We then examined whether GATA1 protein binds to ALAS2int1GATA present in the center of ChIPmini using EMSA (Figure 2A). The WT probe contains ALAS2int1GATA (Figure 1B). The incubation of labeled WT probe with nuclear extracts of K562 cells yielded the retarded band that represents the protein-probe complex (lane 2), whereas this retarded band was undetectable with an excess amount of non-labeled WT probe (lane 3). Moreover, the addition of anti-GATA1 antibody reduced the intensity of the retarded band (lane 4), suggesting that GATA1 protein may bind to the WT probe. In fact, the retarded band was not detected when the labeled probe was incubated with nuclear extracts of mock-transfected HEK293 cells (lane 5). In contrast, the retarded band was observed when the labeled probe was incubated with the nuclear extracts of HEK293 cells expressing FLAG-fused GATA1 (lane 6). Importantly, the retarded band observed in lane 6 was not detectable in the presence of an excess amount of non-labeled probe (lane 7). The formation of the retarded band was partially inhibited by anti-GATA1 antibody (lane 8). Likewise, the inclusion of anti-FLAG antibody (lane 9) resulted in the disappearance of the retarded band and instead generated the super-shifted band (indicated by an asterisk). These results suggest that GATA1 protein binds to the WT probe containing ALAS2int1GATA.
Figure 2.
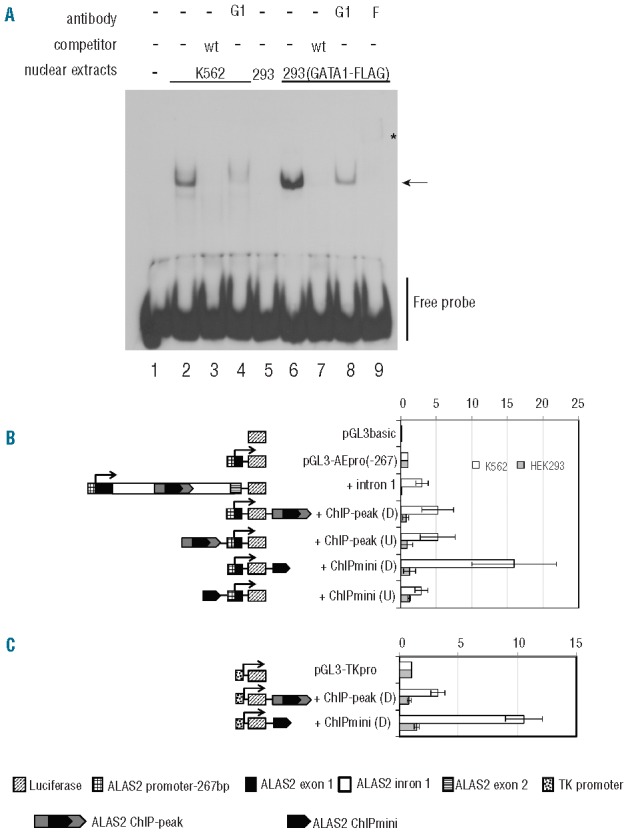
Functional analyses of ChIPmini present in the first intron of the ALAS2 gene. (A) Electrophoretic mobility shift assay (EMSA). Wild-type (wt) probe was incubated with nuclear extracts prepared from K562 cells (lanes 2–4) or HEK293 cells expressing GATA1-FLAG (lanes 6–9). HEK293 cells were transfected with mock vector (lane 5) or FLAG-fused GATA1 expression vector before preparation of nuclear extracts. The protein-probe complex was detected as a retarded band (arrow). An excess amount of unlabeled probe (lanes 3, 7), anti-GATA1 antibody (G1) (lanes 4, 8) or anti-FLAG antibody (F) (lane 9) was included in the reaction mixture. Lane 1 shows the control without nuclear extracts. The asterisk indicates the super-shifted band (lane 9). (B) Functional analysis of ChIPmini as an enhancer for the ALAS2 gene. Details of the fragments for each plasmid, such as intron1, ChIP-peak and ChiIPmini, are described in the Methods section. Each DNA fragment was inserted upstream of the ALAS2 proximal promoter or downstream of luciferase cDNA, indicated as (U) or (D), respectively. Results are expressed as a relative activity compared to that of pGL3-AEpro(−267), and are presented as the mean ± standard deviation (SD) of three independent experiments. (C) Functional analysis of ChIPmini as an enhancer for non-erythroid gene promoter. The enhancer activity of the first intron was examined using the herpes simplex virus TK promoter as a non-erythroid promoter. ChIP-peak or ChIPmini was inserted downstream of the luciferase gene of pGL3-TKpro, yielding pGL3-TKpro+ChIP-peak(D) or pGL3-TKpro+ChIPmini(D). Each of these reporter vectors was introduced into K562 cells or HEK293 cells to measure enhancer activity. Results are expressed as a relative activity compared to that of pGL3-TKpro, and are presented as the mean ± SD of three independent experiments.
Enhancement of ALAS2 promoter activity by the DNA segment containing ALAS2int1GATA
To examine the functional importance of ALAS2int1GATA in the promoter activity of the ALAS2 gene (Figure 2B), we constructed the pGL3-AEpro(−267) vector, in which the expression of firefly luciferase gene is controlled under the proximal promoter of the ALAS2 gene (g.4820_5115). The presence of the first intron of ALAS2 (pGL3-AEpro(−267)+intron1) increased luciferase activity about 3-fold in K562 cells, whereas luciferase activity was decreased to 10% of pGL3-AEpro(−267) in HEK293 cells. When the ChIP-peak, the region determined by ChIP-seq analysis (g.7488_7960),22 was present downstream [+ChIP-peak(D)] or upstream [+ChIP-peak(U)] of the ALAS2 proximal promoter, luciferase activity was increased about 5-fold, irrespective of the location, compared to that of pGL3-AEpro(−267) in K562 cells. Moreover, the presence of the ChIPmini fragment downstream of the luciferase gene [+ChIPmini (D)] resulted in a 16-fold increase of luciferase activity. However, when the same fragment was inserted upstream of the ALAS2 promoter [+ChIPmini(U)], luciferase activity increased only 3-fold. Thus, the enhancer activity of the ChIPmini fragment varies, depending on its location. Moreover, among the constructs examined, the ChIPmini fragment showed maximum enhancer activity downstream of the luciferase gene. The ChIP-peak or ChIPmini fragment downstream of the ALAS2 promoter influenced luciferase activity marginally (0.73- or 1.25-fold, respectively) in HEK293 cells (Figure 2B). These results suggest that the enhancer activity of each fragment containing ALAS2int1GATA is specific to erythroid cells.
To examine whether the erythroid-specific enhancer activity depends on the ALAS2 promoter, we replaced the ALAS2 promoter with the herpes simplex virus TK promoter (Figure 2C). The ChIP-peak and ChIPmini enhanced TK promoter activity 3.4- and 9.8-fold in K562 cells, respectively, whereas they did not enhance TK promoter activity in HEK293 cells. These results indicate that the erythroid-specific enhancer is present in the ChIP-peak and ChIPmini fragments. In addition, the erythroid-specific enhancer is functional in the non-erythroid gene promoter.
Identification of mutations in the first intron of the ALAS2 gene in patients with congenital sideroblastic anemia
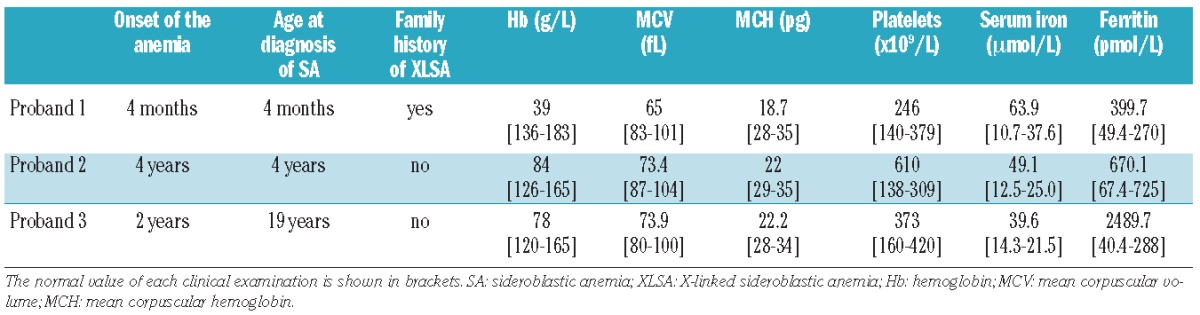
Considering the newly identified enhancer in the first intron of the ALAS2 gene, we examined whether some CSA patients carry the mutation in ChIP-peak or ChIPmini of ALAS2. We determined the nucleotide sequence of the first intron of ALAS2 in 11 probands (eight pedigrees), and found two distinct mutations in the newly identified enhancer region in five Japanese patients (three pedigrees). The clinical features and hematologic status of the probands at diagnosis of the disease are summarized in Table 1.
Table 1.
Hematologic status of each proband at diagnosis of the disease.
Proband 1 in a pedigree with XLSA
The first male Japanese proband was referred to hospital at the age of 3 months to investigate the cause of his pale face. No problems were reported during the birth. Investigations showed microcytic/hypochromic anemia, an increased concentration of serum iron and raised serum ferritin level. Bone marrow aspiration revealed the presence of ring sideroblasts. Two maternal relatives – male cousins of the proband’s mother – have sideroblastic anemia (Figure 3A). The pedigree of this family suggested X chromosome-linked inheritance of the disease. The proband’s anemia was not improved by pyridoxine administration (5 mg/kg/day for 3 months), and the boy required once monthly transfusions of one unit of concentrated red blood cells to maintain an adequate hemoglobin level. At the age of 7 months, this proband died of sepsis caused by alpha-streptococcus.
Figure 3.
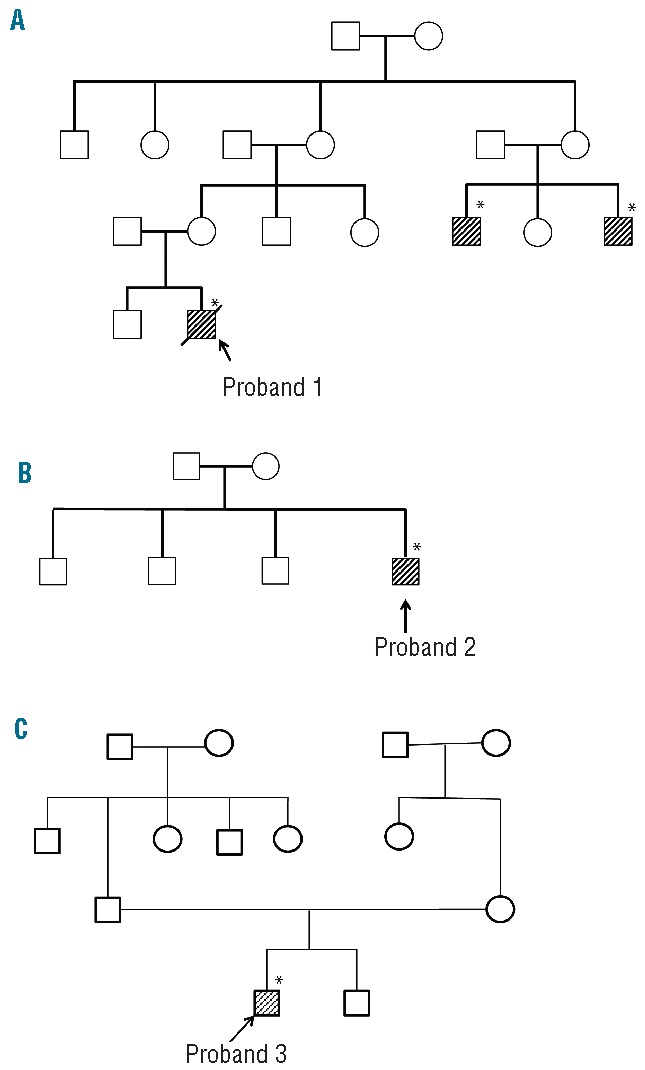
Family trees of three unrelated probands. Family tree of: (A) proband 1 with XLSA, (B) proband 2 with nfCSA, and (C) proband 3 with nfCSA. Shaded boxes indicate affected individuals in each pedigree. Asterisks indicate the individuals in whom a mutation in the first intron of the ALAS2 gene was detected.
Proband 2 with nfCSA
The second male Japanese proband visited hospital at the age of 4 years because of the paleness of his complexion. Investigations showed microcytic/hypochromic anemia, mild thrombocytosis, and a high serum iron concentration with a normal serum ferritin concentration. Bone marrow aspiration revealed the presence of ring sideroblasts (38% of the erythroblasts). Giant platelets were observed in the bone marrow, although dysplasia of the megakaryocytes was not clear. There was no family history of sideroblastic anemia (Figure 3B).
Proband 3 with nfCSA
The third male Japanese proband was noted to have anemia at the age of 2 years, but details are not available. Without any treatment, serum hemoglobin level was maintained at 70 g/L, and increased to 100 g/L at the age of 10. Accordingly, the proband stopped visiting the hospital. At the age of 19, however, the proband was admitted to hospital because of general fatigue. Investigations revealed microcytic, hypochromic anemia with systemic iron overload. The presence of ring sideroblasts was confirmed in his bone marrow by Prussian blue staining (36% of erythroblasts). Although this proband was treated with pyridoxine (150 mg/day) for 8 months, his anemia did not improve. There was no family history of sideroblastic anemia (Figure 3C).
In proband 1 from the pedigree with XLSA (Figure 3A), we identified a single nucleotide mutation (Figure 4, upper panel, g.7863T>C), which alters the core sequence of ALAS2int1GATA in the antisense strand from GATA to GGTA (referred to as “GGTA mutation”). The same mutation of the ALAS2 gene was also identified in two cousins of the proband’s mother, both of whom were diagnosed as having sideroblastic anemia (Figure 3A). Clinical specimens for genetic analysis were not available from either the parents or the elder brother of proband 1.
Figure 4.
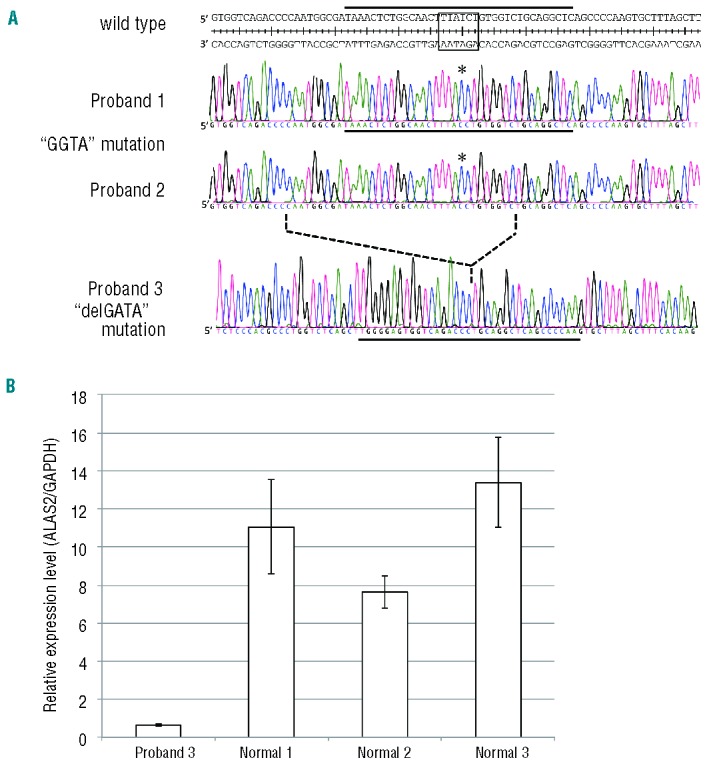
Identification of mutations in the first intron of the ALAS2 gene in a patient with XLSA and two patients with nfCSA. (A) ALAS2 mutations in three probands. Upper, middle and lower panels show the sequences of the flanking regions of ALAS2int1GATA (boxed in the wild-type sequence) in the ALAS2 gene of probands 1, 2 and 3, respectively. Asterisks indicate the T to C transition in the sense strand identified in the ALAS2 gene of proband 1 and proband 2 with CSA. The broken line between the middle and lower panels indicates the deleted region identified in proband 3 with CSA. The solid horizontal bar in each panel indicates the sequence of the sense strand of each probe used for the EMSA (see Figures 3A and 5B). (B) ALAS2 mRNA expression in erythroblasts of proband 3. ALAS2 mRNA levels were determined in purified erythroblasts isolated from proband 3 and three independent normal individuals using real-time PCR. Results are expressed as the mean ± SD of three independent experiments.
The same GGTA mutation was identified at ALAS2int1GATA in proband 2 with CSA (Figure 4, middle panel). There was no known consanguinity between proband 1 and proband 2. Genomic DNA from the parents of proband 2 was not available, because they did not agree to provide their clinical specimens for genetic analysis. Since proband 2 was also noted to have thrombocytosis (Table 1), we searched for a JAK2 mutation in the genomic DNA extracted from the peripheral blood of this patient. However, no V617F mutation or any missense mutation in exon 12, each of which is frequently observed in patients with refractory anemia with ring sideroblasts and thrombocytosis (RARS-T),27 was detected (data not shown). Thus, the GGTA mutation at ALAS2int1GATA may be responsible for the sideroblastic anemia in proband 2.
In proband 3 with CSA, a deletion of 35 bp was identified in the first intron of the ALAS2 gene (Figure 4A, lower panel, g.7836_7870del, referred to as “delGATA mutation”). The delGATA mutation results in the loss of ALAS2int1GATA. However, the delGATA mutation was not identified in the ALAS2 gene of the parents of proband 3 (data not shown). Thus, the delGATA mutation may be a de novo mutation or a somatic mutation. Accordingly, we compared the relative ALAS2 mRNA level in the erythroid progenitor cells isolated from proband’s bone marrow with those of normal subjects. The ALAS2 mRNA level was more than 7-fold lower in the proband’s erythroblasts than in those of three independent, normal subjects (Figure 4B), suggesting that the delGATA mutation may lead to decreased transcription of the ALAS2 gene.
Lastly, we examined the sequence of the region corresponding to g.7513_8165 of the ALAS2 gene, which contains ChIPmini, in 103 healthy, Japanese volunteers (44 males and 59 females, total 162 alleles) using PCR followed by direct sequencing. No mutation was found in this region (data not shown). In addition, no single nucleotide polymorphism was reported in this GATA element, based on the single nucleotide polymorphism database available at the NCBI home page (http://www.ncbi.nlm.nih.gov/snp, current assembly is GRCh37.p5). Thus, the GGTA mutation and delGATA mutation at ALAS2int1GATA may be unique to patients with sideroblastic anemia. Taken together, we suggest that the newly identified mutations at ALAS2int1GATA are responsible for sideroblastic anemia.
The mutation at ALAS2int1GATA impairs GATA1-binding activity and enhancer function
We examined the effect of the GGTA mutation or the delGATA mutation on the binding of GATA1 protein to ALAS2int1GATA using each mutant probe (Figure 5A). The delGATA probe represents the 5′- and 3′-flanking sequences of the deleted 35-bp segment (see Figure 4A). As shown in Figure 5B, the incubation of labeled WT probe with nuclear extracts from HEK293 cells expressing FLAG-fused GATA1 showed a retarded band (lane 3): this band was super-shifted by the addition of anti-FLAG antibody (lane 4), or undetectable with non-labeled WT probe (lane 5), whereas the non-labeled GGTA probe (lane 6) or delGATA probe (lane 7) could not compete for the labeled WT probe. Furthermore, the retarded band was not detectable when labeled GGTA probe (lane 8) or delGATA probe (lane 9) was incubated with the nuclear extracts of HEK293 cells expressing FLAG-fused GATA1. These results suggest that either the GGTA mutation or the delGATA mutation may impair the binding of GATA1 to ALAS2int1GATA.
Figure 5.
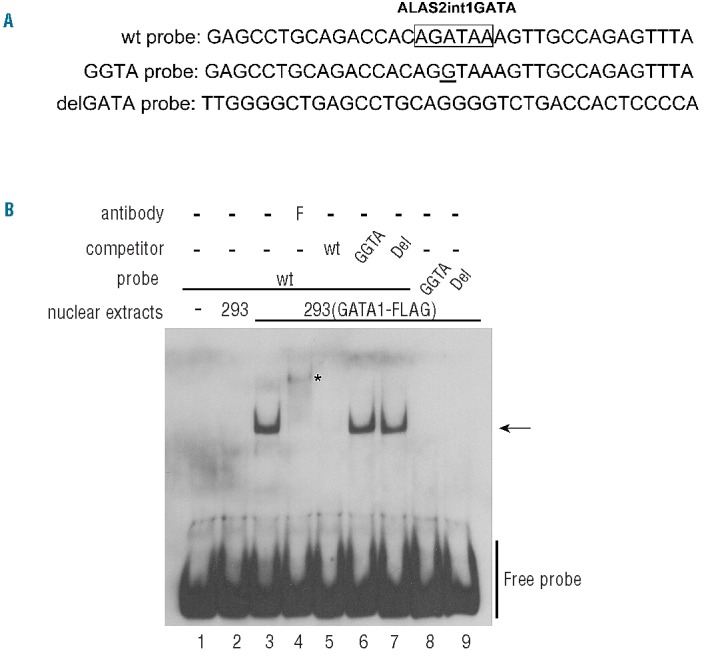
Effects of the mutations of ALAS2int1GATA on GATA1-binding activity. (A) DNA probes used in the EMSA. The nucleotide sequences in the antisense strand of the probes are shown. The position of each probe is also indicated in Figure 1B as the solid horizontal bar. ALAS2int1GATA is boxed in the sequence of the wt probe, and the single nucleotide transition (GGTA mutation) is underlined in the sequence of the GGTA probe. The delGATA probe represents the 5′- and 3′-flanking sequences of the deleted 35-bp segment (see Figure 3B). (B) Effect of each mutation of ALAS2int1GATA on GATA1-binding activity. Wild-type probe (lanes 3–7) or each mutant probe (lanes 8, 9) was incubated with the nuclear extracts prepared from HEK293 cells transfected with the GATA1-FLAG expression vector. An excess amount of unlabeled wild-type probe (lane 5), each of the unlabeled mutant probes (lanes 6, 7), or anti-FLAG antibody (lane 4) was included in the reaction mixture. Lane 2 shows the negative control with nuclear extracts from HEK293 cells transfected with mock vector.
We then examined the influence of the point mutation or deletion of ALAS2int1GATA on the enhancing activity of the first intron of the ALAS2 gene (Figure 6A). The GGTA mutation decreased the enhancing activity of the first intron, ChIP-peak or ChIPmini in K562 cells to 17.0%, 18.5% or 12.9%, respectively, of that of the WT construct. The delGATA mutation decreased the enhancing activity of the first intron of ALAS2, ChIP-peak or ChIPmini in K562 cells to 10.5%, 15.7% or 12.6%, respectively, of that of the WT construct. In contrast, the relative luciferase activity of the construct carrying each mutation was only marginally different from that of WT intron 1, ChIP-peak or ChIPmini in HEK293 cells (Figure 6A), thereby confirming that ALAS2int1GATA functions as an erythroid-specific enhancer.
Figure 6.
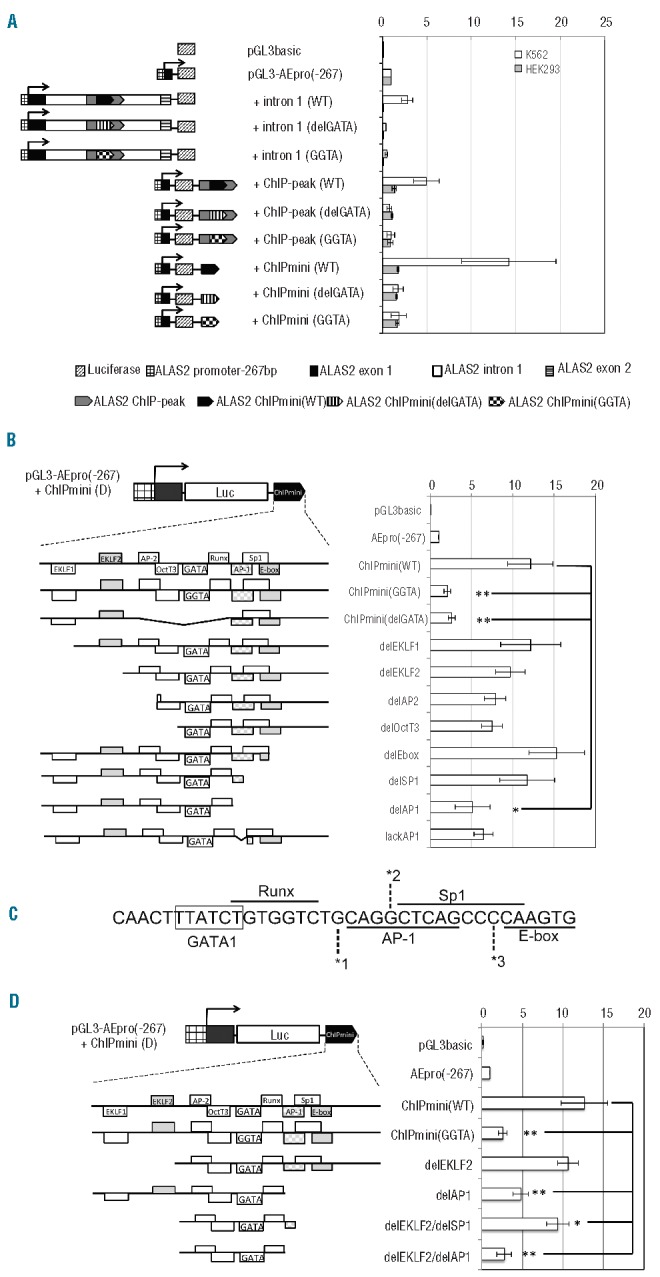
Identification of cis-elements essential for the erythroid-specific enhancer activity of ChIPmini. (A) Effect of each mutation of ALAS2int1GATA on the enhancer activity of ALAS2 ChIPmini. The region corresponding to +intron1, ChIP-peak or ChIPmini, derived from proband 1 or proband 3, was subcloned into pGL3-AEpro(−267) to construct the reporter vector containing the GGTA mutation or the deletion of ALAS2int1GATA, respectively. (B) Effect of the deletion at the 5′- or 3′-flanking region of ALAS2int1GATA on the enhancer activity of ChIPmini. The 5′- and 3′-flanking regions of ALAS2int1GATA contain potential transcription factor-binding sites (cis-elements), and a portion of each flanking region was deleted, as schematically shown. The enhancer activity of each deletion mutant was determined in K562 erythroleukemia cells. (C) The nucleotide sequence of the 3′-flanking region of ALAS2int1GATA. Note that the Sp1 site overlaps the AP-1 site and E-box. Each number, *1, *2 or *3, indicates the nucleotide at the 3′ end of the deletion mutant, delAP1, delSP1 or delE-box, respectively. Thus, delSP1 also lacks the 3′ portion of the AP-1 site. (D) Effect of deletion of the 5′- and 3′-flanking regions of ALAS2int1GATA on the enhancer activity of ChIPmini. The construct, delEKLF2/delSP1, lacks two EKLF sites in the 5′-flanking region and both the Sp1 element and E-box in the 3′-flanking region. The AP-1 element at the 3′-flanking region was deleted from delEKLF2/delSP1, yielding delEKLF2/delAP1. Results are expressed as relative activity compared to that of pGL3-AEpro(−267), and are presented as the mean ± SD of at least three independent experiments.
There are several potential cis-elements at the flanking regions of ALAS2int1GATA, such as EKLF and Sp1, each of which may be involved in the erythroid-specific transcriptional regulation of the ALAS2 gene.16,21 We thus analyzed the roles of these cis-elements in the enhancer activity of ALAS2int1GATA using deletion mutants at the 5′- or 3′-flanking region of ChIPmini, constructed in pGL3-AEpro(−267)+ChIPmini(D). Deletion of the EKLF1 element at the 5′-flanking region or both E-box and Sp1 elements at the 3′-flanking region did not significantly influence the enhancer activity of ChIPmini (Figure 6B). It should be noted that the Sp1 site overlaps with the 3′-portion of the AP-1 site and the 5′-portion of the E-box (Figure 6C). Moreover, deletion at the 5′-flanking region of ChIPmini (“delEKLF2”, “delAP2” and “delOctT3”) marginally decreased the enhancer activity (Figure 6B), but the change was not statistically significant. In contrast, deletion of the AP-1 element at the 3′-flanking region (“delAP1” in Figure 6B) significantly decreased the enhancer activity, by about 40% of the activity of ChIPmini(WT). The significant decrease of enhancer activity was observed only in ChIPmini(GGTA), ChIPmini(delGATA) and delAP1, compared to the activity of ChIPmini(WT) (*P<0.05 and **P<0.01 in Figure 6B). We next constructed another reporter vector that carries an internal deletion of the 5′ portion of the AP-1 element with an intact Sp1 site (“lackAP1” in Figure 6B). Internal deletion of the AP-1 element alone in ChIPmini decreased the enhancer activity, although not to a statistically significant degree. Thus, the entire AP-1 element seems to be important for the enhancer activity of ChIPmini (WT) (Figure 6B).
Consequently, we constructed delEKLF2/delSP1 and delELKLF2/delAP1, each of which lacks EKLF elements at the 5′-flanking region and the Sp1 element or the AP-1 element at the 3′-flanking region, respectively (Figure 6D). The deletion mutant, delEKLF2/delSP1, still retained enhancer activity at about 80% of that of ChIPmini(WT), whereas delEKLF2/delAP1 showed decreased enhancer activity similar to the activity of ChIPmini(GGTA). These data indicate that ALAS2int1GATA and its flanking region, especially the AP-1 element, are critically important for the erythroid-specific enhancer activity of ChIPmini.
Taken together, these results suggest that the ChIPmini region acts as an erythroid-specific enhancer for the ALAS2 promoter, and that both the GGTA mutation and the delGATA mutation represent loss-of-function mutations of ALAS2int1GATA.
Discussion
In the present study, we identified an erythroid-specific enhancer region in the first intron of the human ALAS2 gene (a 130 bp region referred to as ChIPmini), a region which contains ALAS2int1GATA, a functional GATA1-binding site. We also identified the GGTA mutation and the delGATA mutation at ALAS2int1GATA, each of which is associated with XLSA or CSA. Moreover, we confirmed that each mutation diminished the binding of GATA1 transcription factor to ALAS2int1 (Figure 5B) and decreased enhancer activity of ChIPmini (Figure 6A). Thus, the GGTA mutation and delGATA mutation are loss-of function mutations of the ALAS2 gene. In fact, the expression of ALAS2 mRNA in bone marrow erythroblasts was lower in proband 3 (Figure 4B) than in normal controls. Thus, each loss-of function mutation may lead to decreased transcription of the ALAS2 gene, thereby causing sideroblastic anemia in male patients. Such a molecular basis is consistent in part with the lack of pyridoxine responsiveness in these patients (see “Patients” section).
The intronic enhancer, ChIPmini, increased ALAS2 promoter activity most efficiently in erythroid cells when it was present downstream of the promoter (Figure 2B). ChIPmini contains potential cis-acting elements, including two EKLF-binding sites, each of which overlaps with the Sp1-binding site or p300-binding site, AP-2 site, OctT3 site Runx site, AP-1 binding site, Sp1 site, and E-box (Figure 1B). Further analysis using deletion mutants of ChIPmini revealed that the potential AP-1 binding site at the 3′-flanking region might be involved in the erythroid-specific enhancer activity of ChIPmini (Figure 6B). These results suggest that ALAS2int1GATA and its 3′-flanking region are essential for the erythroid-specific enhancer activity of ChIPmini. In fact, EKLF28 and AP-129 are involved in erythroid-specific gene expression. It is interesting that the inclusion of the whole first intron of the ALAS2 gene in a reporter construct resulted in a decrease of ALAS2 promoter activity [11% of pGL3-AEpro(−267)] in non-erythroid HEK293 cells (Figures 2B and 6A). Likewise, the ChIP-peak upstream or downstream of the promoter also reduced the promoter activity in HEK293 cells [73% or 88% of pGL3-AEpro(−267), respectively] (Figure 2B). These results suggest that the first intron of the ALAS2 gene may contain suppressor element(s) in addition to the erythroid-specific enhancer, although the mechanism of the suppression and the relevant region remain elusive.
We have successfully identified a novel erythroid-specific enhancer for ALAS2 expression, and have identified disease-causative mutations of this enhancer in patients with CSA. Despite the fact that about 50 missense or non-sense mutations of the ALAS2 gene have been reported as disease-causative mutations in patients with XLSA,3,30 a mutation in the regulatory region for the transcription of ALAS2 has rarely been reported to date. Ducamp et al. reported a 48-bp deletion of the ALAS2 gene at the proximal promoter region (c.−91_−44del) in a patient with XLSA, and proposed that the identified deletion would cause XLSA, since the level of ALAS2 mRNA in the proband’s bone marrow was lower than that of normal controls.31 In this context, it has been reported that the deleted region contained a functionally important element for ALAS2 transcription.16 Bekri et al. reported a C-to-G transversion at nucleotide −206 (c.−258C>G) from the transcription start site in the proximal region of the human ALAS2 gene in patients with XLSA;24 however, May et al. identified this transversion in normal individuals from South Wales at the rate of 0.05, suggesting that this promoter mutation is a polymorphism.32
In conclusion, we have identified a novel erythroid-specific enhancer in the first intron of the human ALAS2 gene, the enhancer function of which may be directed by GATA1 with other transcription factors, such as EKLF and AP-1 binding proteins. Furthermore, we identified the loss-of-function mutation of ALAS2int1GATA, the GATA element within this enhancer, in five of 11 patients with CSA in whom the gene responsible could not be identified. Thus, the intronic region containing ALAS2int1GATA of the ALAS2 gene should be examined in patients with XLSA or nfCSA in whom the genetic mutation causing the sideroblastic anemia is unknown.
Supplementary Material
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research (C) (to KF) and Health and Labour Sciences Research Grants (to HH and KF). The authors thank Prof. Norio Komatsu (Juntendo University) for examination for the JAK2 mutation. We are also grateful to the Biomedical Research Core of Tohoku University Graduate School of Medicine for allowing us to use various facilities.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic & Molecular Bases of Inherited Disease. New York: McGraw-Hill Medical Publishing Division, 2001:2991–3062 [Google Scholar]
- 2.Cotter PD, Willard HF, Gorski JL, Bishop DF. Assignment of human erythroid delta-aminolevulinate synthase (ALAS2) to a distal subregion of band Xp11.21 by PCR analysis of somatic cell hybrids containing X; autosome translocations. Genomics. 1992;13(1):211–2 [DOI] [PubMed] [Google Scholar]
- 3.Bottomley SS. Sideroblastic Anemias. In: Greer JP, Foerster J, Rogers GM, Paraskevas F, Glader B, Arber DA, et al., eds. Wintrobe’s clinical hematology. 12th ed Philadelphia l London: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2009:835–56 [Google Scholar]
- 4.Ohba R, Furuyama K, Yoshida K, Fujiwara T, Fukuhara N, Onishi Y, et al. Clinical and genetic characteristics of congenital sideroblastic anemia: comparison with myelodysplastic syndrome with ring sideroblast (MDS-RS). Ann Hematol. 2013;92(1):1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harigae H, Furuyama K, Kudo K, Hayashi N, Yamamoto M, Sassa S, et al. A novel mutation of the erythroid-specific gamma-aminolevulinate synthase gene in a patient with non-inherited pyridoxine-responsive sideroblastic anemia. Am J Hematol. 1999;62(2):112–4 [DOI] [PubMed] [Google Scholar]
- 6.Guernsey DL, Jiang H, Campagna DR, Evans SC, Ferguson M, Kellogg MD, et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet. 2009;41(6):651–3 [DOI] [PubMed] [Google Scholar]
- 7.Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110(4):1353–8 [DOI] [PubMed] [Google Scholar]
- 8.Allikmets R, Raskind WH, Hutchinson A, Schueck ND, Dean M, Koeller DM. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum Mol Genet. 1999;8(5):743–9 [DOI] [PubMed] [Google Scholar]
- 9.Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet. 2004; 74(6):1303–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Labay V, Raz T, Baron D, Mandel H, Williams H, Barrett T, et al. Mutations in SLC19A2 cause thiamine-responsive mega-loblastic anaemia associated with diabetes mellitus and deafness. Nat Genet. 1999;22 (3):300–4 [DOI] [PubMed] [Google Scholar]
- 11.Bergmann AK, Campagna DR, McLoughlin EM, Agarwal S, Fleming MD, Bottomley SS, et al. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer. 2010;54(2):273–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zon LI, Youssoufian H, Mather C, Lodish HF, Orkin SH. Activation of the erythropoietin receptor promoter by transcription factor GATA-1. Proc Natl Acad Sci USA. 1991; 88(23):10638–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiba T, Ikawa Y, Todokoro K. GATA-1 transactivates erythropoietin receptor gene, and erythropoietin receptor-mediated signals enhance GATA-1 gene expression. Nucleic Acids Res. 1991;19(14):3843–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans T, Felsenfeld G. The erythroid-specific transcription factor eryf1: a new finger protein. Cell. 1989;58(5):877–85 [DOI] [PubMed] [Google Scholar]
- 15.Whitelaw E, Tsai SF, Hogben P, Orkin SH. Regulated expression of globin chains and the erythroid transcription factor GATA-1 during erythropoiesis in the developing mouse. Mol Cell Biol. 1990;10(12):6596–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Surinya KH, Cox TC, May BK. Transcriptional regulation of the human erythroid 5-aminolevulinate synthase gene. Identification of promoter elements and role of regulatory proteins. J Biol Chem. 1997;272(42):26585–94 [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi M, Nishikawa K, Yamamoto M. Hematopoietic regulatory domain of gata1 gene is positively regulated by GATA1 protein in zebrafish embryos. Development. 2001;128(12):2341–50 [DOI] [PubMed] [Google Scholar]
- 18.Ohneda K, Yamamoto M. Roles of hematopoietic transcription factors GATA-1 and GATA-2 in the development of red blood cell lineage. Acta Haematol. 2002; 108(4):237–45 [DOI] [PubMed] [Google Scholar]
- 19.Weiss MJ, Keller G, Orkin SH. Novel insights into erythroid development revealed through in vitro differentiation of GATA-1 embryonic stem cells. Genes Dev. 1994;8(10):1184–97 [DOI] [PubMed] [Google Scholar]
- 20.Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc Natl Acad Sci USA. 1996;93(22):12355–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Surinya KH, Cox TC, May BK. Identification and characterization of a conserved erythroid-specific enhancer located in intron 8 of the human 5-aminolevulinate synthase 2 gene. J Biol Chem. 1998;273 (27):16798–809 [DOI] [PubMed] [Google Scholar]
- 22.Fujiwara T, O’Geen H, Keles S, Blahnik K, Linnemann AK, Kang YA, et al. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol Cell. 2009;36(4):667–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vargas PD, Furuyama K, Sassa S, Shibahara S. Hypoxia decreases the expression of the two enzymes responsible for producing linear and cyclic tetrapyrroles in the heme biosynthetic pathway. FEBS J. 2008;275(23): 5947–59 [DOI] [PubMed] [Google Scholar]
- 24.Bekri S, May A, Cotter PD, Al-Sabah AI, Guo X, Masters GS, et al. A promoter mutation in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causes X-linked sideroblastic anemia. Blood. 2003;102(2): 698–704 [DOI] [PubMed] [Google Scholar]
- 25.Muto A, Hoshino H, Madisen L, Yanai N, Obinata M, Karasuyama H, et al. Identification of Bach2 as a B-cell-specific partner for small maf proteins that negatively regulate the immunoglobulin heavy chain gene 3′ enhancer. EMBO J. 1998;17(19): 5734–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadirvel S, Furuyama K, Harigae H, Kaneko K, Tamai Y, Ishida Y, et al. The carboxyl-terminal region of erythroid-specific 5-aminolevulinate synthase acts as an intrinsic modifier for its catalytic activity and protein stability. Exp Hematol. 2012;40(6): 477–86 e1 [DOI] [PubMed] [Google Scholar]
- 27.Szpurka H, Tiu R, Murugesan G, Aboudola S, Hsi ED, Theil KS, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood. 2006;108 (7):2173–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller IJ, Bieker JJ. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol Cell Biol. 1993;13(5):2776–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ney PA, Sorrentino BP, McDonagh KT, Nienhuis AW. Tandem AP-1-binding sites within the human beta-globin dominant control region function as an inducible enhancer in erythroid cells. Genes Dev. 1990;4(6):993–1006 [DOI] [PubMed] [Google Scholar]
- 30.Harigae H, Furuyama K. Hereditary sideroblastic anemia: pathophysiology and gene mutations. Int J Hematol. 2010;92(3):425–31 [DOI] [PubMed] [Google Scholar]
- 31.Ducamp S, Kannengiesser C, Touati M, Garcon L, Guerci-Bresler A, Guichard JF, et al. Sideroblastic anemia: molecular analysis of the ALAS2 gene in a series of 29 probands and functional studies of 10 missense mutations. Hum Mutat. 2011;32(6):590–7 [DOI] [PubMed] [Google Scholar]
- 32.May A, Barton C, Masters G, Kingston J, Lawless S, Jenner M. Severe sideroblastic anaemia in an ALAS2 compound heterozygote for −206G, a common polymorphism, and a novel mutation in exon 11 (Lys535del) linked to lack of haemoglobinisation in vitro and ineffective erythropoiesis in vivo. Blood (ASH Annual Meeting Abstracts). 2005;106 (11):3541 [Google Scholar]
- 33.Dou QP, Fridovich-Keil JL, Pardee AB. Inducible proteins binding to the murine thymidine kinase promoter in late G1/S phase. Proc Natl Acad Sci USA. 1991;88(4): 1157–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lagrange T, Kapanidis AN, Tang H, Reinberg D, Ebright RH. New core promoter element in RNA polymerase II-dependent transcription: sequence-specific DNA binding by transcription factor IIB. Genes Dev. 1998;12(1):34–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallagher PG, Sabatino DE, Romana M, Cline AP, Garrett LJ, Bodine DM, et al. A human beta-spectrin gene promoter directs high level expression in erythroid but not muscle or neural cells. J Biol Chem. 1999;274(10):6062–73 [DOI] [PubMed] [Google Scholar]
- 36.Faisst S, Meyer S. Compilation of vertebrate-encoded transcription factors. Nucleic Acids Res. 1992;20(1):3–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rikitake Y, Moran E. DNA-binding properties of the E1A-associated 300-kilodalton protein. Mol Cell Biol. 1992;12(6):2826–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moser M, Imhof A, Pscherer A, Bauer R, Amselgruber W, Sinowatz F, et al. Cloning and characterization of a second AP-2 transcription factor: AP-2 beta. Development. 1995;121(9):2779–88 [DOI] [PubMed] [Google Scholar]
- 39.Gilthorpe J, Vandromme M, Brend T, Gutman A, Summerbell D, Totty N, et al. Spatially specific expression of Hoxb4 is dependent on the ubiquitous transcription factor NFY. Development. 2002;129(16): 3887–99 [DOI] [PubMed] [Google Scholar]
- 40.Nallur GN, Prakash K, Weissman SM. Multiplex selection technique (MuST): an approach to clone transcription factor binding sites. Proc Natl Acad Sci USA. 1996; 93(3):1184–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sorensen KD, Quintanilla-Martinez L, Kunder S, Schmidt J, Pedersen FS. Mutation of all Runx (AML1/core) sites in the enhancer of T-lymphomagenic SL3–3 murine leukemia virus unmasks a significant potential for myeloid leukemia induction and favors enhancer evolution toward induction of other disease patterns. J Virol. 2004;78(23):13216–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spandidos DA, Yiagnisis M, Pintzas A. Human immunodeficiency virus long terminal repeat responds to transformation by the mutant T24 H-ras1 oncogene and it contains multiple AP-1 binding TPA-inducible consensus sequence elements. Anticancer Res. 1989;9(2):383–6 [PubMed] [Google Scholar]
- 43.Mullhaupt B, Feren A, Jones A, Fodor E. DNA sequence and functional characterization of the human and rat epidermal growth factor promoter: regulation by cell growth. Gene. 2000;250(1–2):191–200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.