

Abstract
Complement blockade by eculizumab is clinically effective in hemolytic paroxysmal nocturnal hemoglobinuria. However, the response is variable and some patients remain dependent on red blood cell transfusions. In 72 patients with hemolytic paroxysmal nocturnal hemoglobinuria on eculizumab we tested the hypothesis that response may depend on genetic polymorphisms of complement-related genes. We found no correlation between the complement component C3 genotypes and the need for blood transfusions. On the other hand, we found a significant correlation with the HindIII polymorphism of a complement regulatory gene, the complement receptor 1 (CR1) gene. At this locus two co-dominant alleles are known, of which H (common) is associated with high expression, whereas L (rare) is associated with low expression of CR1 on red blood cells. Patients who still needed blood transfusion on eculizumab accounted for 18% of the H/H homozygotes, 33% of the H/L heterozygotes and 68% of the L/L homozygotes (P=0.016). Thus, patients with paroxysmal nocturnal hemoglobinuria who have the L/L genotype are seven times more likely to be sub-optimal responders to eculizumab. Both in vitro and in vivo we found that the CR1 HindIII genotype correlates with the abundance of paroxysmal nocturnal hemoglobinuria red cells that have bound C3, and with the kinetics of C3 binding. These results are consistent with the notion that by affecting C3 binding the CR1 genotype influences the response to eculizumab treatment, and this emerges as a novel example of pharmacogenetics.
Introduction
Blockade of the distal complement pathway through the use of eculizumab, a monoclonal antibody against complement component 5 (C5), has been a major advance in the clinical management of paroxysmal nocturnal hemoglobinuria (PNH).1,2 Eculizumab effectively controls intravascular hemolysis and its direct consequences in PNH patients, which results in remarkable clinical improvement.1,3 The natural history of the disease, which had already improved before the introduction of eculizumab (compare the data reported by Hillmen et al., 19954 with those reported by de Latour et al., 20085), may have improved further with the use of this agent.6 However, the hematologic response to eculizumab is variable. In some patients who are not transfusion-dependent the average hemoglobin level increases following treatment with the monoclonal antibody; the majority of patients who are transfusion-dependent become transfusion-independent, with or without an increase in their average hemoglobin level, but some patients remain transfusion-dependent, even though their transfusion requirement may decrease.7,8 In addition, in almost all PNH patients on eculizumab a significant fraction of glycosylphosphatidylinositol (GPI)-negative red blood cells (RBC) are opsonized by complement component 3 (C3) (with the previously negative Coombs’ test becoming positive) and the red cells so opsonized may undergo extravascular hemolysis.7 This phenomenon, testified also by a persistent reticulocytosis, may not be clinically relevant in most patients; however, in some patients it may limit the hematologic benefit from eculizumab significantly. In fact, 25–35% of patients still need RBC transfusions2,6,8,9 and the size of the fraction of their own red cells with bound C3 seems to correlate with transfusion requirement.7,10
In view of the above, we hypothesized that one determinant of the variability of response to eculizumab might be the levels of factors that can regulate complement activation. In order to test this hypothesis we analyzed genetic polymorphisms of complement-related genes: specifically, polymorphic alleles of the C3 gene and of the complement receptor 1 (CR1) gene.
Methods
Patients
Seventy-two patients with hemolytic PNH who had received eculizumab treatment were analyzed after a median follow-up of 52 months (range, 11–98 months). Patients with evidence of bone marrow failure (reticulocytes ≤60×109/L, platelets ≤50×109/L, neutrophils ≤1×109/L) were not included in this series because this condition may affect the clinical response to eculizumab regardless of how well hemolysis is controlled. Peripheral blood samples were collected for clinical testing, flow cytometry (GPI-molecules and C3-binding)7 and genotyping after the patients had signed an informed consent form approved by the respective Institutional Review Board. As a criterion of response to eculizumab we used the one that is most stringent and objective: namely RBC transfusion at any time after the first 6 months on eculizumab treatment. Of the 72 patients 60 were transfusion-dependent before eculizumab: of these 60, those who on eculizumab received no further transfusion during follow-up have been classified as good responders; those who on eculizumab received one or more RBC transfusion at any time during follow-up have been classified as suboptimal responders. Twelve patients started eculizumab before having received any RBC transfusions. Of these, the ten patients who on eculizumab remained transfusion-independent have been classified as good responders because their hemoglobin level increased, on average, by 2.2 g/dL; the remaining two patients required RBC transfusions on eculizumab and have been classified as sub-optimal responders. Only one patient, who had hemolytic PNH at the time of starting eculizumab and who had already been classified as a sub-optimal responder, became aplastic during follow-up.
Genotyping
The polymorphism rs2230199 C>G of the C3 gene was investigated by a newly designed tetra-primer amplification refractory mutation system-polymerase chain reaction method.11 Three polymorphisms of the CR1 gene were genotyped by restriction fragment length (RFLP) analysis:12 HindIII RFLP (intron 27); His1208Arg (exon 22); Pro1827Arg (exon 33).
Kinetics of C3 binding in vitro
RBC and sera were promptly separated from peripheral blood freshly collected from PNH patients. The RBC were then incubated with sera containing eculizumab as previously described.13,14 Briefly, a 2% suspension of RBC from PNH patients was incubated at 37°C with a pool of ABO-compatible sera from patients on eculizumab, and the complement alternative pathway was activated by mild acidification (HCl 0.016 M). At serial time points (15, 30, 60, and 120 min) after complement activation the fraction of GPI-negative RBC with newly bound C3 fragments was measured by flow cytometry (AccuriC6, Becton Dickinson, NJ, USA) after staining with anti-CD59 Alexa647 (Mem43, Serotec, UK) and anti-C3d-neoantigen (A250, Quidel, CA, USA); secondary staining was performed with phycoerythrin-labeled polyclonal rabbit-anti-mouse antibodies (Dako Cytomation, Denmark).
Statistical analysis
The Hardy-Weinberg equilibrium was tested with the Pearson χ2 test using the Finetti program (http://ihg2.helmholtz-muenchen.de/cgi-bin/hw/hwa1.pl). The association between genotypes and response was tested by the χ2 test for trend, because these polymorphisms affect gene expression (or gene product activity), and for each one of them the level of expression in heterozygotes is intermediate with respect to that in both homozygotes. Mann-Whitney and Kruskal-Wallis tests were used for continuous variables as appropriate. All statistical tests were two-sided; statistical significance was accepted for P values < 0.05.
Results and Discussion
C3 is central in the complement system.15 The single nucleotide polymorphism rs2230199 C>G of the C3 gene is responsible for the allelic electrophoretic variants, slow (common allele) and fast (rare allele).16,17 This polymorphism of C3 influences the activity of the complement alternative pathway15 and it is known to be associated with certain complement-mediated disorders.18–20 In 72 hemolytic PNH patients on eculizumab the frequencies of the slow and fast alleles of the C3 polymorphism rs2230199 were in Hardy-Weinberg equilibrium. We found no correlation between response to eculizumab and C3 genotype (P=0.939; Table 1).
Table 1.
Association of polymorphisms of the C3 and CR1 genes with hematologic response to eculizumab.
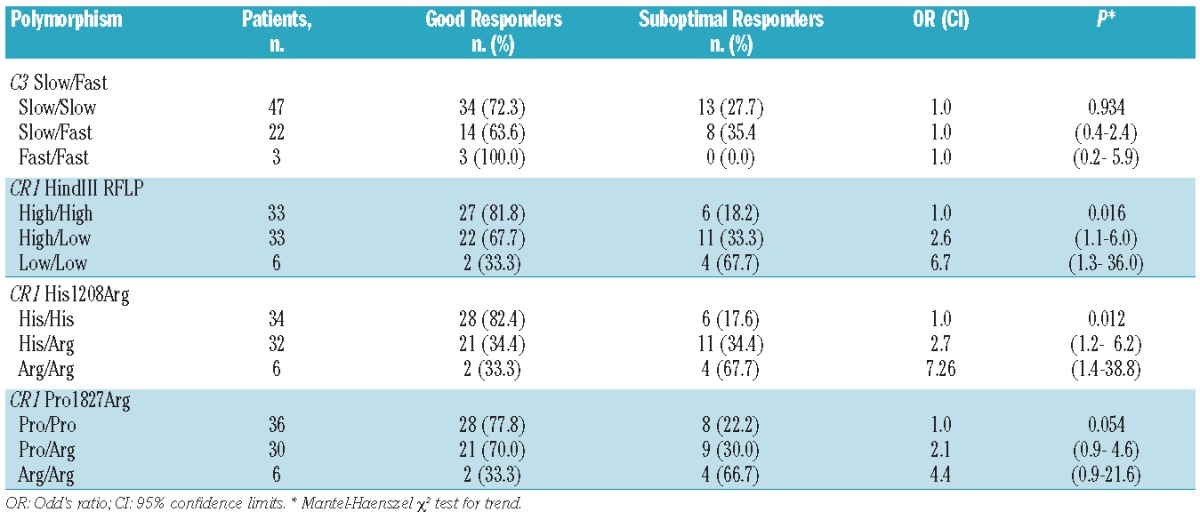
The distribution of the levels of CR1 on RBC is trimodal in the general population.21 The variability of expression of this important cell surface complement regulatory protein is determined by two co-dominant alleles: H (high expression) and L (low expression).22 In Europe and in Asia,12 but not in Africa,23 these two alleles are associated with a HindIII RFLP, which in turn is in strong linkage disequilibrium with a number of other polymorphisms in the CR1 coding sequence, including His1208Arg and Pro1827Arg.12 Although CR1 is not a GPI-linked molecule, it has been suggested that it may play a role in PNH.24–26
In our series of 72 PNH patients the genotype frequencies of these CR1 polymorphisms were in Hardy-Weinberg equilibrium. Since only five of our patients are of African descent, a high level of linkage disequilibrium among the three polymorphisms was expected: in fact, only in three patients was the common CR1 HindIII RFLP allele not associated with the common CR1 Pro1827Arg allele, and in only one of these it was not associated with the common CR1 His1208Arg allele. Thus, the frequencies of the rare alleles were almost identical: 0.31 for the HindIII RFLP and for His1208Arg; 0.29 for Pro1827Arg. When we considered the response of individual patients to eculizumab, we found that the proportion of sub-optimal responders was significantly higher in patients who were heterozygotes for the CR1 HindIII polymorphism (genotype H/L) and even higher in those who were homozygous for the rare allele (genotype L/L) (P=0.016, Table 1). This correlation, as expected, is also significant for the two closely linked polymorphisms His1208Arg and Pro1827Arg (Table 1). It is important to note that the assessment of response to eculizumab, on which this result is based, was substantiated over a median follow-up of more than 4 years (minimum 1 year).
In principle the CR1 HindIII polymorphism might influence blood transfusion requirement in PNH regardless of eculizumab therapy. However, this is not the case: in patients with CR1 HindIII genotypes H/H, H/L and L/L the mean pre-eculizumab hemoglobin levels were 8.3, 8.2 and 8.0 g/dL, respectively (P=0.76) and the mean numbers of packed RBC units/month were 0.6, 0.8 and 0.8, respectively (P=0.38). Interestingly, the percentage of patients not requiring RBC transfusions before eculizumab was higher (33%) among patients with the L/L genotype than among those with the H/H or H/L genotype (15%). The different response of patients with the three genotypes to eculizumab was not related to a difference in the control of intravascular hemolysis: in fact, the levels of lactate dehydrogenase were similar in good responders and in sub-optimal responders: 1.05 and 1.18 times the upper limit of normal levels, respectively (P=0.11). In addition, patients on eculizumab with CR1 HindIII genotypes H/H, H/L and L/L had similar levels of lactate dehydrogenase: 1.05, 1.13 and 1.10 times the upper limit of normal levels, respectively (P=0.80). Thus, in this series the CR1 HindIII genotype was not associated with significantly different clinical features, but rather with the way patients respond to eculizumab.
Our findings might be explained by considering the multiple functions of CR1 within the complement system.27 Indeed, by binding C3b and C4b, CR1 enhances the decay of the C3 and C5 convertases;28 and since CR1 is a cofactor of factor I it can also help inactivating C3b and C4b, thus tuning both the alternative and the classical complement activation pathways.29,30 In addition, CR1 plays a role in the clearance of immune complexes and in phagocytosis.31–33 Thus, the lower levels of CR1 expression on the red cell surface associated with the H/L and L/L genotypes22 are expected to result in increased complement activation and C3 binding on PNH red cells.
In order to test this hypothesis we investigated the kinetics of C3 binding to GPI-negative red cells in vitro13,14 from patients with the three CR1 HindIII genotypes. When GPI-negative red cells were exposed to activated complement in the presence of eculizumab, we promptly detected C3-positive PNH red cells, the percentage of which increased with time.14 The rate of this increase was highest for red cells with the L/L genotype, lowest for red cells with the H/H genotype, and intermediate for red cells with the heterozygous genotype (Figure 1). These in vitro findings suggest that the density of CR1 on the surface of red cells modulates the binding of C3 fragments to the GPI-negative red cells when C5 is blocked by eculizumab. These data are in good agreement with the observation that in our patients on eculizumab the CR1 HindIII genotypes correlated with the fraction of GPI-negative red cells with bound C3 in vivo: H/H (n=20) 21.4±19.9%, H/L (n=16) 29.1 ± 16.2% and L/L (n=4) 46.3±19.5% (P=0.032).
Figure 1.
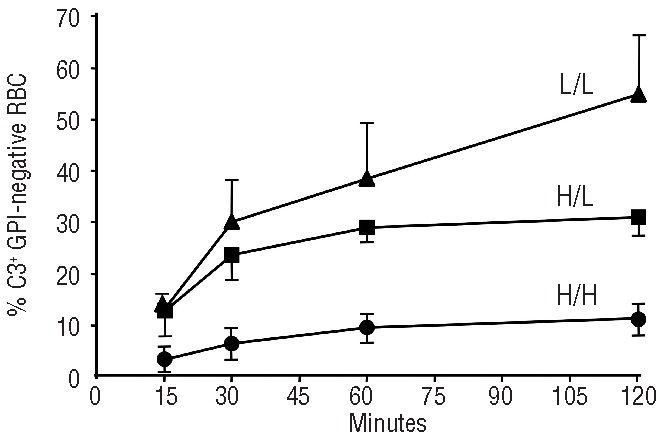
The CR1 genotype is a major determinant of the rate of in vitro C3 binding to GPI-negative red cells. In each patient this time course experiment was carried out twice. H/H: homozygotes for the high expression allele of the CR1 HindIII RFLP polymorphism. L/L: homozygotes for the low expression allele of the CR1 HindIII RFLP polymorphism. H/L= heterozygotes for the CR1 HindIII RFLP polymorphism. The standard deviation is shown for each experimental point. When eculizumab-containing sera were added to red cells, the slope of novel C3 binding to GPI-negative red cells was significantly different depending on the CR1 genotype. For the comparison of H/H (n=6) vs. H/L (n=3), P=0.0425; for the comparisonvs H/L (n=3) vs. L/L (n=2), P=0.0173; for the comparison H/H (n=6). L/L (n=2), P<0.0001. Given a certain CR1 genotype these differences were not the result of eculizumab treatment: indeed, in three patients with the H/H genotype who were on eculizumab, the rate of C3 binding to GPI-negative red cells was very similar to that seen in three patients with the H/H genotype who were not on eculizumab (P=0.87).
During eculizumab treatment abrupt complement activation by intercurrent infection or inflammation may sporadically overcome C5 blockade resulting, especially in patients with low levels of CR1, in occasional episodes of intravascular hemolysis, irrespective of the plasma level of eculizumab.9 However, more commonly, when C5 is effectively blocked, low CR1 levels on red cells would continuously facilitate and increase C3 binding on the RBC surface because the decay of C3 convertase is slower. Thus, there will be an increased proportion of C3-opsonized red cells that can be removed by phagocytosis, resulting, in PNH patients with these CR1 genotypes, in a higher rate of chronic extravascular hemolysis.9 This relationship between the response to eculizumab and the CR1 genotype is in keeping with our current understanding of the pathophysiology of PNH on eculizumab treatment.7,9
Given the small number of L/L homozygotes in our series we must be guarded in drawing definitive conclusions: larger prospective studies will be needed. Nevertheless, the concordance between clinical, biological and in vitro data indicates that the CR1 polymorphism may predict the hematologic response to eculizumab. Patients on eculizumab, even when they remain transfusion-dependent, benefit considerably from this treatment in terms of reduced risk of thrombosis and of subjective symptoms, and thus in their quality of life. Therefore, although our data indicate that PNH patients with the CR1 HindIII RFLP L/L genotype are 6.7 times more likely to be sub-optimal responders to eculizumab than patients with the H/H genotype, this certainly does not disqualify them from eculizumab treatment. Rather, the interaction between the CR1 polymorphism and eculizumab, a novel example of pharmacogenetics, may help to explain the pattern of response to eculizumab. This is important, particularly in view of the development of new complement-targeting agents for the management of PNH.13
Acknowledgments
We thank Luca Boni for statistical advice. Preliminary data on part of this work were reported at the 54th Annual Meeting of the American Society of Hematology, December 8–11, 2012 in Atlanta, GA, USA.
Footnotes
Funding
The work reported in this manuscript was funded in part by grants from the “Associazione Italiana per la Ricerca sul Cancro – AIRC” (LL, RN), from the “Aplastic Anemia and MDS Foundation” (AMR) and by a start-up grant from the “Istituto Toscano Tumori” (RN).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355 (12):1233–43 [DOI] [PubMed] [Google Scholar]
- 2.Luzzatto L, Gianfaldoni G, Notaro R. Management of paroxysmal nocturnal haemoglobinuria: a personal view. Br J Haematol. 2011;153(6):709–20 [DOI] [PubMed] [Google Scholar]
- 3.Kanakura Y, Ohyashiki K, Shichishima T, Okamoto S, Ando K, Ninomiya H, et al. Safety and efficacy of the terminal complement inhibitor eculizumab in Japanese patients with paroxysmal nocturnal hemoglobinuria: the AEGIS clinical trial. Int J Hematol. 2011;93(1):36–46 [DOI] [PubMed] [Google Scholar]
- 4.Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253–8 [DOI] [PubMed] [Google Scholar]
- 5.de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008;112(8): 3099–106 [DOI] [PubMed] [Google Scholar]
- 6.Kelly RJ, Hill A, Arnold LM, Brooksbank GL, Richards SJ, Cullen M, et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011;117(25): 6786–92 [DOI] [PubMed] [Google Scholar]
- 7.Risitano AM, Notaro R, Marando L, Serio B, Ranaldi D, Seneca E, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113(17):4094–100 [DOI] [PubMed] [Google Scholar]
- 8.Dezern AE, Dorr D, Brodsky RA. Predictors of hemoglobin response to eculizumab therapy in paroxysmal nocturnal hemoglobinuria. Eur J Haematol. 2013;90(1):16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luzzatto L, Risitano AM, Notaro R. Paroxysmal nocturnal hemoglobinuria and eculizumab. Haematologica. 2010;95(4): 523–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hill A, Rother RP, Arnold L, Kelly R, Cullen MJ, Richards SJ, et al. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematologica. 2010;95(4):567–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peruzzi B, Serra M, Pescucci C, Sica M, Lastraioli S, Rondelli T, et al. Easy genotyping of complement C3 ‘slow’ and ‘fast’ allo-types by tetra-primer amplification refractory mutation system PCR. Mol Cell Probes. 2010;24(6):401–2 [DOI] [PubMed] [Google Scholar]
- 12.Xiang L, Rundles JR, Hamilton DR, Wilson JG. Quantitative alleles of CR1: coding sequence analysis and comparison of haplotypes in two ethnic groups. J Immunol. 1999;163(9):4939–45 [PubMed] [Google Scholar]
- 13.Risitano AM, Notaro R, Pascariello C, Sica M, del Vecchio L, Horvath CJ, et al. The complement receptor 2/factor H fusion protein TT30 protects paroxysmal nocturnal hemoglobinuria erythrocytes from complement-mediated hemolysis and C3 fragment. Blood. 2012;119(26):6307–16 [DOI] [PubMed] [Google Scholar]
- 14.Sica M, Pascariello C, Rondelli T, Risitano A, RN Kinetics of complement protein 3 (C3) binding to PNH (GPI-negative) erythrocytes under complement blockade by eculizumab. Haematologica. 2010;95(s3):53820378576 [Google Scholar]
- 15.Botto M, Kirschfink M, Macor P, Pickering MC, Wurzner R, Tedesco F. Complement in human diseases: Lessons from complement deficiencies. Mol Immunol. 2009;46(14): 2774–83 [DOI] [PubMed] [Google Scholar]
- 16.Alper CA, Propp RP. Genetic polymorphism of the third component of human complement (C′3). J Clin Invest. 1968;47(9):2181–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Botto M, Fong KY, So AK, Koch C, Walport MJ. Molecular basis of polymorphisms of human complement component C3. J Exp Med. 1990;172(4):1011–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rambausek M, van den Wall Bake AW, Schumacher-Ach R, Spitzenberg R, Rother U, van Es LA, et al. Genetic polymorphism of C3 and Bf in IgA nephropathy. Nephrol Dial Transplant. 1987;2(4):208–11 [PubMed] [Google Scholar]
- 19.Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M, Kirschfink M, et al. Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol. 2005;16(5):1392–403 [DOI] [PubMed] [Google Scholar]
- 20.Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007;39(10):1200–1 [DOI] [PubMed] [Google Scholar]
- 21.Wilson JG, Wong WW, Schur PH, Fearon DT. Mode of inheritance of decreased C3b receptors on erythrocytes of patients with systemic lupus erythematosus. N Engl J Med. 1982;307(16):981–6 [DOI] [PubMed] [Google Scholar]
- 22.Wilson JG, Murphy EE, Wong WW, Klickstein LB, Weis JH, Fearon DT. Identification of a restriction fragment length polymorphism by a CR1 cDNA that correlates with the number of CR1 on erythrocytes. J Exp Med. 1986;164(1):50–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowe JA, Raza A, Diallo DA, Baby M, Poudiougo B, Coulibaly D, et al. Erythrocyte CR1 expression level does not correlate with a HindIII restriction fragment length polymorphism in Africans; implications for studies on malaria susceptibility. Genes Immun. 2002;3(8):497–500 [DOI] [PubMed] [Google Scholar]
- 24.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Deficiency of an erythrocyte membrane protein with complement regulatory activity in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci USA. 1983;80(17):5430–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross GD, Yount WJ, Walport MJ, Winfield JB, Parker CJ, Fuller CR, et al. Disease-associated loss of erythrocyte complement receptors (CR1, C3b receptors) in patients with systemic lupus erythematosus and other diseases involving autoantibodies and/or complement activation. J Immunol. 1985;135(3):2005–14 [PubMed] [Google Scholar]
- 26.Roberts WN, Wilson JG, Wong W, Jenkins DE, Jr, Fearon DT, Austen KF, et al. Normal function of CR1 on affected erythrocytes of patients with paroxysmal nocturnal hemoglobinuria. J Immunol. 1985;134(1):512–7 [PubMed] [Google Scholar]
- 27.Khera R, Das N. Complement receptor 1: disease associations and therapeutic implications. Mol Immunol. 2009;46(5):761–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iida K, Nussenzweig V. Complement receptor is an inhibitor of the complement cascade. J Exp Med. 1981;153(5):1138–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross GD, Lambris JD, Cain JA, Newman SL. Generation of three different fragments of bound C3 with purified factor I or serum. I. Requirements for factor H vs CR1 cofactor activity. J Immunol. 1982;129(5):2051–60 [PubMed] [Google Scholar]
- 30.Medof ME, Nussenzweig V. Control of the function of substrate-bound C4b-C3b by the complement receptor Cr1. J Exp Med. 1984;159(6):1669–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshida K, Yukiyama Y, Miyamoto T. Interaction between immune complexes and C3b receptors on erythrocytes. Clin Immunol Immunopathol. 1986;39(2):213–21 [DOI] [PubMed] [Google Scholar]
- 32.Cosio FG, Shen XP, Birmingham DJ, Van Aman M, Hebert LA. Evaluation of the mechanisms responsible for the reduction in erythrocyte complement receptors when immune complexes form in vivo in primates. J Immunol. 1990;145(12):4198–206 [PubMed] [Google Scholar]
- 33.Craig ML, Bankovich AJ, Taylor RP. Visualization of the transfer reaction: tracking immune complexes from erythrocyte complement receptor 1 to macrophages. Clin Immunol. 2002;105(1):36–47 [DOI] [PubMed] [Google Scholar]