

Abstract
Expression of the immediate early response gene X-1 (IEX-1, IER3) is diminished significantly in hematopoietic stem cells in a subgroup of patients with early stage myelodysplastic syndromes, but it is not clear whether the deregulation contributes to the disease. The current study demonstrates increased apoptosis and a concomitant decrease in the number of hematopoietic stem cells lacking this early response gene. Null mutation of the gene also impeded platelet differentiation and shortened a lifespan of red blood cells. When bone marrow cells deficient in the gene were transplanted into wild-type mice, the deficient stem cells produced significantly fewer circulating platelets and red blood cells, despite their enhanced repopulation capability. Moreover, after exposure to a non-myeloablative dose of radiation, absence of the gene predisposed to thrombocytopenia, a significant decline in red blood cells, and dysplastic bone marrow morphology, typical characteristics of myelodysplastic syndromes. These findings highlight a previously unappreciated role for this early response gene in multiple differentiation steps within hematopoiesis, including thrombopoiesis, erythropoiesis and in the regulation of hematopoietic stem cell quiescence. The deficient mice offer a novel model for studying the initiation and progression of myelodysplastic syndromes as well as strategies to prevent this disorder.
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous array of disorders, collectively characterized by impaired and clonally restricted hematopoiesis, dysplastic bone marrow (BM) and peripheral blood cell morphology, and a tendency to progress into acute myeloid leukemia (AML). Analyses of somatic mutations and colony assays have implicated hematopoietic stem cells (HSCs) or common myeloid progenitors as the cells of origin of MDS. It is postulated that patients with MDS may lose normal HSCs over time as a result of defective self-renewal and gradual accumulation of MDS HSCs.1,2 In accordance with this, a variety of genes responsible for HSC self-renewal, differentiation, and quiescence are found either mutated or aberrantly expressed in MDS patients, and contributions of some of these genes to a limited hematopoietic reservoir are corroborated by the development of an MDS-like phenotype in corresponding genetically engineered mice.1,3 In addition, abnormal ultra-structures of mitochondria are frequently identified in MDS patients,4 implying that dysfunction or inadequate activity of mitochondria may be also part of the etiology of a subgroup of MDS,5,6 which is consistent with a high incidence of the disease in the elderly, as mitochondria degenerate with aging. Evidence in support of this etiology has recently begun to emerge.5,6
The immediate early response gene X-1 (IEX-1), also known as IER3, p22/PRG1, Dif-2 and gly96, is a stress-inducible gene.7 Our previous investigations showed that IEX-1 was involved in delicate regulation of mitochondrial F1Fo-ATP synthase/ATPase activity,8 a multi-protein complex that converts electron energy to bioenergy ATP in mitochondrial respiratory chain, which is responsible for 90% ATP synthesis in a cell. One of the inhibitory subunits of the complex, named IF1 that inhibits ATPase of this complex, is targeted by IEX-1 for degradation via an as yet unidentified mitochondrial protease.8,9 Hence, overexpression of IEX-1 reduces the level of IF1 protein expression, increases ATP synthase/ATPase activity, and sustains the mitochondrial inner membrane Δψm at a phosphorylating status, thus protecting cells from mitochondrion-dependent apoptosis.8,10,11 In contrast, lack of IEX-1 stabilizes IF1 and decreases the activity of F1Fo-ATPase, concurrent with increased production of reactive oxygen species (ROS).8 Apart from regulation of the mitochondrial activity, IEX-1 also participates in multiple cell signaling pathways such as ERK signaling prolongation, nuclear Mcl-1 accumulation, and DNA repair in cells under stress.12,13 Strikingly, clinical studies have shown deregulation of IEX-1 expression in approximately 60% of MDS patients.14 It was among the most down-regulated genes in CD34+ stem cells isolated from MDS patients compared to healthy donors.15,16 The decrease is particularly apparent in the early stage/low-risk group of MDS patients, often concomitant with excessive intramedullary apoptosis in association with hematopoietic failure.14 However, it is not known whether IEX-1 deregulation is a bystander event or whether it contributes significantly to MDS pathogenesis in these patients.
The present study shows that null mutation of IEX-1 perturbs HSC quiescence and impairs maturation of platelets and red blood cells (RBCs). Upon BM transplantation or non-myeloablative radiation, changes resembling MDS occurred, including thrombocytopenia, a trend toward anemia, and granulocyte dysplasia. These observations underscore an indispensable role for IEX-1 in maintenance of HSC quiescence as well as in multiple differentiation steps along thrombopoiesis and erythropoiesis. IEX-1-deficient mice confer a novel model for investigating how mitochondrial dysfunction may cause MDS and how to prevent or slow down the disease.
Methods
Mice
IEX-1 knockout (KO) mice on mixed 129Sv/C57BL/6 background were generated by gene-targeting deletion in our laboratory, as previously described.17 The KO mice were bred with wild type (WT) C57BL/6 (B6) for 10 generations to obtain IEX-1 KO B6 mice. WT B6 mice and mice on 129Sv/B6 background were derived from breeding as above and used as controls. Cognate BL/6.SJL-PtprcaPep3/BoyJ (CD45.1) or PepJ mice were obtained from Jackson Laboratory. All experimental mice and BM donor mice were used at 6–8 weeks of age. Animals were maintained in the pathogen-free animal facilities of Massachusetts General Hospital in compliance with institutional guidelines. All animal studies were approved by the Subcommittee on Research Animal Care of the institute. Extended methods can be found in the Online Supplementary Appendix.
Results
Perturbation of HSC quiescence in the absence of IEX-1
IEX-1 KO mice display no gross developmental defect except for modest hypertension at 2-months of age or older,17,18 in agreement with its function required primarily in response to stress. Given the significant loss of IEX-1 reported in patients with early stage MDS,14,16 we examined the status of LSK (Lineage- Sca-1+ c-Kit+) stem cells in IEX-1 KO mice. Loss of IEX-1 led to a significant decline in the proportion of LSK cells, in comparison with WT mice (0.10±0.07% vs. 0.24±0.06 %; P<0.01) (Figure 1A). Apparently, the decline contradicted increased cycling of the cells, as shown in Figure 1B and C, in which relatively higher proportions of KO LSK cells entered G1 and G2/S/M cycling phases than WT counterparts, concurrent with a reduced percentage of the cells at a Go or quiescent phase. We thus examined whether the decline was ascribed to increased apoptosis of the cells, because apoptosis in HSCs was abnormally high in many MDS patients.19 Increased apoptosis of IEX-1 KO over WT BM cells was confirmed by apoptosis-specific terminal deoxynucleotydyl transferase-mediated dUTP nick end labeling or TUNEL (11.6±0.7% vs. 2.2±0.4%; P<0.01), which detected apoptosis at both early and late stages. Early apoptotic marker Annexin V staining further revealed that apoptosis was increased by more than 50% in the BM (6.9±0.5% vs. 4.5±0.3%; P<0.01) (Figure 1D) or by greater than 160% in LSK cells (4.5±0.6% vs. 1.7±0.2%; P<0.001) in the absence versus the presence of IEX-1 (Figure 1E), suggesting that IEX-1 affected LSK cells more than other BM cells. The result explains a reduced proportion of LSK cells in IEX-1 KO mice compared to WT mice. In spite of a reduced percentage of LSK stem cells, there was no significant difference in the total number of LSK cells between these two strains of mice. Presumably, a relatively high level of apoptosis was well offset by correspondingly high cycling of KO LSK cells in the mice (Figure 1B and C). The results resemble clinical findings that showed a higher than normal proliferation that is often accompanied by a significant increase in apoptosis of HSCs in MDS patients.20 Apart from LSK cells, we did not observe any difference in long- or short-term LSK cell populations, downstream common myeloid progenitors (CMP), common lymphocyte progenitors (CLP), megakaryocyte-erythrocyte progenitors (MEP), or peripheral blood counts (Online Supplementary Figure S1). Moreover, ex vivo differentiation of BM cells failed to demonstrate any significant difference in the production of multipotential progenitors that generate myeloid lineage cells including granulocytes (G), erythrocytes (E), monocytes (M), and megakaryocytes (M), also called GEMM, and granulocyte-monocyte (GM) or erythroid (E) progenitors in hematopoietic colony forming culture (Online Supplementary Figure S1B), confirming previous findings.14
Figure 1.
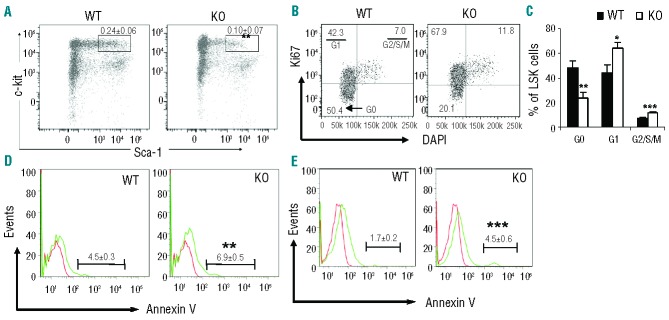
Analysis of frequency, proliferation and apoptosis of LSK cells. (A) Flow cytometric analysis of LSK cells. BM cells were isolated from wild type (WT) and IEX-1 KO mice and subjected to flow cytometric analysis after staining with antibodies against various lineages, c-Kit, and Sca-1. The numbers in the plot are mean percentages ± standard deviation (SD) of LSK cells relative to total BM cells (n = 7). (B and C) Cell cycle analysis within LSK cells using DAPI and anti-Ki67 antibody. The numbers in the plot are percentages of LSK cells at G0, G1 and G2/S/M phases as indicated and data from 7 mice are summarized in (C). (D and E) FACS histograms of apoptotic cells in total BM (D) and LSK cells (E). The numbers in the plot are mean percentages ± SD of Annexin-V+ cells in total BM cells or LSK cells. *, ** and ***, P<0.05, 0.01 or 0.001, respectively, in the presence versus absence of IEX-1.
To address whether this cycling abnormality was an intrinsic defect of HSCs, rather than BM stromal cells, LSK cells sorted from IEX-1 KO and WT animals were labeled with vital dyes DiD (red) or DiI (blue), mixed at 1:1 ratio, and injected into the tail vein of non-irradiated WT mice, followed by intravital imaging of fluorescently labeled cells in the calvarium bone.21 The imaging study allows us to track HSC cycling in a subnormoxic O2 level in a physiological microenvironment of the BM, which cannot be recapitulated by in vitro culture.22 IEX-1 KO LSK cells divided more frequently than WT counterparts, appearing as two cells in close proximity to each other marked in a circle (Figure 2A). Among 200 randomly selected cells traced in 25 imaging stacks, an average 23±7% KO LSK cells were divided compared to 10.5±3% WT LSK cells dividing when tracked seven days after adoptive transfer (Figure 2B). The increased cycling of IEX-1-deficent LSK cells in a non-irradiated BM microenvironment confirmed an autonomous and intrinsic abnormality of the cells.
Figure 2.
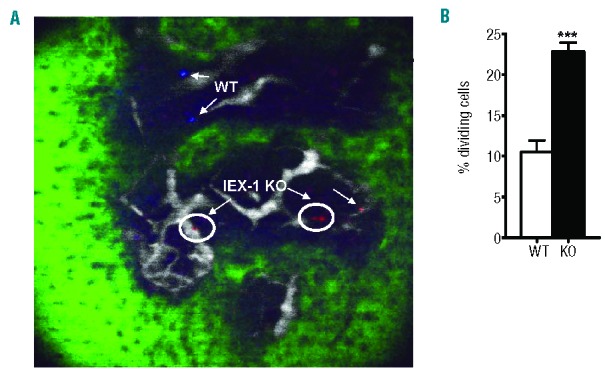
Increased cycling of IEX-1 KO LSK cells in non-irradiated WT mice. WT and KO LSK cells were labeled with DiI (blue) or DiD (red) dye, respectively, mixed in equal numbers, and i.v. injected into non-irradiated WT mice. Intravital microscopy was used to scan the region of mouse calvari-um within the frontal bones on day 7 after the transfer. Shown in (A) are two blue WT cells, two dividing red IEX-1 KO cells (circle), and one non-dividing IEX-1 KO cell. Green and white fluorescent areas are bone and vasculature, respectively. Average percentages ± SD of dividing cells were determined in 25 imaging stacks with a total of 200 randomly selected cells counted (B). ***, P< 0.001 in the presence or absence of IEX-1.
Ineffective RBC and platelet production from IEX-1 KO BM cells
Serial BM transplant assays were next used to address the long-term self-renewal ability of KO stem cells, in which KO or WT BM cells (CD45.2) were transferred into lethally irradiated cognate mice (PepJ, CD45.1) and CD45.1 BM (PepJ) cells were reciprocally transferred into irradiated CD45.2 WT or KO recipients. No differences in the levels of peripheral donor chimerism were noticed in any of the groups up to three months post transplantation (data not shown). Then, a second serial transplantation was carried out similarly in lethally irradiated recipients, and peripheral donor chimerism again showed no significant difference after three months (Figure 3A). However, KO donor BM chimerism in PepJ recipients was significantly increased at this time (59% vs. 42%; P<0.01) (Figure 3B). Despite this increase in donor BM chimerism, platelet and RBC counts were unexpectedly diminished below a WT level in PepJ recipients of KO BM cells after three months of BM transplant (P<0.01) (Figure 3C and D). The difference in platelets was not seen in one month post-BM transfer (Figure 3C) and, instead, a slight increase in RBCs was observed in the mice at this early time point (Figure 3D), presumably due to increased BM engraftment in the mice (Figure 3B). All mice receiving WT or PepJ BM cells, regardless of whether or not the recipients expressed IEX-1, gave rise to a normal level or above, defined by a horizontal dash line, of circulating platelets or RBCs in either one or three months post-TBI (Figure 3C and D), ruling out any effect of BM stroma on the described abnormality of KO LSK cells. A slight decrease in RBCs in WT mice receiving PepJ BM cells was without statistical significance (Figure 3D). White blood cell counts remained unaffected, with no significant differences between KO donor groups and WT controls (data not shown). The results suggest insufficient production of platelets and RBCs by KO BM cells, reiterating a myeloid specific defect caused by a loss of IEX-1.
Figure 3.
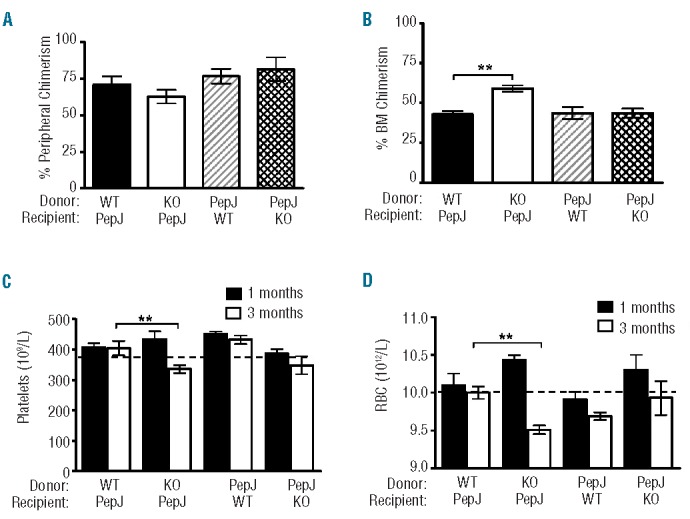
Enhanced BM engraftment with peripheral hematopoietic deficiencies. Secondary BM transplant was carried out by transfer of IEX-1 KO (KO) or WT BM cells (CD45.2) into lethally irradiated cognate PepJ mice (CD45.1) or PepJ BM cells reciprocally into WT or KO recipients. Peripheral (A) and BM (B) chimerism was analyzed 3 months later as percent donor cells based on CD45.1 versus CD45.2 expression. Platelet (C) and RBC (D) counts were evaluated in one (filled) and three months (unfilled) post-BM transplantation. A normal level of platelets and RBCs in the periphery is indicated by a horizontal dash level. Data are presented as mean ± SD, n= 5 mice per group. **, P<0.01, compared in PepJ recipients receiving WT vs. KO donor BM cells.
Loss of IEX-1 shortens RBC lifespan
The conflicting finding that successful BM engraftment concurred with decreased RBC and platelet counts in recipients of KO BM cells prompted us to investigate a potential role for IEX-1 in erythropoiesis and thrombopoiesis. We first recorded the kinetics of RBC turnover in KO and WT mice following in vivo biotin labeling of RBC populations. The biotin-labeled RBCs were tracked with a flourescein-avidin marker in blood samples collected weekly for six weeks. Flow cytometric quantification of the biotin-labeled cells remaining in circulation revealed that absence of IEX-1 shortened the mean half-life of KO RBCs to eight days from the 15 days normally seen in WT RBCs (Figure 4A). A similarly fast turnover of IEX-1 KO RBCs relative to WT RBCs was also observed when biotin-labeled RBCs were administered into WT mice (data not shown), concluding an intrinsic defect in KO RBCs. We next determined whether early uptake of RBCs by macrophages was responsible for the fast RBC turnover by examining CD47 expression on RBCs, a common self-marker of erythrocytes whose aging/senescence induced loss results in macrophage clearance.23 It was found that CD47 expression was significantly lower on KO RBCs than WT RBCs (Figure 4B and C). The observation suggests that lack of IEX-1 alters RBC maturation and reduces CD47 expression on RBCs, resulting in their early clearance by macrophages.
Figure 4.
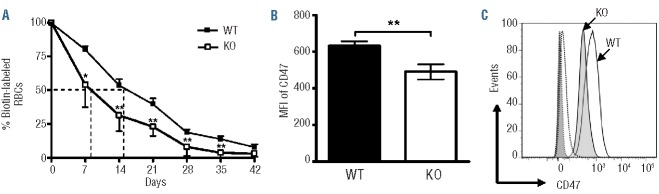
Abnormal RBCs in IEX-1 KO mice. (A) Fast turnover of IEX-1 KO RBCs. WT and KO mice were administered a biotin labeling agent, after which blood samples were obtained at indicated days and incubated with flourescein-labeled avidin and phycoerythrin-conjugated TER119 antibody, followed by flow cytometry to identify biotin+ RBCs. The sample collected 2 h after biotin labeling is arbitrarily set as 100% and a half-life of labeled RBCs is indicated with dashed lines. (B and C) Reduced CD47 expression on RBCs lacking IEX-1. The mean fluorescence intensity (MFI) of CD47 was analyzed with flow cytometry following staining with anti-CD47 and TER-119 antibodies (B). Histogram of CD47 expression was attained on gated TER-119+ cells as illustrated in (C). *and **, P<0.05 or 0.01, respectively, in the presence or absence of IEX-1 and n = 6 mice in each group.
Defective platelet formation in IEX-1 KO mice
We then investigated a possible role for IEX-1 in megakaryocyte and platelet production. Megakaryocytes were quantified in BM using a megakaryocyte specific marker CD41 and no significant difference was found between WT and KO mice (Figure 5A). There was no difference either in the platelet life-span in the absence or presence of IEX-1 when assessed with in vivo biotinylation assays (Online Supplementary Figure S2). However, when the same number of megakaryocytes prepared from KO or WT mice were fluorescently labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE), and transferred intravenously into WT mice, KO megakaryocytes produced significantly fewer platelets than WT megakaryocytes, especially during peaking time points of platelet shedding, as measured by percentages of CFSE+ platelets in the blood of WT recipients during the experimental course of ten days (Figure 5B).
Figure 5.
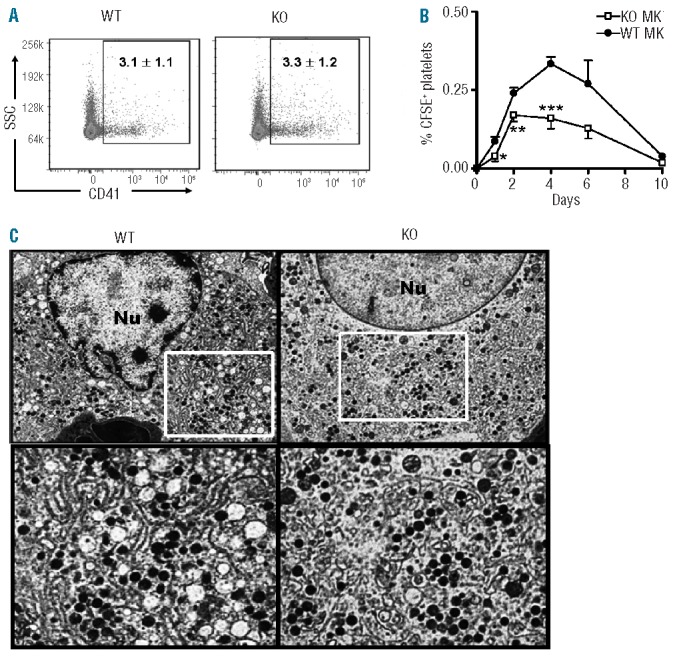
Functional and morphological abnormalities in KO megakaryocytes. (A) Representative FACS plots of CD41+ megakaryocytes prepared from 8 week-old WT and IEX-1 KO BM cells. (B) Insufficient production of platelets from IEX-1 KO megakaryocytes. CFSE-stained megakaryocytes of indicated genotypes were transferred into WT cognate mice at 5×105 cells/mouse. The resultant platelet production was evaluated at indicated days and expressed as percentages ± SD of CFSE+ platelets relative to total platelets in circulation. *, **, and ***, P<0.05, 0.01 or 0.001, respectively, in the presence or absence of IEX-1. Data in A and B were collected from 2 independent experiments each with 5 mice per group. (C) Transmission electron microscopy (TEM) of megakaryocytes. BM cells were isolated from 8-week old WT and KO mice and subjected to TEM sample processing. Shown are the finely structured DMS of a mature WT megakaryocyte (left) and abnormal DMS of a mature KO megakaryocyte (right). Original magnification 6,000× in the upper panel, in which highlighted areas are enlarged in the bottom panels.
To gain a better understanding of what might be causing a reduction in platelet formation, megakaryocytes isolated from WT and KO mice were prepared for ultrastructural analysis by transmission electron microscopy (TEM). A striking abnormality was identified in the demarcation membrane system (DMS) in KO megakaryocytes compared to WT ones (Figure 5C). DMS are the microtubule dense delineating systems responsible for creating platelet territories in mature megakaryocytes during thrombopoiesis, and defects within this intricate system have been noted in multiple thrombocytopenia-inclusive diseases.24 KO megakaryocytes appeared to have short, dilated DMS patterns giving rise to irregular and fewer well-delineated platelet territories, a phenotype potentially responsible for reduced platelet production.25,26 In contrast, WT megakaryocytes developed a distinct and well-developed microtubule DMS pattern required for effective thrombopoiesis. Notably, despite the aberrant DMS, KO megakaryocytes were able to form platelets, albeit at a lesser efficacy (Figure 5B).
A single dose of non-myeloablative radiation induces MDS in IEX-1 KO mice
The aforementioned perturbation of HSC quiescence, insufficient thrombopoiesis, and accelerated RBC turnover suggest a pre-MDS phenotype of IEX-1 KO mice. The mice may adaptively maintain a physiological level of circulating RBCs and platelets through both intra- and extramedullary compensation mechanisms. However, upon acute stress that requires increasing hematopoiesis, the mice may develop MDS. We thus mimicked a clinical setting to acutely stress the mice with 3 Gy total body irradiation (TBI), a non-myeloablative dose commonly used in multiple cancer treatments in humans. Within the first month post-TBI, both WT and KO mice exhibited a similar decrease in the number of circulating platelets (Figure 6A), after which, however, only WT mice recovered and completely normalized their platelet production to a pre-TBI level in three months post-TBI. KO mice continued a low level of platelet counts and developed irreversible thrombocytopenia with platelet counts less than 300 × 109/L throughout the whole experimental period (Figure 6A). Thrombocytopenia in these KO mice was functionally confirmed by a significant increase in bleeding time measured at every other month starting at two months post-TBI. Figure 6B showed one of the bleeding time studies performed in eight months post-TBI. In addition, KO mice exhibited a progressive decline in RBC counts from six to eight months post-TBI (Figure 6C). Similar to secondary BM transplantation, no differences were detected in white blood cell counts between these two strains of mice in all time points examined (data not shown), again in agreement with a myeloid specific defect in the absence of IEX-1.
Figure 6.

IEX-1 KO mice develop MDS after total body irradiation (TBI). Eight-week-old WT and IEX-1 KO mice were treated with a single low (3Gy) dose of radiation and platelet (A) and RBC (C) counts in circulation were monitored monthly for a total of 8 months post-TBI. Thrombocytopenia in the mice was functionally corroborated by prolonged bleeding times at 8 months post-TBI, in which the time in seconds (s) to the cessation of blood flow was recorded (B). Representative results of increased reticulocytes in IEX-1 KO mice after TBI are shown in comparison with non-radiated mice (D). Reticulocytes containing residual ribosomal RNA were readily recognized by new methylene blue staining, marked with a white star (E). Increased Howell-Jolly bodies (F) and extramedullary hematopoiesis indicated by splenomegaly (G) were demonstrated only in IEX-1 KO mice post-TBI. Granulocytic dysplasia in irradiated IEX-1 KO mice was shown by a hyposegmented neutrophil with clumped chromatin (Pseudo-Pelger-Huet cell) (H). Symbols represent individual mice and horizontal lines indicate the mean in B, D, F, and G. n= 12 for IEX-1 KO and 8 for WT mice in A and C. Original magnification 100× for E and 200× for H.
Blood smears confirmed an increase of reticulocytes in KO mice but not in WT mice, indicative of a regenerative response to the decrease in RBCs and/or an accelerated release of RBCs from BM (Figure 6D). The increased release of reticulocytes began at six months post-TBI and continued thereafter, concurrent with a decrease in RBC counts in the mice (Figure 6C). Reticulocytes are immature, polychromatophilic RBCs visible by new methylene blue staining (Figure 6E). In addition, the frequency of Howell-Jolly bodies in peripheral blood rose alongside splenomegaly only in KO mice post-irradiation (Figure 6F and G). Howell-Jolly bodies are basophilic DNA nuclear remnants within RBCs and are commonly seen in an increased number in patients with MDS, various anemias and other MDS murine models.27 Myeloid dysplasia was also noticed in the peripheral blood in a majority of irradiated KO mice, presented as Pseudo-Pelger-Huet cells (Figure 6H). Pseudo-Pelger-Huet cells are characterized by hyposegmentation of granulocyte nuclei with coarsely clumped chromatin, and are considered to be important characteristics of myelodysplasia.28
Abnormalities in the BM and spleen of irradiated KO mice
Within the BM, total myeloid blast counts were comparable in KO and WT mice regardless of whether or not the mice were irradiated (Online Supplementary Figure S3A). However, the number of macrophages was significantly elevated within both the splenic and BM compartments (Figure 7A). One possibility for this phenomenon could include the need for increased phagocytic activity in environments of pathologically increased cell turnover, such as increased intramedullary apoptosis characteristics of MDS.29 No significant myelofibrosis was noted in the BM of either WT or KO mice (data not shown).
Figure 7.

LSK exhaustion and compromised progenitor function in IEX-1 KO mice after TBI. (A) An increase in the proportion of macrophages (MØ) marked as F4/80+ cells in the BM and spleen of IEX-1 KO mice compared to WT mice after eight months of TBI. (B) A marked decrease in the percentages of LSK cells in KO mice in comparison with WT mice after eight months of TBI as analyzed by flow cytometry as Figure 1A. (C) Increased cell cycling of KO LSK cells determined similarly as Figure 1B. (D) A decreased LSK pool in IEX-1 KO BM after TBI. Relative percentages of long- and short-term LSK cells as a total LSK cell population were analyzed in irradiated WT and KO BM on the basis of CD135 and CD34 expression as detailed in Methods. (E) Increased erythroblasts within the BM and spleen of irradiated KO mice, quantified as percentages ± SD of TER119+/CD71+ cells relative to a total TER119+ cells. (F) Colony forming assays of BM cells isolated from radiated KO (unfilled) and WT (filled) mice. Shown are the colony numbers of mixed multi-lineage (GEMM), granulocyte-monocyte (GM), and erythroid (E) progenitors per 2×104 BM cells. N=6 for each culture condition. (G) IEX-1 KO megakaryocytes increase in number in KO BM compared to WT BM controls after TBI. Each symbol represents data from individual mice and horizontal lines indicate the mean in (G). *, ** and ***, P<0.05, 0.01 or 0.001, respectively, in the presence versus absence of IEX-1, and n= 12 for IEX-1 KO mice or 8 for WT mice for all studies except for colony forming cultures (F).
The percentage of total LSK cells was reduced remarkably in the absence versus the presence of IEX-1 in eight months post-TBI (Figure 7B). These KO LSK cells were found exiting quiescence in a significantly higher number than WT LSK cells (Figure 7C), resulting in a significant decline in long-term LSK population with a concomitant increase in short-term LSK cells in irradiated KO mice, a finding previously unseen in Control KO mice (Figure 7D). In spite of increased cycling, CMP, MEP, and CLP populations did not differ significantly in percentages or morphology (Online Supplementary Figure S3B and data not shown), confirming no overt defect in the differentiation of HSCs into these progenitors in the absence of IEX-1. There were no differences either in the counts of basophilic, polychromatic or orthochromatic erythroblasts in the presence or absence of IEX-1, prior to or after irradiation (data not shown).
Yet, proportions of TER119+/CD71+ erythroblastic cells were significantly higher in KO BM than in WT BM (Figure 7E). The erythroid dysplasia was further suggested by the appearance of ringed sideroblasts in a majority of bone marrow brushings prepared from irradiated KO mice (Figure 8A). These ringed sideroblasts were completely lacking in non-irradiated KO BM, although coarse iron deposits were presented around erythroblasts in Control mice (Figure 8A). Neither iron deposits nor sideroblasts were presented in BM brushings prepared from WT mice irrespective of radiation (Figure 8A). The BM erythroid dysplasia occurred in parallel to increasing extramedullary erythropoiesis, with a relatively higher level of CD71+TER119+ erythroblasts in irradiated KO spleens than WT counterparts (Figure 7E). Moreover, internuclear bridging and binucleated erythroid precursors were readily seen in histopathology studies of irradiated KO spleens but not in irradiated WT spleens (Figure 8C). These are typical histopathological features of erythroid dysplasia in association with refractory anemia, a syndrome of MDS.30 To corroborate whether the impairment resulted from an intrinsic defect in HSCs lacking IEX-1, BM cells were isolated from irradiated WT and KO mice and differentiated into myeloid progenitors in hematopoietic colony-forming culture (Figure 7F). BM cells of irradiated KO mice showed a significant decrease in the ability to form multi-potential myeloid and erythroid colonies compared to WT counterparts (Figure 7F). Taken together, these results suggest increased erythropoiesis within both BM and spleens in an attempt to compensate insufficient erythropoiesis of IEX-1 KO HSCs in irradiated mice.
Figure 8.

Histopathological examination of BM and spleens after TBI. BM brushings prepared from WT and KO mice without radiation (none) and eight months post-radiation (irradiated) were stained with an HT20 Iron staining kit (Sigma) (A). Ringed sideroblasts were seen only in irradiated KO BM (yellow triangles, bottom right) and coarse iron deposits in non-irradiated KO BM (green triangles, upper right) in (A). Moreover, internuclear bridges (yellow arrows) and binucleated erythroid precursors (white stars) were readily recognized in H&E staining of the spleen prepared from KO mice compared to WT spleens in eight months post-TBI (C). One of the internuclear bridges is enlarged in the middle and binucleated erythroid precursors in the white rectangle are magnified in the lower right corner in C (green arrow). H&E staining of sections prepared from WT and IEX-1 KO BM showed increased megakaryocytes in irradiated IEX-1 KO mice (right bottom) compared to non-irradiated mice (right upper) or non-irradiated or irradiated WT controls (left panels) (B). Megakaryocytes are marked by filled triangles and original magnification 20X.
Besides erythropoiesis, we observed a significantly higher number of CD41+ megakaryocytes sequestered in the BM of KO mice than of WT mice post-TBI (Figure 7G). The increment in the number of megakaryocytes in KO BM was also readily recognized in a histological study in which megakaryocytes were stained relatively light and larger than surrounding cells (Figure 8B). The number of megakaryocytes was significantly greater in irradiated KO BM than in non-irradiated KO Controls or in WT BM with or without irradiation (Figure 8B). These BM sequestered megakaryocytes appeared to undergo endomitosis normally on the basis of ploidy distribution (Online Supplementary Figure S4). The increased number of megakaryocytes within the BM of irradiated KO mice may be ascribed to a positive feedback regulation to counteract thrombocytopenia in the mice. There was no increase in megakaryocytes within the spleen (Online Supplementary Figure S5), in contrast to extramedullary erythropoiesis, which explains a more severe decline in the number of circulating platelets than RBCs in irradiated KO mice (Figure 6A and C).
Discussion
IEX-1 KO HSCs undergo cell cycling and apoptosis at a level greater than WT HSCs, an abnormality frequently described in patients with early MDS.20 The KO mice also produce RBCs with a shortened life-span and platelets at a relatively lower level. The insufficiency in maintenance of RBC and platelet counts appears to be well compensated by extra- and intra-medullary erythropoiesis and intramedullary thrombopoiesis under a steady condition. Yet, upon acute stress, shown here through BM transplantation and radiation, IEX-1 KO mice developed irreversible thrombocytopenia, a trend towards anemia, increased RBC inclusions, splenomegaly, and granulocyte dysplasia reminiscent of MDS. Moreover, a severe reduction in LSK cell reservoir, particularly long-term LSK cells, was identified after eight months of radiation, presumably ascribed to acceleration of HSC cycling and differentiation in an attempt to counteract the insufficient formation of platelets and RBCs in the stressed mice. These findings argue strongly that diminished IEX-1 expression in patients at early stage MDS is unlikely to be a bystander event, but rather a significant contributing factor to MDS pathogenesis, although further investigation is required to validate the findings in humans.
The most important finding of this investigation is that null mutation of IEX-1 not only perturbs HSC homeostasis but also impairs both thrombopoiesis and erythropoiesis. This finding is consistent with an indispensable role for IEX-1 in the regulation of the mitochondrial respiratory chain activity, which is a key evolutionarily conserved cellular function. There is considerable evidence to suggest that a mutation affecting a key conserved cellular function can perturb multiple differentiation steps involved in hematopoiesis, leading to MDS development.31 For instance, mice receiving BM cells containing a N-terminal missense mutation of runt-related transcription factor1 (RUNX1) develop an-MDS-like phenotype.32 RUNX1 (also called AML1) is the most frequently mutated gene in AML patients and encodes for a transcription factor essential for HSC differentiation. Several genes that regulate ribosomal biogenesis are also implicated in the pathogenesis of MDS, which are located within chromosome 5q31–q32 region that is deleted in 5q-MDS.33 More recently, an Xist-deficient mouse model demonstrated myeloid maturation defects followed by the age-dependent loss of LT-LSK cells, giving rise to an MDS-like syndrome.34 The Xist RNA is required to trigger X chromosome inactivation so as to equalize expression of a group of genes between the sexes. Similarly, transcriptional RNA splicing abnormalities and epigenetic changes are found to associate with acceleration of HSC exhaustion and MDS pathogenesis.33 Given the importance of mitochondrial respiratory chain in a variety of cellular functions,8 it is not surprising that lack of IEX-1 impairs several key differentiation processes in hematopoiesis and perturbs HSC quiescence, predisposing to MDS upon stress.
Although IEX-1 mutations or re-arrangements are rarely detected in MDS patients, IEX-1 expression is diminished in CD34+ stem cells in a large portion of early stage/low-risk MDS patients, concurrent with high apoptotic rates in the cells.14 Severely diminished IEX-1 expression was correlated with a shorter survival time of the patients.15,16 IEX-1 expression can be potentially down-regulated in MDS patients by several mechanisms, one of which would be hypermethylation of IEX-1 promoter, considering well-documented DNA hypermethylation in MDS.35 Secondly, IEX-1 transcription is regulated by ERK-dependent phosphorylation of AML1. Point mutations that impair AML1 function are frequently found in MDS patients and can affect IEX-1 transcription, as multiple consensus-binding sites for AML1 have been identified in IEX-1 promoter.36 Finally, IEX-1 is co-ordinately controlled by a dozen of transcriptional factors including NF-κB, p53, c-Myc, Sp1, p300, Ap1, etc., and alteration in any of these transcriptional factors caused by either DNA hypermethylation or mutations could adversely affect IEX-1 expression.7
Another important finding of the investigation is that a pre-MDS phenotype of IEX-1 KO mice is not detectable by merely examining the numbers of circulating RBCs and platelets, due to extra- and intra-medullary compensation mechanisms. Moreover, null mutation of IEX-1 does not fully abrogate, but reduces the differentiation of platelets and RBCs. Seeking similar phenotypes in MDS patients, such as increased RBC turnover or ineffective platelet formation, may lead to earlier clinical diagnosis and prevention or delay of an onset of the disease. Finally, the study suggests that targeting multiple differentiation steps may be required to cure the disease.
Supplementary Material
Acknowledgments
The authors would like to thank Margaret Sherwood, Jenny Zhao and Danny Cao for their help in the microscopy and flow cytometry during this project, and the the MGH-Center for Comparative Medicine Clinical Pathology Laboratory for assistance with hematology analyses.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work is supported by the National Institutes of Health Grants CA158756, AI089779, and DA028378 to MW and HL097748 to CPL.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Nimer SD. MDS: a stem cell disorder--but what exactly is wrong with the primitive hematopoietic cells in this disease? Hematology Am Soc Hematol Educ Program. 2008;43–51 [DOI] [PubMed] [Google Scholar]
- 2.Pang WW, Pluvinage JV, Price EA, Sridhar K, Arber DA, Greenberg PL, et al. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc Natl Acad Sci USA. 2013;110(8):3011–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lane SW, Sykes SM, Al Shahrour F, Shterental S, Paktinat M, Lo CC, et al. The Apc(min) mouse has altered hematopoietic stem cell function and provides a model for MPD/MDS. Blood. 2010;115(17):3489–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van de Loosdrecht AA, Brada SJ, Blom NR, Hendriks DW, Smit JW, van den BE, et al. Mitochondrial disruption and limited apoptosis of erythroblasts are associated with high risk myelodysplasia. An ultrastructural analysis. Leuk Res. 2001;25(5):385–93 [DOI] [PubMed] [Google Scholar]
- 5.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454 (7201):232–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watson AS, Mortensen M, Simon AK. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle. 2011;10(11): 1719–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu MX. Roles of the stress-induced gene IEX-1 in regulation of cell death and oncogenesis. Apoptosis. 2003;8(1):11–8 [DOI] [PubMed] [Google Scholar]
- 8.Shen L, Zhi L, Hu W, Wu MX. IEX-1 targets mitochondrial F1Fo-ATPase inhibitor for degradation. Cell Death Differ. 2009;16(4): 603–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, et al. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8(1):13–25 [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Schlossman SF, Edwards RA, Ou CN, Gu J, Wu MX. Impaired apoptosis, extended duration of immune responses, and a lupus-like autoimmune disease in IEX-1-transgenic mice. Proc Natl Acad Sci USA. 2002;99(2):878–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez S, Perez-Perez MM, Hernando E, Serrano M, Cordon-Cardo C. p73beta-Mediated apoptosis requires p57kip2 induction and IEX-1 inhibition. Cancer Res. 2005;65(6):2186–92 [DOI] [PubMed] [Google Scholar]
- 12.Pawlikowska P, Leray I, de Laval B, Guihard S, Kumar R, Rosselli F, et al. ATM-dependent expression of IEX-1 controls nuclear accumulation of Mcl-1 and the DNA damage response. Cell Death Differ. 2010;17(11): 1739–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia J, Ye Y, Arranz V, Letourneux C, Pezeron G, Porteu F. IEX-1: a new ERK substrate involved in both ERK survival activity and ERK activation. EMBO J. 2002;21 (19):5151–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steensma DP, Neiger JD, Porcher JC, Keats JJ, Bergsagel PL, Dennis TR, et al. Rearrangements and amplification of IER3 (IEX-1) represent a novel and recurrent molecular abnormality in myelodysplastic syndromes. Cancer Res. 2009;69(19):7518–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hofmann WK, de Vos S, Komor M, Hoelzer D, Wachsman W, Koeffler HP. Characterization of gene expression of CD34+ cells from normal and myelodys-plastic bone marrow. Blood. 2002;100(10): 3553–60 [DOI] [PubMed] [Google Scholar]
- 16.Prall WC, Czibere A, Grall F, Spentzos D, Steidl U, Giagounidis AA, et al. Differential gene expression of bone marrow-derived CD34+ cells is associated with survival of patients suffering from myelodysplastic syndrome. Int J Hematol. 2009;89(2):173–87 [DOI] [PubMed] [Google Scholar]
- 17.Shahid M, Shen L, Seldin DC, Lu B, Ustyugova IV, Chen X, et al. Impaired 3′,5′-Cyclic Adenosine Monophosphate-Mediated Signaling in Immediate Early Responsive Gene X-1-Deficient Vascular Smooth Muscle Cells. Hypertension. 2010; 56(4):705–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sommer SL, Berndt TJ, Frank E, Patel JB, Redfield MM, Dong X, et al. Elevated blood pressure and cardiac hypertrophy after ablation of the gly96/IEX-1 gene. J Appl Physiol. 2006;100(2):707–16 [DOI] [PubMed] [Google Scholar]
- 19.Parker JE, Mufti GJ. The role of apoptosis in the pathogenesis of the myelodysplastic syndromes. Int J Hematol. 2001;73(4):416–28 [DOI] [PubMed] [Google Scholar]
- 20.Lin CW, Manshouri T, Jilani I, Neuberg D, Patel K, Kantarjian H, et al. Proliferation and apoptosis in acute and chronic leukemias and myelodysplastic syndrome. Leuk Res. 2002;26(6):551–9 [DOI] [PubMed] [Google Scholar]
- 21.Lo CC, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, et al. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457(7225):92–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10(8):858–64 [DOI] [PubMed] [Google Scholar]
- 23.Olsson M, Oldenborg PA. CD47 on experimentally senescent murine RBCs inhibits phagocytosis following Fcgamma receptor-mediated but not scavenger receptor-mediated recognition by macrophages. Blood. 2008;112(10):4259–67 [DOI] [PubMed] [Google Scholar]
- 24.Strassel C, Eckly A, Leon C, Petitjean C, Freund M, Cazenave JP, et al. Intrinsic impaired proplatelet formation and micro-tubule coil assembly of megakaryocytes in a mouse model of Bernard-Soulier syndrome. Haematologica. 2009;94(6):800–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckly A, Strassel C, Freund M, Cazenave JP, Lanza F, Gachet C, et al. Abnormal megakaryocyte morphology and proplatelet formation in mice with megakaryocyte-restricted MYH9 inactivation. Blood. 2009; 113(14):3182–9 [DOI] [PubMed] [Google Scholar]
- 26.Strassel C, Nonne C, Eckly A, David T, Leon C, Freund M, et al. Decreased thrombotic tendency in mouse models of the Bernard-Soulier syndrome. Arterioscler Thromb Vasc Biol. 2007;27(1):241–7 [DOI] [PubMed] [Google Scholar]
- 27.Putz G, Rosner A, Nuesslein I, Schmitz N, Buchholz F. AML1 deletion in adult mice causes splenomegaly and lymphomas. Oncogene. 2006;25(6):929–39 [DOI] [PubMed] [Google Scholar]
- 28.Swerdlow SH. WHO Classification of tumors of haematopoietic and lymphoid tissues. 4 ed Lyon; IARC Press, 2008 [Google Scholar]
- 29.Titius BR, Thiele J, Schaefer H, Kreipe H, Fischer R. Ki-S1 and proliferating cell nuclear antigen expression of bone marrow macrophages. Immunohistochemical and morphometric study including reactive (inflammatory) myelitis, secondary aplastic anemia, AIDS, myelodysplastic syndromes and primary (idiopathic) osteomyelofibrosis. Acta Haematol. 1994;91(3):144–9 [DOI] [PubMed] [Google Scholar]
- 30.Suttie AW. Histopathology of the spleen. Toxicol Pathol. 2006;34(5):466–503 [DOI] [PubMed] [Google Scholar]
- 31.Slape CI, Saw J, Jowett JB, Aplan PD, Strasser A, Jane SM, et al. Inhibition of apoptosis by BCL2 prevents leukemic transformation of a murine myelodysplastic syndrome. Blood. 2012;120(12):2475–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe-Okochi N, Kitaura J, Ono R, Harada H, Harada Y, Komeno Y, et al. AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood. 2008;111(8): 4297–308 [DOI] [PubMed] [Google Scholar]
- 33.Lindsley RC, Ebert BL. Molecular pathophysiology of myelodysplastic syndromes. Annu Rev Pathol. 2013;8:21–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, et al. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. 2013;152(4):727–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rush LJ, Plass C. Alterations of DNA methylation in hematologic malignancies. Cancer Lett. 2002;185(1):1–12 [DOI] [PubMed] [Google Scholar]
- 36.Hamelin V, Letourneux C, Romeo PH, Porteu F, Gaudry M. Thrombopoietin regulates IEX-1 gene expression through ERK-induced AML1 phosphorylation. Blood. 2006;107(8):3106–13 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.