

Abstract
Myeloid leukemia of Down syndrome has a better prognosis than sporadic pediatric acute myeloid leukemia. Most cases of myeloid leukemia of Down syndrome are characterized by additional cytogenetic changes besides the constitutional trisomy 21, but their potential prognostic impact is not known. We, therefore, conducted an international retrospective study of clinical characteristics, cytogenetics, treatment, and outcome of 451 children with myeloid leukemia of Down syndrome. All karyotypes were centrally reviewed before assigning patients to subgroups. The overall 7-year event-free survival for the entire cohort was 78% (±2%), with the overall survival rate being 79% (±2%), the cumulative incidence of relapse 12% (±2%), and the cumulative incidence of toxic death 7% (±1%). Outcome estimates showed large differences across the different cytogenetic subgroups. Based on the cumulative incidence of relapse, we could risk-stratify patients into two groups: cases with a normal karyotype (n=103) with a higher cumulative incidence of relapse (21%±4%) than cases with an aberrant karyotype (n=255) with a cumulative incidence of relapse of 9% (±2%) (P=0.004). Multivariate analyses revealed that white blood cell count ≥20×109/L and age >3 years were independent predictors for poor event-free survival, while normal karyotype independently predicted inferior overall survival, event-free survival, and relapse-free survival. In conclusion, this study showed large differences in outcome within patients with myeloid leukemia of Down syndrome and identified novel prognostic groups that predicted clinical outcome and hence may be used for stratification in future treatment protocols.
Introduction
Children with Down syndrome (DS) have an increased risk of developing leukemia, including acute myeloid leukemia (AML) and acute lymphoblastic leukemia.1,2 These children develop a unique type of AML referred to as myeloid leukemia of Down Syndrome (ML-DS), which is recognized as a separate entity in the new World Health Organization classification of leukemias.3 ML-DS is characterized by a low diagnostic white blood cell (WBC) count, myelofibrosis with a low number of leukemic blasts in the marrow,3 mostly French-American-British (FAB) M7 morphology, young age at diagnosis (it occurs almost exclusively in children <5 years old), and superior clinical outcome when treated with reduced intensity chemotherapy protocols without stem cell transplantation.4–10 ML-DS patients have an increased risk of side effects, hence there is a delicate balance between anti-leukemic efficacy and treatment-related toxicity. Drug resistance profiles showed that ML-DS blasts are particularly sensitive to various chemotherapeutic drugs in vivo and in vitro,11,12 which enables dose reduction.
Somatic mutations in the gene encoding for the transcription factor GATA1, localized on the X chromosome (Xp11.2), are pathognomonic for ML-DS.13,14 This transcription factor regulates the differentiation of megakaryocytes and erythrocytes. Mutations mainly occur in exon 2 and lead to the truncated protein GATA1s, and are unique to each patient.15,16
Age has been recognized as a prognostic factor in ML-DS, with an inferior outcome in the limited number of children aged over 4 years.17 In fact, it has been proposed that DS children who present over 4 years of age are in fact suffering from sporadic AML occurring in a child with DS, rather than from ‘true’ ML-DS.18 In addition, ML-DS patients with a history of transient myeloproliferative disease have a significantly better outcome than children with ML-DS without documented transient myeloproliferative disease.19 Until now, no other prognostic factors have been identified in ML-DS.
The leukemic blasts from the majority of patients with ML-DS (72%) show additional cytogenetic changes apart from the constitutional trisomy 21.20 A previous international-BFM study, performed by Forestier et al., showed that the most frequent gains involved chromosomes 8 (27%), 21 (23%), 11 (8.1%), and 19 (7.4%), whereas chromosomes X (3.2%; only females), 5 (1.5%), and 7 (2.2%) were commonly monosomic. The most frequent partial imbalances were duplication 1q (16%), deletion 7p (10%), and deletion 16q (7.4%).20 However, the potential clinical impact of these cytogenetic abnormalities is not known and has not been well studied, mainly due to the small numbers of patients in individual series.9,10,20–22
In current treatment protocols of non-DS pediatric AML patients, stratification is based on cytogenetics and response to therapy.23 In ML-DS, no prognostic cytogenetic groups have yet been identified, nor any other prognostic factors allowing a risk-stratified approach.
We, therefore, conducted a large international study of clinical and outcome data including cytogenetic records from children with ML-DS collected from 13 collaborative study groups. Our aim was to identify differences in outcome related to cytogenetics and clinical characteristics in ML-DS. This was approached by analyzing differences in the cumulative incidence of relapse (CIR), reflecting leukemia resistance, and hence avoiding the influence of toxic (non-leukemic) events on survival estimates. This may result in risk-group stratification and risk-group directed therapy for these patients in the future. In addition, we compared the outcome of ML-DS patients in the different cytogenetic groups with that of non-DS AML patients from the same era treated on AML-BFM regimens as a reference cohort.
Methods
Patients
Data on 451 patients with ML-DS were collected from 13 collaborative study groups participating in the International AML-BFM Study Group. For comparison, a reference cohort of non-DS AML patients (n=543) from the same treatment era, kindly provided by the AML-BFM Study Group, was used. This study was approved by the Institutional Review Boards in accordance with local legislation and guidelines.
Patients were eligible if diagnosed between January 1, 1995 and January 1, 2005. Patients who were not treated with curative intent from diagnosis were excluded. The data collected at diagnosis comprised karyotype, sex, age, white blood cell (WBC) count, hemoglobin level, platelet count, immunophenotypic data and FAB morphology. In addition, we collected data on treatment, such as therapy protocol, including stem-cell transplantation, and all events during follow-up. Only patients between 6 months and up to 5 years of age were included in the analyses. Patients with transient myeloproliferative disease were excluded. Patients were treated in national or collaborative group AML trials.
Cytogenetic results
All karyotypes were provided after review by a national collaborative group, and centrally reviewed by two cytogeneticists (EF, BJ). Fluorescence in situ hybridization analyses were not performed routinely. Of the 451 cases, karyotypes were available for 358 (79%). As there was no a priori knowledge on the prognostic impact of the various cytogenetic groups in ML-DS, the classification of the cases was based on the premise that all groups should be mutually exclusive, i.e. each patient was included only once. Only groups that were sufficiently large (≥5 cases) were analyzed in more detail to allow meaningful statistical analyses.
The numerically largest group was formed of 103 patients (29%) with a normal karyotype (NK). Another entity that was readily delineated consisted of 49 cases with trisomy 8 (14% of all cases), either as a single abnormality (n=16), or with additional cytogenetic aberrations (n=33). Next, a group of 82 cases (23%) with losses of chromosome 5/7 material (excluding those with +21) was distinguished. Other smaller groups consisted of 28 cases (6%) with a gain of chromosome 21 (in addition to +21c); 14 cases (4%) with a duplication of chromosome 1q; and 9 cases (3%) with a deletion of chromosome 16q. Finally, a group of 73 cases (20%) remained, harboring other aberrations that could not be sub-categorized further (Figure 1 and Online Supplementary Figure S1).
Figure 1.
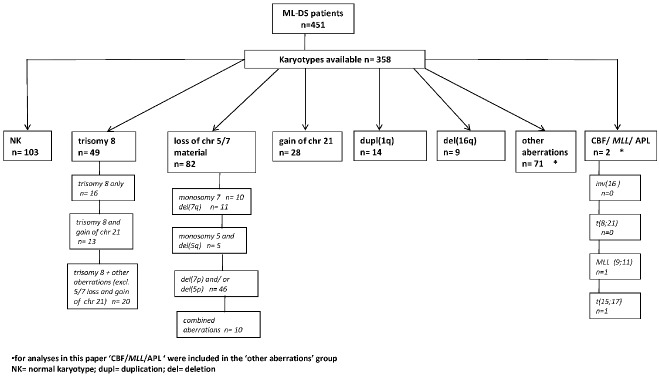
Hierarchy of cytogenetic groups within ML-DS delineated in the present study.
Statistical analyses
Continuous variables were categorized according to cut-off points; age < or ≥3 years, WBC count < or ≥20×109 and Ara-C < or ≥20,000 mg/m2. The χ2 or Fisher exact test was used to compare discrete variables among groups; the Mann-Whitney U test was used for continuous variables. All P values are descriptive and explorative, and were considered statistically significant if ≤0.05. Statistical analyses were performed using SAS software (SAS-PC, Version 9.1).
More details on the methods are provided in the Online Supplementary Material.
Results
Clinical characteristics
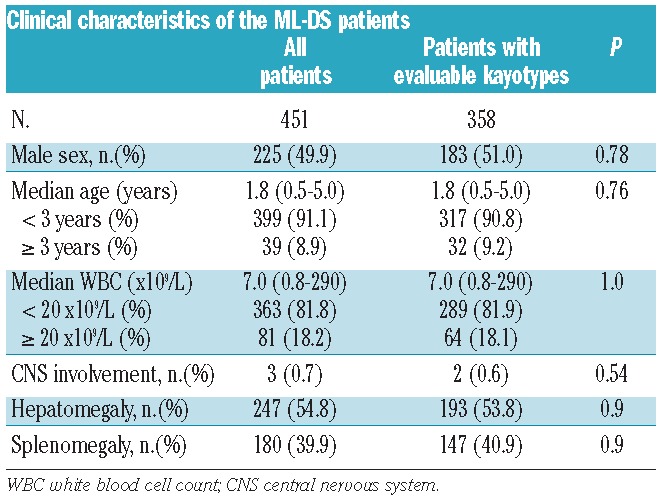
The median age of all ML-DS patients (n= 451) was 1.8 years (range, 6 months − 5.0 years) and the median WBC count was 7.0×109/L (range 0.8 – 290×109/L). The male -female distribution was almost equal (49.9% versus 50.1%). Only two (0.5%) patients had central nervous system involvement. The characteristics of the entire cohort of patients are presented in detail in Table 1.
Table 1.
Clinical characteristics of the ML-DS patients.
The median follow-up of survivors was 4.9 years. Forty-three percent (192 patients) received therapy reduction, or were treated with adjusted DS treatment protocols. Outcome parameters did not differ significantly between these groups. Six patients were also treated with irradiation: three patients received central nervous system irradiation, whereas the radiation target was not specified for the three other patients.
Ninety-two percent of all patients reached complete remission. The 7-year event-free and overall survival rates of all included 451 patients were 78% (± 2%) and 79% (± 2%), respectively. The 7-year CIR was 12% (± 2%), and cumulative incidence of toxic death was 7% (± 1%) (Figure 2). Of all patients with evaluable karyotypes (n=358), the complete remission rate was 92% and the 7-year event-free and overall survival rates were 77% (± 2%) and 79% (± 2%), respectively. The 7-year CIR was 13% (± 2%), and the cumulative incidence of toxic death was 7% (±1%) (Figure 3). There were no statistically significant differences between these two groups when comparing various outcome estimates. We, therefore, conclude that there was no selection bias between the entire study population and the subgroup with informative karyotypes.
Figure 2.
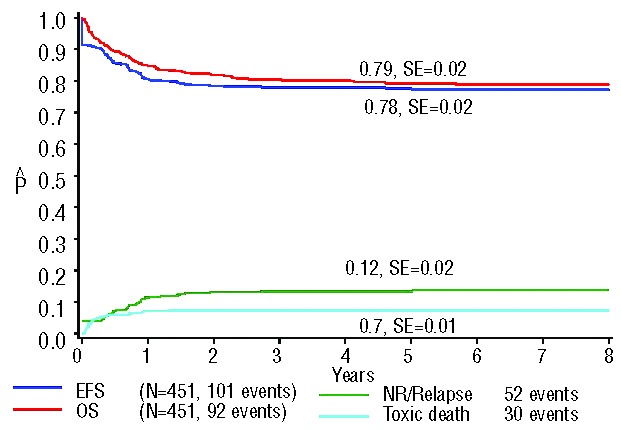
Survival curves of all 451 ML-DS patients included in this study. The 7-year overall survival (OS) was 79% (±2%); the 7-year event-free survival (EFS) 78% (±2%); the 7-year cumulative incidence of relapse was 12% (±2%); and the cumulative incidence of toxic death at 1.5 years from diagnosis was 7% (±1%). NR: non-remitters.
Figure 3.
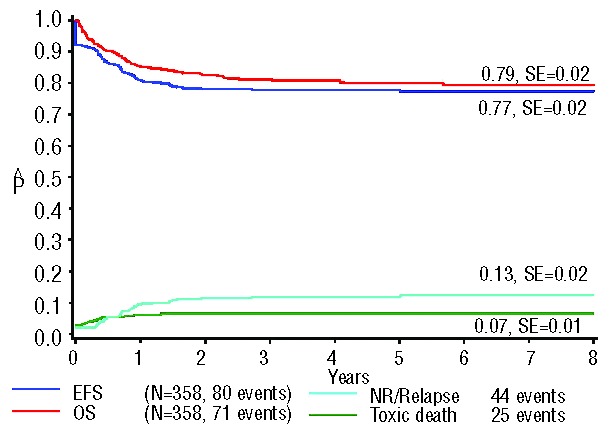
Survival curves of the 358 ML-DS patients with informative karyotypes. The 7-year overall survival (OS) was 79% (±2%); the 7-year event-free survival (EFS) 77% (±2%); the 7-year cumulative incidence of relapse was 13% (±2%); and the cumulative incidence of toxic death at 1.5 years from diagnosis was 7% (±1%). NR: non-remitters.
In total 25 (5.5%) patients were transplanted in first complete remission. One patient underwent autologous stem cell transplantation, three patients were transplanted with a graft from an allogeneic HLA sibling, and three patients received a matched family donor transplant; these specifications were not known for any of the other patients. Forty percent of all transplanted patients died (10/25), half of them due to the leukemia.
Outcome of cytogenetic subgroups
There were no significant differences in the frequency distribution of the various cytogenetic subgroups between the collaborative groups apart from the French cohort, which consisted of a relatively large proportion of NK ML-DS cases. This, however, did not influence the outcome estimates significantly, so there was no study group effect in the overall results.
Interestingly, outcome estimates differed largely across the different cytogenetic subgroups (Figure 4). An overview of all outcome estimates per subgroup is given in Table 2.
Figure 4.

Survival curves for ML-DS patients (n=358) based on their cytogenetic status. (A) Event-free survival (EFS). (B) Overall survival (OS). (C) Cumulative incidence (CI) of relapse. (D) Cumulative incidence of toxic death. Assignment to groups was based on cytogenetic status, as identified after central review.
Table 2.
Survival estimates per cytogenetic subgroup.
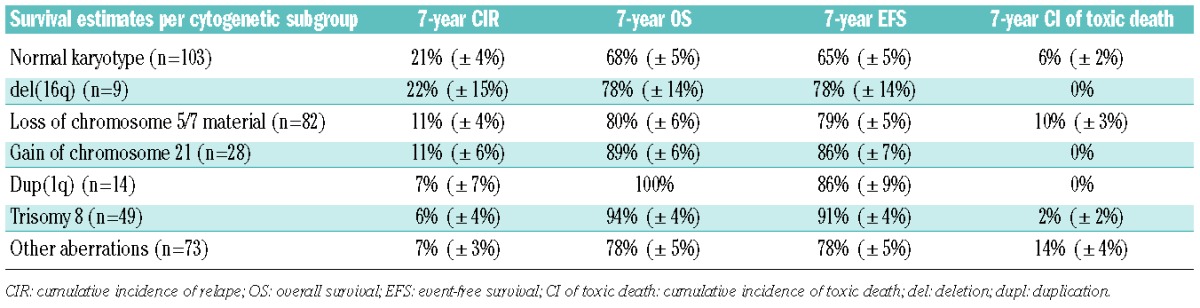
Based on the CIR estimates, patients could be divided into groups with a high CIR (> 20%), comprising those with NK and del(16q) (n=112), and a low CIR (< 20%), comprising all other patients (n=246). Since the former group consisted of NK cases (92%), with only nine cases with del(16q) with two events, we decided to perform further analyses comparing the NK cases (29%) with all cases with aberrant karyotypes (71%). Clinical characteristics did not differ between these two groups (Table 3). The rate of complete remission was significantly lower in NK ML-DS than in cases with an aberrant karyotype (87% versus 96%; P<0.01). The NK patients had significantly worse survival outcomes: 7-year CIR of 21% (± 4%) versus 9% (± 2%) (P=0.004), 7-year overall survival of 68% (± 5%) versus 84% (± 2%) (P=0.0008), and 7-year event-free survival of 65% (± 2%) versus 82% (± 5%) (P=0.0005). The cumulative incidence of toxic death was not significantly different between NK ML-DS and patients with aberrant karyotypes: 6% (± 2%) versus 7% (± 2%) (P=0.58) (Figure 5). Regarding the rate of complete remission, a significantly small proportion of NK-patients than patients with aberrant karyotypes reached complete remission (87% versus 96%; P< 0.01).
Table 3.
Clinical characteristics of the NK ML-DS vs. all other cases (with aberrant karyotypes).
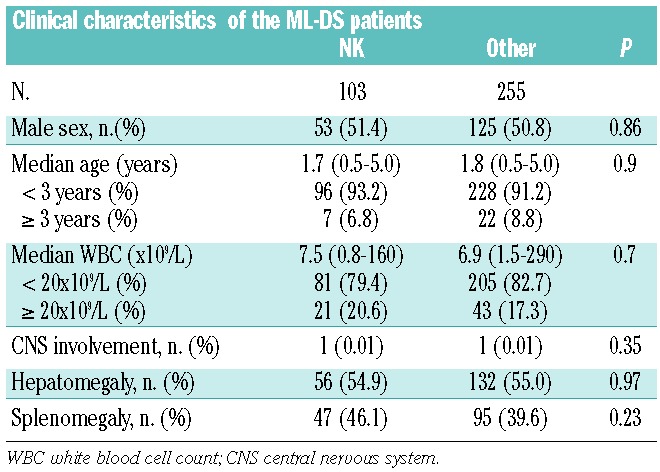
Figure 5.

Survival curves for ML-DS patients (n=358) based on their cytogenetic status, divided into NK ML-DS patients (n=103) versus patients with aberrant karyotypes (n=255). (A) Event-free survival (EFS). (B) Overall survival (OS). (C) Cumulative incidence (CI) of relapse. (D) Cumulative incidence of toxic death. Assignment to groups was based on cytogenetic status, as identified after central review.
Chromosome 7 aberrations
Given the presence of a large number of cases with chromosome 7 abnormalities in our ML-DS cohort and the specific prognostic relevance of chromosome 7 abnormalities in non-DS AML, we focused on this group separately.24 ML-DS patients with chromosome 7 aberrations did not have significantly different survival parameters compared to all other patients (P=0.63). This group was further subdivided into cases with a monosomy 7 (n=10) and those with a del(7q) (n=11). Patients with monosomy 7 tended to have worse survival estimates than patients with a del(7q), but this was not statistically significant: 7-year event-free survival 67% (± 14%) versus 81% (± 10%) (P=0.40), 7-year overall survival 59% (± 17%) versus 88% (± 8%) (P=0.2), and 7-year CIR 9% (± 9%) versus 20% (± 14%) (P=0.36) (Online Supplementary Figure S2).
Regarding the five patients with chromosome 5 aberrations, four of them were alive after at least 4 years of follow up, although one of them suffered from severe infections during treatment. One of them died 2 months after diagnosis due to sepsis in induction; this patient also had a congenital heart defect.
Other prognostic factors
Patients with high WBC counts (≥20×109/L tended to have a worse 7-year event-free survival rate than patients with a lower WBC count (< 20×109/L): 79% (± 2%) versus 70% (± 5%); (P=0.047). However, this did not translate into a significant difference in 7-year overall survival [80% (± 2%) versus 73% (± 5%); P=0.07]. This was due to the occurrence of events in the induction phase; the complete remission rate was significantly lower in patients with high WBC counts (93% versus 81%; P=0.007). The 7-year cumulative incidence of toxic death and CIR did not differ significantly: cumulative incidence of toxic death 6% (± 3%) versus 7% (± 1%) (P=0.83) and CIR 16% (± 4%) versus 10% (± 2%) (P=0.1) (Online Supplementary Figure S3).
In addition, after evaluating various cut-off points for age, patients aged <3 years had significantly better 7-year event-free survival and CIR than had patients aged ≥3 years [event-free survival 78% (± 2%) versus 65% (± 7%) (P=0.04) and CIR 11% (± 2%) versus 21% (± 6%) (P=0.05)] (Online Supplementary Figure S4). This was also due to events in induction, with a higher borderline statistically significant complete remission rate for patients aged <3 years (93% versus 84%; P=0.08). The cumulative incidence of toxic death was not significantly different between these two age groups [7% (± 1%) versus 5% (± 3%)(P=0.58)] nor was the overall survival rate [80% (± 2%) versus 69% (± 7%)(P=0.10)].
Immunophenotyping
ML-DS cases positive for the lymphoid co-expression marker CD7 (n=187/221) had a borderline better event-free survival rate [79% (± 3%) versus 64% (± 8%); P=0.054] (Online Supplementary Figure S5). However, no significant differences were seen for overall survival, CIR or cumulative incidence of toxic death. Expression of CD56 (neural cell adhesion molecule) (n=92/169) was not significantly associated with any of the outcome estimates (Online Supplementary Figure S6), whereas CD34 (expressed on early hematopoietic cells) positive cases (n=94/221) had a worse event-free survival [70% (± 5%) versus 82% (± 3%); P=0.049] and a higher CIR (16 ± 4% versus 7 ± 2%; P=0.04) than CD34-negative cases (Online Supplementary Figure S7).
Treatment
No significant differences in outcome estimates, CIR or cumulative incidence of toxic death were seen between groups treated with different cumulative dosages of anthracyclines and etoposide. Patients treated with higher cumulative dosages of cytarabine (≥20 g/m2) had a significantly better 7-year event-free survival [84% (± 3%) versus 75% (± 3%); P=0.043] and a trend towards a better 7-year overall survival [85% (± 3%) versus 77% (± 3%); P=0.056] than patients treated with lower doses (<20 g/m2) (Online Supplementary Figure S8). There was also a trend for lower 7-year CIR in patients treated with higher doses [7% (± 2%) versus 14% (± 2%); P=0.06]. The cumulative incidence of toxic death was significantly lower in the patients treated with higher cytarabine doses (≥ 20 g/m2) [2% (± 1%) versus 9% (± 2%); P=0.02].
Forty-three percent of all patients received therapy reduction or were treated with adjusted DS treatment protocols. Overall, no differences in outcome estimates were found between patients given reduced therapy and those who received standard therapy.
Multivariate analyses
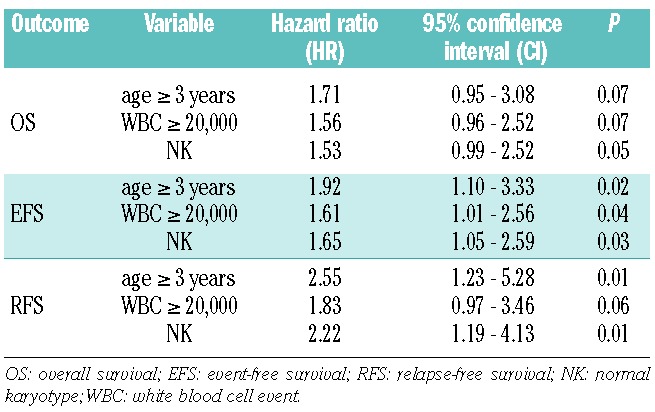
Cox regression analysis of survival estimates from diagnosis revealed both age ≥3 years and WBC counts ≥20×109/L were independent predictors for poor event-free survival (see Table 4), but not for overall survival. In addition, NK independently predicted for poor overall survival [hazard ratio (HR)= 1.53 and P=0.05)], event-free survival (HR= 1.65; P=0.03) and for relapse-free survival (HR= 2.22; P=0.01). Age ≥3 years was also an independent predictor for a lower relapse-free survival with a HR= 2.55 (P=0.01).
Table 4.
Multivariate analysis of survival parameters of survival of ML-DS patients.
Discussion
In this collaborative study we analyzed a large international series of ML-DS cases with the aim of identifying differences in outcome related to cytogenetic features that could enable risk group stratification and risk-adapted therapy for ML-DS patients in the future. The results underscore the importance of international collaboration in the investigation of rare diseases or groups.
It was confirmed that overall outcome for ML-DS was superior to that of AML in non-DS children, with 7-year overall survival and event-free survival rates of 78% and 79%, respectively, in ML-DS, compared to 62% and 50% for non-DS AML patients from the same era treated on AML-BFM regimens as a reference cohort (both P<0.001; see Online Supplementary Figure S9). Of interest, the overall and event-free survival estimates were superimposed in ML-DS, suggesting that most relapsed patients could not be salvaged. However, it is unknown whether these patients were treated with curative intent at relapse. Although there is great concern about toxic mortality in ML-DS, in the present series relapse was more frequent than treatment-related mortality, with cumulative incidences of 12% and 7%, respectively. The relapse frequency of 12% is remarkably low when compared with that in non-DS AML patients from the AML-BFM study group from the same era, who had a CIR of 42% (P<0.001). However, the cumulative incidence of toxic death was similar between DS and non-DS children: 7% and 5%, respectively (P=0.12). The reasonable balance between toxic death and leukemia relapse in ML-DS may be due to the fact that treatment reduction was more frequently applied than in older studies in which higher toxic death rates in ML-DS were reported.6,7,25
Non-random cytogenetic aberrations that are common in non-DS pediatric AML, such as core-binding factor [CBF; t(8;21)], MLL-rearrangements and t(15;17), were identified in single cases only in our ML-DS cohort, which is in line with previous studies.20
The salient finding in the present study was that NK ML-DS patients had poorer survival parameters compared to ML-DS cases with aberrant karyotypes, and that NK independently predicted for poor clinical outcome. NK may, therefore, be used for treatment stratification in future treatment ML-DS protocols. In the NK ML-DS cases, the complete remission rate was significantly lower, and relapse (CIR 21%) determined prognosis to a greater extent than cumulative toxic death (6%). Hence, in this subgroup no further therapy reduction should be applied, whereas until now the increase in survival in ML-DS patients has mainly been achieved through the application of reduced-intensity chemotherapy protocols.4,6,8,9 In fact, treatment intensification may even be needed. In order to reduce the number of induction failures a double induction based, for instance, on day 15 bone marrow blasts may be considered in patients with residual demonstrable leukemia. In addition, detection of GATA1-mutations using real-time quantitative polymerase chain reaction analysis may be feasible as a marker for minimal residual disease in the nearby future,26 but is not routinely used yet. Alternative methods for detecting minimal residual disease include flow cytometry or reverse-transcription polymerase chain reaction for the WT1 gene.27 Increasing the cumulative doses of cytarabine, for example, may be of benefit during consolidation and intensification, as the CIR was lower in patients treated with higher doses. Recently, stem cell transplantation in ML-DS was reviewed but transplant-related mortality (24%) was significantly higher in this setting than in non-DS AML,28 so its use should be limited to patients who do not attain sufficient remission or as salvage therapy at relapse.
Understanding the underlying biology of NK ML-DS may reveal potential new treatment targets. Non-DS pediatric NK AML cases are characterized by various abnormalities, including overexpression of specific genes (MN1, BAALC, and ERG),29 but also single gene mutations such as FLT3-ITD, WT1, NPM1, and CEBPA,30–32 as well as cryptic translocations.33 We recently showed that the abnormalities mentioned above are absent or rare in (NK-) ML-DS.34 Hence, the underlying biology of NK ML-DS needs to be studied in more detail, for example by using novel techniques such as whole genome sequencing.
Non-DS pediatric AML with a trisomy 8 is classified in an intermediate-risk group.35 In the present study, we showed, in a direct comparison, that the outcome estimate of ML-DS patients with trisomy 8 is significantly better that those of non-DS AML patients with trisomy 8 (CIR of 6% versus 62%; P<0.0001) (Online Supplementary Figure S10). Apparently, an additional copy of chromosome 8 has biologically different consequences in ML-DS compared to non-DS AML.
Monosomy 7 is known to be a poor prognostic factor in non-DS pediatric AML, as shown in another international-BFM collaborative study.35 Outcome was significantly worse in patients with a loss of the whole chromosome (monosomy 7) than in patients with a del(7q).24,35 In our ML-DS series, such differences were not observed, but numbers were small. Comparing ML-DS and non-DS AML patients revealed that ML-DS patients with monosomy 7 and/or del(7q) had a remarkably lower CIR (14% versus 52%; P=0.003) (Online Supplementary Figure S11). Thus chromosome 7 aberrations do not seem to have the same implications in ML-DS as in non-DS pediatric AML.
Interestingly, most chromosome 5/7 losses in ML-DS involved the p-arms rather than the q-arms. This is in contrast to non-DS AML, in which 5q and 7q losses are much more common and also prognostically relevant.35
Regarding the treatment of ML-DS patients, we have no clear explanation for the fact that the cumulative incidence of toxic death was significantly lower in the patients treated with higher doses of cytarabine. A hypothesis could be that due to concern for toxicity these patients received different and more intensive supportive care. We did not find any differences in outcome estimates between ML-DS patients treated with therapy reduction and those who received standard therapy, although it should to be mentioned that exact details of treatment reduction, individual treatment protocols, protocol adherence or individual adaptations of therapy were not available given the retrospective nature of this study.
In terms of prognostic factors other than cytogenetics, Klusmann et al. reported that ML-DS patients with a history of transient myeloproliferative disease had a significantly better outcome than children with ML-DS without documented transient myeloproliferative disease,19 but unfortunately we were not able to collect data on whether ML-DS was preceded by transient myeloproliferative disease.
Age ≥3 years and high WBC count (>20×109) were identified in the present study as independent predictors of poor outcome (event-free survival) in ML-DS, which is in concordance with the findings of previous studies.9 This is mainly explained by the fact that there is a low(er) complete remission rate in these groups. These variables are also known from non-DS pediatric AML studies, in which older age and high WBC predict for poor outcome.14 Regarding age in ML-DS, it has been proposed that DS children who present over 4 years of age do in fact suffer from sporadic AML occurring in a child with DS, rather than from a ‘true’ ML-DS.18 For this reason we used the age cut-off in our inclusion criteria, to avoid ‘contamination’ with non-GATA1 mutated AML cases in DS children. In addition, AML in children with DS older than 4 years of age is exceedingly rare.18
A limitation of this collaborative study is that there was a wide variation in treatment intensity. Although all included patients were treated on collaborative treatment protocols, almost half of the patients received therapy according to protocols or risk arms specifically designed for DS patients and/or treatment reductions were made in standard protocols. These factors may have biased the study results.
In conclusion, this study showed that NK predicts a poor clinical outcome in ML-DS. As the incidence of relapse is higher than that of treatment-related mortality in these cases, further therapy reduction is not indicated in this group; in fact, treatment intensification may be needed. On the other hand, treatment reduction may be feasible in ML-DS cases with aberrant karyotypes. Such treatment stratification needs to be confirmed in prospective clinical studies. As the prognosis of high-risk NK ML-DS patients cannot be explained by the presence of known mutations in non-DS NK AML, the biological background must be elucidated to identify potential novel targets for therapy.
Supplementary Material
Acknowledgments
This study was a Berlin-Frankfurt-Münster Study Group- collaboration. The project of MB was funded by the Sophia Foundation, and the Stichting Kinderen Kankervrij (KIKA, project n. 15). The Japanese part of the study was partially supported by a Grant-in-Aid from the Ministry of Health, Labour and Welfare of Japan. The Nordic part of the study was supported by the Swedish Cancer Society, the Swedish Childhood Cancer Foundation, and the Swedish Research Council. The authors would like to thank all of the above mentioned bodies for the funds.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000;355(9199):165–9 [DOI] [PubMed] [Google Scholar]
- 2.Zwaan MC, Reinhardt D, Hitzler J, Vyas P. Acute leukemias in children with Down syndrome. Pediatr Clin North Am. 2008;55 (1):53–70 [DOI] [PubMed] [Google Scholar]
- 3.Hasle H, Niemeyer CM, Chessells JM, Baumann I, Bennett JM, Kerndrup G, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17(2):277–82 [DOI] [PubMed] [Google Scholar]
- 4.Creutzig U, Reinhardt D, Diekamp S, Dworzak M, Stary J, Zimmermann M. AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia. 2005;19(8):1355–60 [DOI] [PubMed] [Google Scholar]
- 5.Creutzig U, Ritter J, Vormoor J, Ludwig WD, Niemeyer C, Reinisch I, et al. Myelodysplasia and acute myelogenous leukemia in Down’s syndrome. A report of 40 children of the AML-BFM Study Group. Leukemia. 1996;10(11):1677–86 [PubMed] [Google Scholar]
- 6.Gamis AS, Woods WG, Alonzo TA, Buxton A, Lange B, Barnard DR, et al. Increased age at diagnosis has a significantly negative effect on outcome in children with Down syndrome and acute myeloid leukemia: a report from the Children’s Cancer Group Study 2891. J Clin Oncol. 2003;21(18):3415–22 [DOI] [PubMed] [Google Scholar]
- 7.Lange BJ, Kobrinsky N, Barnard DR, Arthur DC, Buckley JD, Howells WB, et al. Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children’s Cancer Group Studies 2861 and 2891. Blood. 1998;91(2): 608–15 [PubMed] [Google Scholar]
- 8.Rao A, Hills RK, Stiller C, Gibson BE, de Graaf SS, Hann IM, et al. Treatment for myeloid leukaemia of Down syndrome: population-based experience in the UK and results from the Medical Research Council AML 10 and AML 12 trials. Br J Haematol. 2006;132(5):576–83 [DOI] [PubMed] [Google Scholar]
- 9.Zeller B, Gustafsson G, Forestier E, Abrahamsson J, Clausen N, Heldrup J, et al. Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol. 2005;128(6):797–804 [DOI] [PubMed] [Google Scholar]
- 10.Zipursky A, Thorner P, De Harven E, Christensen H, Doyle J. Myelodysplasia and acute megakaryoblastic leukemia in Down’s syndrome. Leuk Res. 1994;18(3): 163–71 [DOI] [PubMed] [Google Scholar]
- 11.Taub JW, Huang X, Matherly LH, Stout ML, Buck SA, Massey GV, et al. Expression of chromosome 21-localized genes in acute myeloid leukemia: differences between Down syndrome and non-Down syndrome blast cells and relationship to in vitro sensitivity to cytosine arabinoside and daunorubicin. Blood. 1999;94(4):1393–400 [PubMed] [Google Scholar]
- 12.Zwaan CM, Kaspers GJ, Pieters R, Hahlen K, Janka-Schaub GE, van Zantwijk CH, et al. Different drug sensitivity profiles of acute myeloid and lymphoblastic leukemia and normal peripheral blood mononuclear cells in children with and without Down syndrome. Blood. 2002;99(1):245–51 [DOI] [PubMed] [Google Scholar]
- 13.Mundschau G, Gurbuxani S, Gamis AS, Greene ME, Arceci RJ, Crispino JD. Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood. 2003;101(11):4298–300 [DOI] [PubMed] [Google Scholar]
- 14.Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32(1):148–52 [DOI] [PubMed] [Google Scholar]
- 15.Hitzler JK, Zipursky A. Origins of leukaemia in children with Down syndrome. Nat Rev Cancer. 2005;5(1):11–20 [DOI] [PubMed] [Google Scholar]
- 16.Kanezaki R, Toki T, Terui K, Xu G, Wang R, Shimada A, et al. Down syndrome and GATA1 mutations in transient abnormal myeloproliferative disorder: mutation classes correlate with progression to myeloid leukemia. Blood. 2010;116(22): 4631–8 [DOI] [PubMed] [Google Scholar]
- 17.Sorrell AD, Alonzo TA, Hilden JM, Gerbing RB, Loew TW, Hathaway L, et al. Favorable survival maintained in children who have myeloid leukemia associated with Down syndrome using reduced-dose chemotherapy on Children’s Oncology Group trial A2971: a report from the Children’s Oncology Group. Cancer. 2012;118(19): 4806–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasle H, Abrahamsson J, Arola M, Karow A, O’Marcaigh A, Reinhardt D, et al. Myeloid leukemia in children 4 years or older with Down syndrome often lacks GATA1 mutation and cytogenetics and risk of relapse are more akin to sporadic AML. Leukemia. 2008;22(7):1428–30 [DOI] [PubMed] [Google Scholar]
- 19.Klusmann JH, Creutzig U, Zimmermann M, Dworzak M, Jorch N, Langebrake C, et al. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood. 2008;111(6):2991–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forestier E, Izraeli S, Beverloo B, Haas O, Pession A, Michalova K, et al. Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood. 2008;111(3):1575–83 [DOI] [PubMed] [Google Scholar]
- 21.Litz CE, Davies S, Brunning RD, Kueck B, Parkin JL, Gajl Peczalska K, et al. Acute leukemia and the transient myeloproliferative disorder associated with Down syndrome: morphologic, immunophenotypic and cytogenetic manifestations. Leukemia. 1995;9(9):1432–9 [PubMed] [Google Scholar]
- 22.Kaneko Y, Rowley JD, Variakojis D, Chilcote RR, Moohr JW, Patel D. Chromosome abnormalities in Down’s syndrome patients with acute leukemia. Blood. 1981;58(3):459–66 [PubMed] [Google Scholar]
- 23.Kaspers GJ, Zwaan CM. Pediatric acute myeloid leukemia: towards high-quality cure of all patients. Haematologica. 2007;92(11):1519–32 [DOI] [PubMed] [Google Scholar]
- 24.Hasle H, Alonzo TA, Auvrignon A, Behar C, Chang M, Creutzig U, et al. Monosomy 7 and deletion 7q in children and adolescents with acute myeloid leukemia: an international retrospective study. Blood. 2007;109(11):4641–7 [DOI] [PubMed] [Google Scholar]
- 25.Ravindranath Y. Down syndrome and acute myeloid leukemia: the paradox of increased risk for leukemia and heightened sensitivity to chemotherapy. J Clin Oncol. 2003;21(18):3385–7 [DOI] [PubMed] [Google Scholar]
- 26.Pine SR, Guo Q, Yin C, Jayabose S, Levendoglu-Tugal O, Ozkaynak MF, et al. GATA1 as a new target to detect minimal residual disease in both transient leukemia and megakaryoblastic leukemia of Down syndrome. Leuk Res. 2005;29(11):1353–6 [DOI] [PubMed] [Google Scholar]
- 27.Hasle H, Lund B, Nyvold CG, Hokland P, Ostergaard M. WT1 gene expression in children with Down syndrome and transient myeloproliferative disorder. Leuk Res. 2006;30(5):543–6 [DOI] [PubMed] [Google Scholar]
- 28.Hitzler JK, He W, Doyle J, Cairo M, Camitta BM, Chan KW, et al. Outcome of Transplantation for acute myelogenous leukemia in children with down syndrome. Biol Blood Marrow Transplant. 2013;19(6): 893–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metzeler KH, Dufour A, Benthaus T, Hummel M, Sauerland MC, Heinecke A, et al. ERG expression is an independent prognostic factor and allows refined risk stratification in cytogenetically normal acute myeloid leukemia: a comprehensive analysis of ERG, MN1, and BAALC transcript levels using oligonucleotide microarrays. J Clin Oncol. 2009;27(30):5031–8 [DOI] [PubMed] [Google Scholar]
- 30.Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, Balgobind BV, Arentsen-Peters ST, Alders M, et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood. 2009;113(23):5951–60 [DOI] [PubMed] [Google Scholar]
- 31.Hollink IH, Zwaan CM, Zimmermann M, Arentsen-Peters TC, Pieters R, Cloos J, et al. Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia. 2009;23(2):262–70 [DOI] [PubMed] [Google Scholar]
- 32.Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, Zimmermann M, Peeters JK, Valk PJ, et al. Characterization of CEBPA mutations and promoter hypermethylation in pediatric acute myeloid leukemia. Haematologica. 2011;96(3):384–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, Pratcorona M, Abbas S, Kuipers JE, et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood. 2011;118 (13):3645–56 [DOI] [PubMed] [Google Scholar]
- 34.Blink M, van den Heuvel-Eibrink MM, de Haas V, Klusmann JH, Hasle H, Zwaan CM. Low frequency of type-I and type-II aberrations in myeloid leukemia of Down syndrome, underscoring the unique entity of this disease. Haematologica. 2012;97(4): 632–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.von Neuhoff C, Reinhardt D, Sander A, Zimmermann M, Bradtke J, Betts DR, et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol. 2010;28(16):2682–9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.