

Abstract
The introduction of multiagent treatment protocols has led to a remarkable increase in survival rates for children diagnosed with acute lymphoblastic leukemia, yet for a subpopulation of patients, resistance to chemotherapeutics remains an obstacle to successful treatment. Here we investigate the role of the mitochondrial (or intrinsic) apoptosis pathway in modulating the onset and outcomes of childhood acute lymphoblastic leukemia. Cell death is a highly regulated process that plays an essential role in regulating cell homeostasis, particularly in tissues with high intrinsic proliferating capacity such as the hematopoietic system. Following the underlying paradigm that cis-acting genetic variation can influence disease risk and outcomes by modulating gene expression, we performed a systematic analysis of the proximal promoter regions of 21 genes involved in apoptosis. Using gene reporter assays, we show that promoter variations in 11 intrinsic apoptosis genes, including ADPRT, APAF1, BCL2, BAD, BID, MCL1, BIRC4, BCL2L1, ENDOG, YWHAB, and YWHAQ, influence promoter activity in an allele-specific manner. We also show that correlated promoter variation and increased expression of MCL1 is associated with reduced overall survival among high-risk patients receiving higher doses of corticosteroid, suggesting that increased expression of this anti-apoptosis gene could lead to reduced cell death and influence treatment response in a disease- and dose-responsive manner.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common cancer among children and is likely caused by multiple genetic and environmental factors. However, few established genetic risk factors for ALL have been identified. Following our previous work and the hypothesis that variation in genes involved in core biological processes such as oxidative stress response,1,2 DNA repair,3,4 and cell-cycle control5,6 contribute to increased susceptibility to childhood ALL, we set out to investigate whether deregulation of the tightly regulated apoptosis process could also influence disease risk and/or outcome. Apoptosis plays an essential role in the regulation of cell homeostasis, particularly in tissues with high intrinsic proliferating capacity and high cell turnover rate such as the hematopoietic system.7,8 Multiple distinct signaling pathways regulate apoptosis; however, two pathways have been mostly implicated in hematologic malignancies: the mitochondrial (or intrinsic) pathway and the death receptor (or extrinsic) pathway.8 Briefly, activation of the intrinsic pathway is initiated by mitochondrial damage that leads to release of apoptogenic factors, such as cytochrome c, Smac/Diablo, apoptosis-inducing factor (AIF), Omi/HtrA2, endonuclease G, caspase-2, and caspase-9 from the mitochondrial intermembrane space.9 Release of these factors is mediated by Bcl-2 family proteins that either suppress or promote changes in mitochondrial membrane permeability.10 Twenty-four Bcl-2 family members have been identified as either pro-apoptotic (e.g. Bax, Bak, BCL2L1/Bcl-XS, Bid, and Bad) or anti-apoptotic (e.g. Bcl-2 and BCL2L1/Bcl-XL).11 Upon release, cytochrome c forms the apoptosome complex with the adaptor molecule APAF1, ATP, and caspase-9,12 which in turn, activates the caspase-3 effector caspase cascade.13 Initiation of the extrinsic apoptosis pathway involves ligand-induced aggregation of death receptors and activation of procaspase-8 or procaspase-10 within the death-inducing signaling complex (DISC). Activated procaspase-8 or procaspase-10 is released into the cytoplasm, where it induces activation of downstream effector caspases. Both the intrinsic and extrinsic apoptotic pathways converge at the level of caspase-3 activation, which functions as a common death effector molecule.14
Differential expression of a number of key apoptosis genes has been observed in children with ALL15 and sequence variation in apoptosis gene promoters has been linked to various types of cancers,16–19 including leukemia,15,20 suggesting that increased growth and/or survival advantage of tumor cells caused by inherited polymorphisms in these genes could serve as a common mechanism for carcinogenesis. Variation in regulatory DNA sequences has been studied less intensively, despite evidence that cis-acting variation may explain up to 25–35% of inter-individual differences in gene expression.21 Given that deregulated expression of key apoptosis genes might contribute to cancer onset and progression as well as affect treatment responses and disease outcome, we postulated that functional cis-acting regulatory DNA variants in genes encoding components of the intrinsic/mitochondrial apoptosis pathway might be determinants of risk and/or disease outcome of childhood ALL.
Here we present a systematic analysis of the proximal promoter regions of 21 genes involved in apoptosis and their putative association with childhood ALL. Using gene reporter assays, we show that promoter single nucleotide polymorphisms (pSNP) and/or promoter haplotypes in 11 intrinsic apoptosis genes, including ADPRT, APAF1, BCL2, BAD, BID, MCL1, BIRC4, BCL2L1, ENDOG, YWHAB, and YWHAQ, influence promoter activity in an allele-specific manner. Through a case-control association study on a cohort of children with ALL, we also show that, although none of these SNP is associated with ALL susceptibility, the anti-apoptotic gene MCL1 was significantly associated with overall survival in this cohort.
Methods
Study subjects
Our cohort consisted of 321 childhood pre-B ALL patients and 329 healthy controls. Parental DNA was also collected and was available for 203 of the probands. Study subjects were all French-Canadians of European descent from the established Quebec Childhood ALL (QcALL) cohort.22 Incident cases were diagnosed in the Division of Hematology-Oncology at the CHU Sainte-Justine Hospital, Montreal, Canada, between October 1985 and November 2006. The study sample comprised 190 males and 131 females with a median age of 4.3 years. The healthy controls, 182 males and 146 females, were a group of French-Canadian neonates and adults recruited at the CHU Sainte-Justine Hospital.
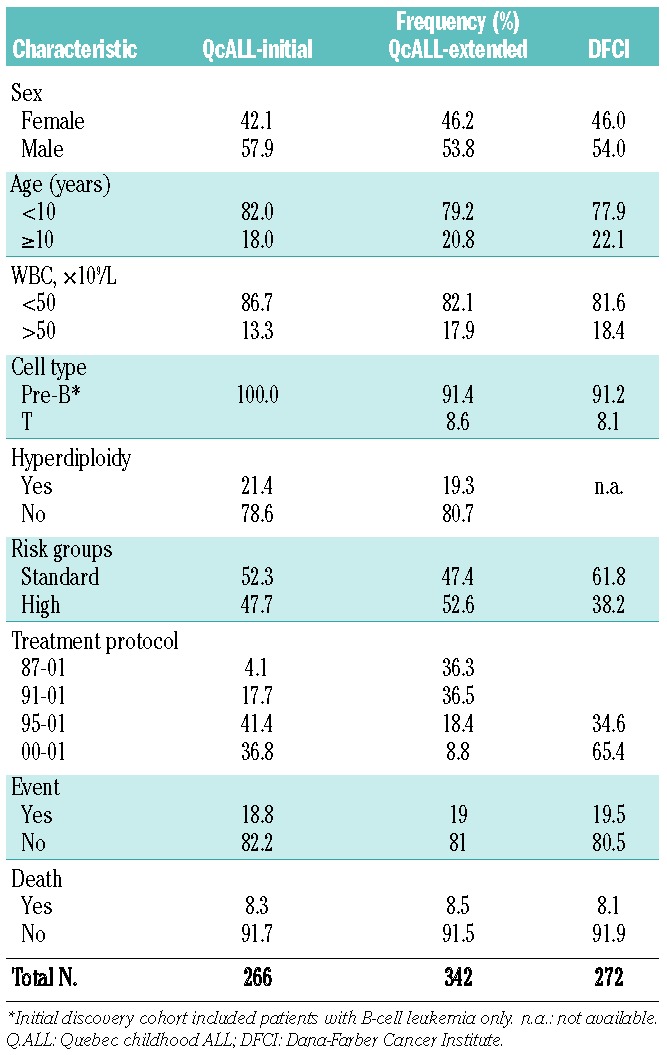
The polymorphism significantly associated with ALL outcome was subsequently analyzed in an extended cohort consisting of 342 childhood QcALL patients (Table 1). Events occurred in 65 children (19% of patients) including six cases in whom induction failed, two patients who died with no prior relapse, 25 patients who relapsed during treatment (or a few months following treatment) and 34 patients whose relapse occurred at a later period. In 23 cases relapse occurred more than once. The analyses were also performed in an independent replication cohort from the Dana-Farber Cancer Institute composed of 272 Caucasians.23 The Institutional Review Boards approved the research protocol and informed consent was obtained from all participating individuals and/or their parents.
Table 1.
Characteristics of ALL patients in the discovery and validation cohorts.
Identification of promoter single nucleotide polymorphisms in apoptosis genes
To identify pSNP in candidate genes selected based on their involvement in the mitochondrial apoptosis pathway, we directly sequenced the promoters of 21 intrinsic apoptosis genes (ADRPT, APAF1, BAD, BAG1, BAG3, BAG4, BAX, BCL2, BCL2A1, BCL2L1, BCL2L11, BCL2L10, BID, BIRC4, BIRC5, CASP3, CASP6, ENDOG, MCL1, YWHAB, and YWHAQ) in a SNP discovery cohort consisting of 20 randomly selected unrelated healthy individuals and 20 randomly selected pre-B ALL patients from the QcALL cohort.
Gene reporter assays
For each of the 21 intrinsic apoptosis genes, haplotype-specific fragments corresponding to ~2.0 kb of the proximal promoter region were amplified by polymerase chain reaction from genomic DNA of known genotype/haplotype to generate reporter constructs representative of the major haplotypic alleles (Table 2 and Online Supplementary Table S2). Constructs were generated by cloning the promoter haplotypes of interest into the pGL3-basic promoterless firefly luciferase reporter vector (Promega) as previously described.24 We sequenced the chosen clones to confirm the identity and integrity of each genotype/haplotype prior to transfection. Three clones for each construct were pooled and used to transfect HeLa, JEG-3, and HepG2 cells together with the pRL-SV40 plasmid containing the Renilla luciferase gene, used as a positive control of transfection. The promoterless pGL3-basic plasmid was used as the negative control. Reporter activities were normalized to the activity of the internal pRL-SV40 control and expressed as relative luciferase expression (mean±SD in 3 replicates). For a subset of selected genes for which haplotype-specific constructs showed positive promoter activity, Jurkat cells were used to further confirm the observed effect in a human leukemia cell line.
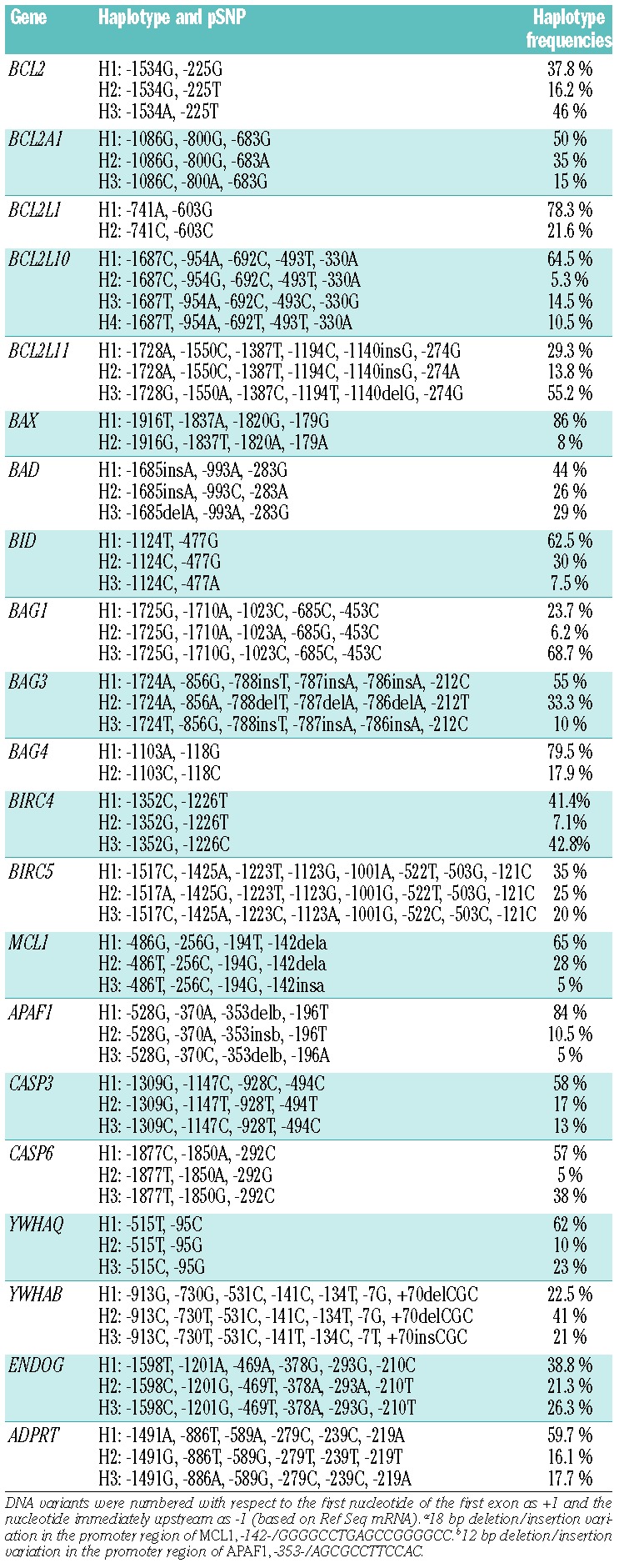
Table 2.
Promoter haplotypes studied in the gene reporter assays.
Genotyping and quality control checks of promoter single nucleotide polymorphisms
pSNP with differential allele-specific promoter activity in the gene reporter assays were further genotyped among the 321 childhood pre-B ALL patients, 329 healthy controls, and 203 parental pairs from the QcALL cohort, using the LuminexMAP/Autoplex Analyzer CS1000 system (Perkin Elmer, Waltham, MA, USA).
Results
Reporter assays reveal allele-specific promoter activity in ADPRT, APAF1, BAD, BCL2, BCL2L1, BID, BIRC4, ENDOG, MCL1, YWHAB, and YWHAQ
By directly re-sequencing genomic DNA of 20 unrelated healthy individuals and 20 pre-B ALL patients (n=80 chromosomes) of European descent, we identified a total of 88 SNP (Online Supplementary Table S1), including 85 that were known (dbSNP build 135), located in the proximal promoter of 21 genes involved in the intrinsic/mitochondrial apoptosis pathway: ADPRT, APAF1, BAD, BAG1, BAG3–4, BAX, BCL2, BCL2A1, BCL2L1, BCL2L10, BCL2L11, BID, BIRC4–5, CASP3, CASP6, ENDOG, MCL1, YWHAQ and YWHAB. All pSNP were in Hardy-Weinberg equilibrium and, after eliminating those that had minor allele frequencies <5% (n= 2), 86 pSNP were retained for further investigation. Promoter haplotype phase and corresponding frequencies (based on 80 chromosomes from the SNP discovery cohort) are given in Table 2.
To assess the functional significance of these 86 pSNP on promoter activity, 2.0 kb promoter haploptypes, corresponding to the major haplotypes (frequency > 5%) of each gene, were subcloned into the promoterless pGL3-basic vector. Transient transfection experiments were carried out for each of the resulting 61 haplotype-specific constructs (Table 3 and Online Supplementary Table S2). Assuming a conservative definition of promoter activity as a 1.5-fold increase in the activity of the ‘highly expressed allele’ over the expression of the pGL3-basic vector,25 most constructs showed promoter activity in at least one of the cell lines tested (Table 3). Significant fold difference (P <0.05) in either the HeLa, Jeg-3 or HepG2 cell line was considered as a criterion for functionality, at least in vitro. We observed significant differences in promoter activity associated with specific promoter haploptypes in 11 apoptosis genes (ADPRT, APAF1, BAD, BCL2, BCL2L1, BID, BIRC4, ENDOG, MCL1, YWHAB, and YWHAQ), suggesting that these promoter haploptypes and corresponding pSNP could function to modulate expression of these genes. However, for several constructs (e.g. BCL2A1, BCL2L10, BIRC5, and CASP3) we did not observe any promoter activity in the three cell lines tested (Online Supplementary Table S2).
Table 3.
Results of the gene reporter assays for the haplotypes showing promoter-specific allelic activity.
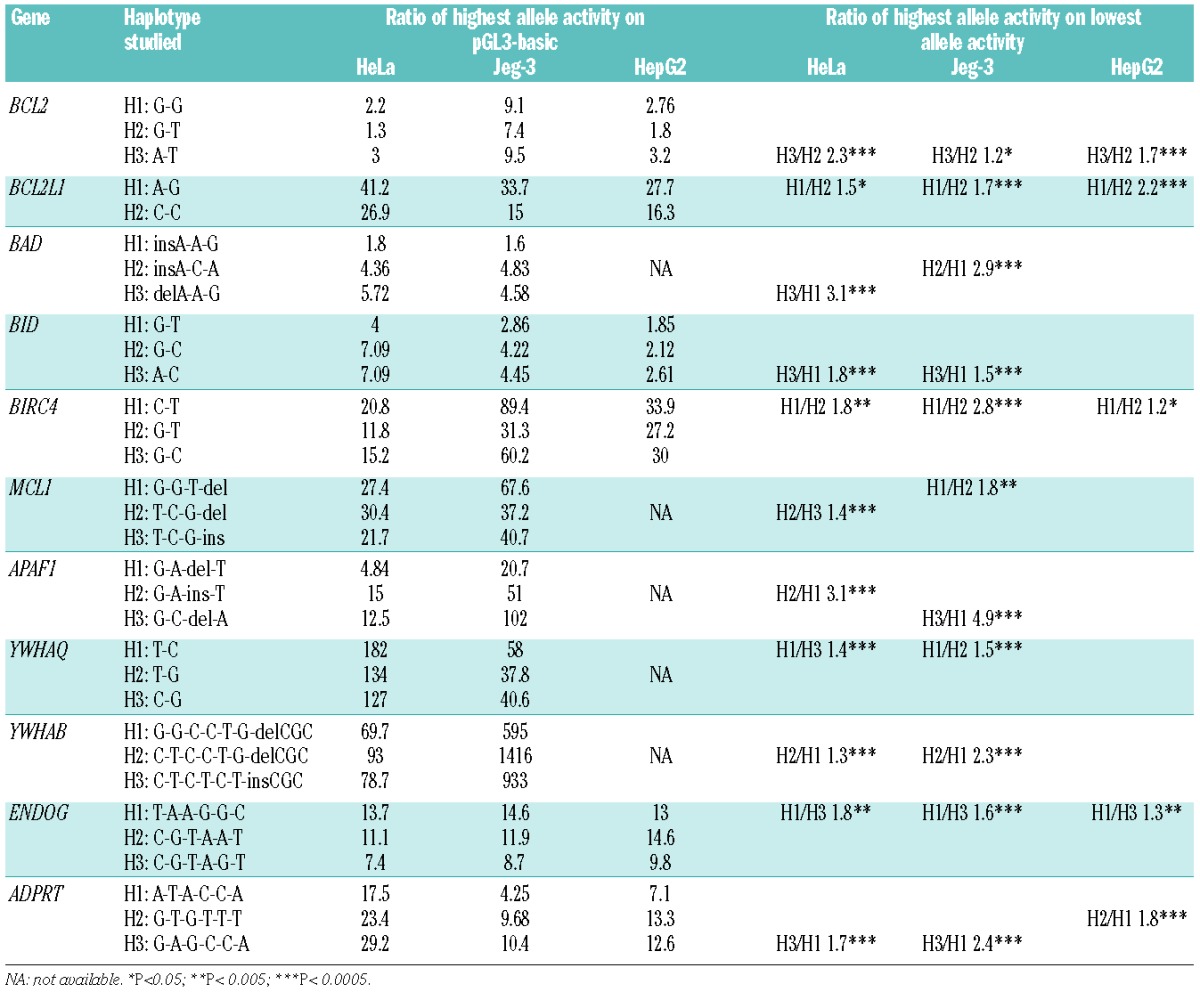
Furthermore, although we did detect promoter activity for genes BAG1, BAG3, BAG4, BAX, BCL2L11, and CASP6, there were no allele-specific differences for any of the haplotypes tested (Online Supplementary Table S2). For the four pro-apoptosis genes (ADPRT, APAF1, BAD, and BID), luciferase gene expression controlled by BID promoter haplotypes BID-H1, BID-H2 and BID-H3 showed differential expression when transfected into HepG2, HeLa and JEG-3 cells (Table 2), with BID-H3 showing the highest promoter activity (BID-H3/Basic 2.6, 7.1, and 4.5-fold difference, respectively). We found significant allele-specific promoter activity differences between BID-H3 and BID-H1 haplotypes in all three cell lines tested (BID-H3/BID-H1 1.4, 1.7, and 1.5-fold difference, respectively) (Table 2). Interestingly, for BAD we found that promoter activity was cell-specific, which is indicative of different modes of regulation in the cell lines tested. For example, BAD-H3 had the highest promoter activity (5.7-fold) in HeLa cells, whereas haplotype BAD-H2 had the highest activity in JEG-3 (4.8-fold). Similar results were observed with the APAF1 haplotypes, with different allele-specific promoter activity in HeLa and JEG-3 cells. In HeLa cells, the APAF1-H2 haplotype showed highest promoter activity (15-fold) and an allele-specific promoter activity between APAF1-H2/APAF1-H1 of 3.1-fold; however, in JEG-3 cells, haplotype APAF-H3 had the strongest activity with an APAF1-H3/APAF1-H1 difference of 4.9-fold. ADPRT showed significant increases in promoter activity for haplotype ADPRT-H3 compared to ADPRT-H1 in both HeLa (1.7-fold) and JEG-3 (2.4-fold) cells; however, in HepG2 cells ADPRT-H2 had significantly higher promoter activity (ADPRT-H2/ADPRT-H1 1.8-fold difference).
We also observed allele-specific promoter activity for all anti-apoptotic genes (BCL2, BCL2L1, BIRC4, ENDOG, MCL1, YWHAB, and YWHAQ) (Table 3). BCL2, BCL2L1, and ENDOG showed uniform allele-specific differences in HeLa, JEG-3 and HepG2 cells, whereas BIRC4, MCL1, YWHAB, and YWHAQ showed more cell-type specific promoter activity. For example, the luciferase levels driven by the BCL2-H3 haplotype were higher than the levels produced by BCL2-H2 in HeLa, JEG-3, and HepG2 cells (BCL2-H3/BCL2-H2 2.3, 1.2, and 1.7-fold differences, respectively) (Table 3). For MCL1, MCL1-H2 had highest promoter activity in HeLa cells (MCL1-H2/MCL1-H3 1.4-fold difference), while MCL1-H1 had the highest activity in JEG-3 cells (MCL1-H1/MCL1-H2 1.8-fold). Using the Jurkat leukemia cell line, we further confirmed that MCL1-H2 has the highest promoter activity in this cell type (MCL1-H2/MCL1-H3 1.6-fold difference) (Figure 1). The BCL2L1 haplotype BCL2L1-H1 had the highest luciferase activity across all constructs tested (BCL2L1-H1/BCL2L1-H2 1.5, 1.7, and 2.2-fold in difference in HeLa, JEG-3 and HepG2 cells, respectively). Thus, we found significant variability in allele-specific promoter activity in vitro for the intrinsic apoptosis genes ADPRT, APAF1, BCL2, BAD, BID, MCL1, BIRC4, BCL2L1, ENDOG, YWHAB, and YWHAQ, which could suggest functional consequences of promoter variation on gene expression and gene function, leading to deregulation of the intrinsic apoptosis pathway and potentially modifying disease risk and outcome.
Figure 1.
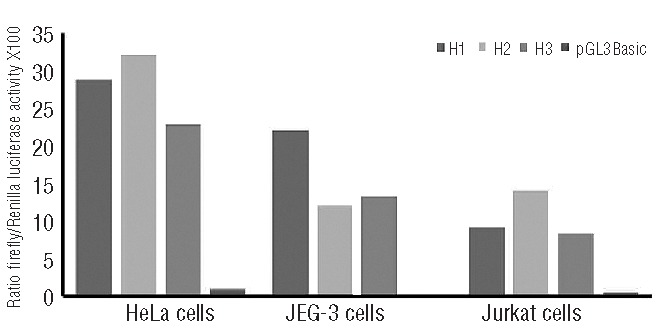
Gene reporter assays to evaluate the functional impact of the common MCL1 promoter haplotypes. Relative luciferase activity of MCL1 promoter haplotype constructs was measured following transient transfection in HeLa, JEG-3 and Jurkat cells. The empty promoter-less pGL3-Basic vector was used as a negative control. Results are expressed as a ratio of Firefly/Renilla activity multiplied by 100. P values are calculated from four replicates with unpaired Student’s t-test. Statistically significant differences: H2/H3 for HeLa (P<0.0005) and H1/H2 for Jeg-3 (P<0.005) (see also Table 3).
Promoter single nucleotide polymorphisms and haplotypes and the risk of childhood pre-B-cell acute lymphoblastic leukemia
To assess the impact of the pSNP/promoter haploptypes with allele-specific promoter activity on disease risk, we performed an association study using a case-control design to test for genotypic as well as haplotypic associations with childhood pre-B ALL. A total of 18 pSNP, in nine genes identified to be associated with changes in promoter activity, were tested for associations. The estimated odds ratios and 95% confidence intervals (95% CI) for the corresponding alleles and genotypes at these positions are given in Online Supplementary Tables S3 and S4. The allele and genotype frequencies for the variant positions in genes ADPRT, APAF1, BAD, BCL2, BID, BIRC4, MCL1, YWHAQ, and YWHAB did not differ significantly between patients and controls (Online Supplementary Tables S3 and S4), indicating that these pSNP alone do not modify the risk of childhood pre-B ALL, at least in our cohort. To further capture the genetic variability within these promoter regions, haplotypes were reconstructed in the case-control cohort (Online Supplementary Table S5) and tested for association with childhood ALL. However, despite their effects on promoter activity, we did not find any evidence of association between the individual haplotypes tested and the risk of developing pre-B ALL.
MCL1 genotypes influence overall survival in high-risk pre-B-cell acute lymphoblastic leukemia patients
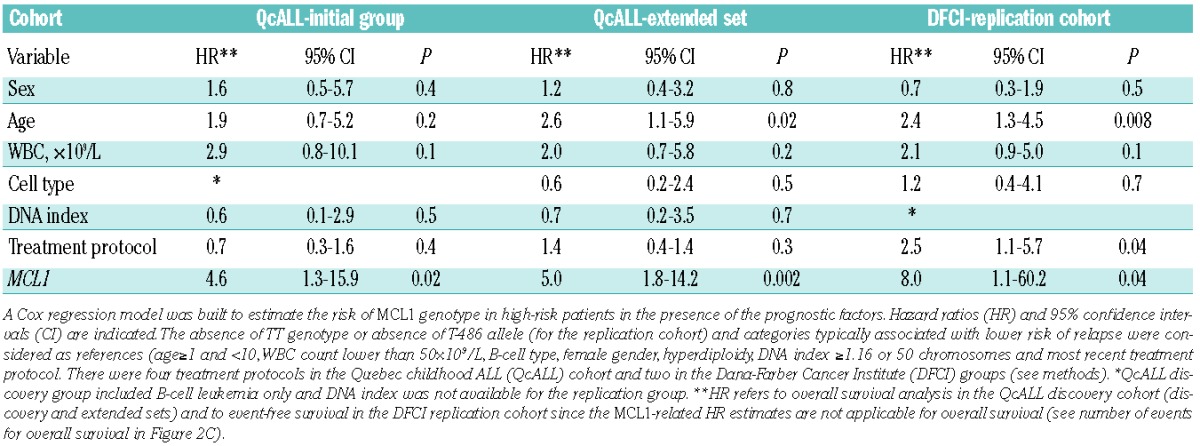
Given that none of the pSNP/promoter haploptypes was found to be associated with disease risk, we assessed the impact of the promoter variants on disease outcome. Survival analysis between the 18 pSNP and childhood ALL revealed no significant associations with either event-free or overall survival when all patients (266 patients; for whom genotype and outcome data were available) were analyzed. No association with prognostic factors or treatment protocol was seen. Because disease severity and progression, and treatment intensity differ between risk groups, we performed the analysis with ALL outcome following stratification of patients based on risk (high or standard) and found significant associations of MCL1 genotypes with overall survival in high-risk patients (Figure 2A). Patients with MCL1 -486TT genotype had reduced overall survival compared with individuals carrying the G allele (hazard ratio, HRTT= 4.0, 95% CI=1.3–12.5; P=0.01). Decreased overall survival was also observed in individuals who were homozygous for MCL1 -256C (HRCC=4.4, 95% CI=1.4–13.8; P=0.006) and MCL1 -194G (HRGG=3.9, 95% CI=1.2–12.4; P=0.01) alleles. Haplotype analysis further demonstrated that the associations observed at the three individual MCL1 loci are not independent and reflect a single association signal deriving from haplotype MCL1-H2. All associations remained significant with the inclusion of typical prognostic factors in the multivariate Cox regression analysis (Table 4). We further analyzed the MCL1 -486 polymorphism in the extended QcALL discovery cohort consisting of 342 childhood ALL patients. A similar reduction in overall survival was observed for TT individuals (HRTT= 4.1, 95% CI=1.6–10.3; P=0.001) (Figure 2B) and remained significant in the multivariate Cox regression model (Table 4) and following multiple testing corrections (false discovery rate = 11%). The MCL1 -486 variation was subsequently analyzed in the Dana-Farber Cancer Institute replication cohort, showing reductions in both event-free and overall survival in patients with the T-486 allele (P=0.03 and P=0.04, respectively; Figure 2C and Table 4).
Figure 2.
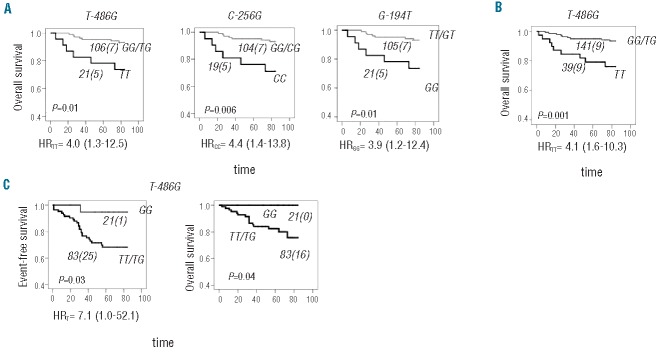
Overall survival in high-risk ALL patients according to MCL1 genotypes. (A) Overall survival (OS) curves are illustrated for patients with (black line) or without (gray line) TT-486, CC-256 and GG-194 MCL1genotypes in the left, middle and right panel, respectively. (B) An association between MCL1 TT-486 and reduced OS is shown in the extended QcALL discovery cohort. (C) An association between MCL1 T-486 and reduced OS and event-free survival was found in the Dana-Farber Cancer Institute replication cohort. The number of ALL patients in each curve (with the number of cases with an event in brackets) is indicated next to the curve. The difference in OS between patients with and without indicated genotypes was estimated by the log-rank test and the P value is indicated on each plot. Genotype-related risk carriers, expressed (when applicable) as a univariable hazard ratio (HR) with 95% confidence interval (CI), are indicated below each plot.
Table 4.
Survival analysis in ALL patients in relation to MCL1 G to T variations – multivariate models.
Discussion
In this study, we investigated whether deregulation of the highly orchestrated intrinsic apoptosis pathway could influence childhood ALL risk and/or outcome. Using gene reporter assays, we found that 32 promoter haplotypes, and 40 corresponding pSNP, in ADPRT, APAF1, BCL2, BAD, BID, MCL1, BIRC4, BCL2L1, ENDOG, YWHAB, and YWHAQ influenced promoter activity in an allele-specific manner. The observed up- or down-regulation of these anti-apoptosis and pro-apoptosis genes, associated with allele-specific promoter variation, could correlate with increased survival and proliferation potential of leukemia cells in patients and affect disease onset and/or outcome. For example, decreased levels of expression of the pro-apoptosis gene ADPRT have been shown to decrease drug-induced apoptosis, conferring resistance in human leukemia cells.26–28 In patients with chemoresistant acute myeloid leukemia, up-regulation of anti-apoptotic BIRC genes and down-regulation of pro-apoptotic APAF1 have been detected and were associated with dysregulation of the apoptosis machinery;29 furthermore, leukemia patients expressing high levels of the anti-apoptotic gene BCL2L12 have been shown to be three times more likely to relapse or die than patients with low levels.30 The relation between the expression of apoptosis genes and in vitro31,32 or in vivo response33–35 has been extensively studied in ALL. However, to the best of our knowledge, this is the largest systematic analysis of the role of regulatory variation in modulating expression of apoptosis genes and influencing childhood ALL onset and progression. The limitations of the study include the lack of analysis in the promoter region beyond 2 kb and the large number of polymorphisms and genes analyzed implying the necessity for multiple testing correction.
Although we did observe gene expression changes linked to promoter variation in a number of key apoptosis genes, we did not find any of the corresponding pSNP/promoter haploptypes to be significantly associated with susceptibility to ALL. This may be due to the limited statistical power of our sample size, or because the variants may not act alone but rather in combination with other variants, in this or other related pathways, through epistatic interactions to influence disease risk. We did, however, identify a statistically significant association between the anti-apoptotic gene MCL1 and overall survival in our childhood ALL cohort. Homozygosity for the variant alleles at positions MCL1 -486T>G, -256G>C, -194T>G was predictive of decreased overall survival among high-risk patients. Our reporter assays clearly highlight that the same MCL1-H2, carrying variant alleles -486T, -256C, -194G, has higher promoter activity in HeLa and Jurkat cells, compared to the major H1 haplotype that carries the common -486G, -256G, -194T alleles. The association of -486T T>G (used as a tag SNP in further analysis) was confirmed in the extended QcALL discovery and Dana-Farber Cancer Institute replication cohorts. The association with event-free survival was also observed in the replication cohort. These results suggest that promoter variation in MCL1 could result in higher MCL1 gene expression and subsequently increased survival of cancer cells thereby influencing treatment response and overall survival. The absence of an association with event-free survival in the discovery cohort and the difference in gene dosage effect between the discovery and replication cohorts might be due to differences in cumulative corticosteroid doses, type of corticosteroid or heterogeneity of the event, since not all patients with an event are necessarily comparable in terms of response to treatment or disease progression. Indeed, the distribution of events between the MCL1-positive and -negative groups was more comparable for overall survival than for event-free survival. The number of events in the event-free and overall survival analyses was similar in the MCL1-positive group, but different in the MCL1-negative group: the overall survival analysis showed a higher frequency of cases with multiple relapses (55% versus 36%) and no cases with late relapse (versus 27% in event-free survival). Larger cohorts of patients are needed to address differences related to subtypes of events.
MCL1 is gaining attention as a potential therapeutic target in cancer as it has been implicated in both tumorigenesis and chemoresistance, and in particular MCL1-dependent leukemias have been shown to be more sensitive to a wide range of chemotherapeutic agents.36 There is also evidence that MCL1 is important for the survival of myeloma cells,37 and in murine bone marrow viral infection models, MCL1 was found to accelerate both myeloid and lymphoid malignancies.38,39 Furthermore, the presence of an MCL1 promoter insertion was found to correlate with increased RNA and protein levels in leukemia and was associated with poor survival in a cohort of patients with chronic lymphocytic leukemia,40 further suggesting that regulatory variants in MCL1 can influence disease outcome. Importantly increased expression of MCL1 has also been associated with prednisolone resistance in B-lineage ALL cells.41 Differential expression of this Bcl-2 family member may contribute to the apoptotic blockage that is observed upstream of the mitochondria in prednisolone-resistant ALL cells.41 Furthermore, the fact that the mTOR inhibitor rapamycin reverses glucocorticoid resistance via MCL1 modulation confirms that MCL1 overexpression is essential for corticosteroid-related resistance in leukemia.42 We observed an association between high-activity MCL1 haplotypes and reduced overall survival in high-risk patients who received higher doses of corticosteroid compared to the standard-risk group, suggesting that this association could be corticosteroid related. Failure to achieve the required effect of corticosteroid therapy in patients presenting with less favorable disease characteristics could lead to disease progression and dismal outcomes. Indeed, six of nine MCL1-H2 homozygous patients with lower overall survival in the extended cohort had resistant disease as evidenced by multiple relapses or induction failure: six patients died of disease progression, two patients died from bone marrow transplant complications following relapse and one patient died from treatment complications with no prior relapse. This study provides the first evidence that pSNP in MCL1 could lead to dysregulation of the intrinsic apoptosis process in ALL cells, and influence patients’ response to treatment, suggesting that MCL1 is an important factor for the clinical response in childhood ALL. Furthermore, given the complex action of corticosteroids, it is possible that other genetic factors may also play a role, concomitantly or through gene-gene interactions, in modulating disease outcome.
The fact that we identified a significant association of MCL1 promoter variants with overall survival following risk-based stratification may be explained by the presence of modifying effects of corticosteroids acting in conjunction with more aggressive presenting disease. Our results provide evidence that MCL1 plays a role in disease progression, drug resistance and/or corticosteroid-induced apoptosis. The observed functional effects of the promoter polymorphisms/haplotypes on ADPRT, APAF1, BCL2, BAD, BID, MCL1, BIRC4, BCL2L1, ENDOG, YWHAB and YWHAQ expression warrant further investigation in larger cohorts of children with ALL.
Supplementary Material
Acknowledgments
The authors are indebted to all the patients and their parents who consented to participate in this study.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This study was supported by research funds provided by the Canadian Institutes for Health Research, as well as Genome Quebec and Genome Canada. DS holds the François-Karl Viau Chair in Pediatric Oncogenomics. Dana-Farber Cancer Institute ALL treatment protocols are supported by the National Cancer Institute/NIH grant CA068484.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Krajinovic M, Labuda D, Richer C, Karimi S, Sinnett D. Susceptibility to childhood acute lymphoblastic leukemia: influence of CYP1A1, CYP2D6, GSTM1, and GSTT1 genetic polymorphisms. Blood. 1999;93 (5):1496–501 [PubMed] [Google Scholar]
- 2.Krajinovic M, Lamothe S, Labuda D, Lemieux-Blanchard E, Theoret Y, Moghrabi A, et al. Role of MTHFR genetic polymorphisms in the susceptibility to childhood acute lymphoblastic leukemia. Blood. 2004;103(1):252–7 [DOI] [PubMed] [Google Scholar]
- 3.Infante-Rivard C, Krajinovic M, Labuda D, Sinnett D. Parental smoking, CYP1A1 genetic polymorphisms and childhood leukemia (Quebec, Canada). Cancer Causes Control. 2000;11(6):547–53 [DOI] [PubMed] [Google Scholar]
- 4.Mathonnet G, Krajinovic M, Labuda D, Sinnett D. Role of DNA mismatch repair genetic polymorphisms in the risk of childhood acute lymphoblastic leukaemia. Br J Haematol. 2003;123(1):45–8 [DOI] [PubMed] [Google Scholar]
- 5.Healy J, Belanger H, Beaulieu P, Lariviere M, Labuda D, Sinnett D. Promoter SNPs in G1/S checkpoint regulators and their impact on the susceptibility to childhood leukemia. Blood. 2007;109(2):683–92 [DOI] [PubMed] [Google Scholar]
- 6.Sherborne AL, Hosking FJ, Prasad RB, Kumar R, Koehler R, Vijayakrishnan J, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010;42(6):492–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wickremasinghe MI, Thomas LH, Friedland JS. Pulmonary epithelial cells are a source of IL-8 in the response to Mycobacterium tuberculosis: essential role of IL-1 from infected monocytes in a NF-kappa B-dependent network. J Immunol. 1999;163(7):3936–47 [PubMed] [Google Scholar]
- 8.Schimmer AD, Hedley DW, Penn LZ, Minden MD. Receptor- and mitochondrial-mediated apoptosis in acute leukemia: a translational view. Blood. 2001;98(13): 3541–53 [DOI] [PubMed] [Google Scholar]
- 9.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6(5):513–9 [DOI] [PubMed] [Google Scholar]
- 10.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13(15): 1899–911 [DOI] [PubMed] [Google Scholar]
- 11.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21(1): 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90(3):405–1 [DOI] [PubMed] [Google Scholar]
- 13.Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, et al. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192(4):571–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424 [DOI] [PubMed] [Google Scholar]
- 15.Holleman A, den Boer ML, de Menezes RX, Cheok MH, Cheng C, Kazemier KM, et al. The expression of 70 apoptosis genes in relation to lineage, genetic subtype, cellular drug resistance, and outcome in childhood acute lymphoblastic leukemia. Blood. 2006;107(2):769–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sibley K, Rollinson S, Allan JM, Smith AG, Law GR, Roddam PL, et al. Functional FAS promoter polymorphisms are associated with increased risk of acute myeloid leukemia. Cancer Res. 2003;63(15):4327–30 [PubMed] [Google Scholar]
- 17.Wang LE, Cheng L, Spitz MR, Wei Q. Fas A670G polymorphism, apoptotic capacity in lymphocyte cultures, and risk of lung cancer. Lung Cancer. 2003;42(1):1–8 [DOI] [PubMed] [Google Scholar]
- 18.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119(5):591–602 [DOI] [PubMed] [Google Scholar]
- 19.Dong D, Gao X, Zhu Z, Yu Q, Bian S, Gao Y. A 40-bp insertion/deletion polymorphism in the constitutive promoter of MDM2 confers risk for hepatocellular carcinoma in a Chinese population. Gene. 2012;497(1):66–70 [DOI] [PubMed] [Google Scholar]
- 20.Kim DH, Xu W, Ma C, Liu X, Siminovitch K, Messner HA, et al. Genetic variants in the candidate genes of the apoptosis pathway and susceptibility to chronic myeloid leukemia. Blood. 2009;113(11):2517–25 [DOI] [PubMed] [Google Scholar]
- 21.Lo HS, Wang Z, Hu Y, Yang HH, Gere S, Buetow KH, et al. Allelic variation in gene expression is common in the human genome. Genome Res. 2003;13(8):1855–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Healy J, Richer C, Bourgey M, Kritikou EA, Sinnett D. Replication analysis confirms the association of ARID5B with childhood B-cell acute lymphoblastic leukemia. Haematologica. 2010;95(9):1608–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rousseau J, Gagne V, Labuda M, Beaubois C, Sinnett D, Laverdiere C, et al. ATF5 polymorphisms influence ATF function and response to treatment in children with childhood acute lymphoblastic leukemia. Blood. 2011;118(22):5883–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belanger H, Beaulieu P, Moreau C, Labuda D, Hudson TJ, Sinnett D. Functional promoter SNPs in cell cycle checkpoint genes. Hum Mol Genet. 2005;14(18):2641–8 [DOI] [PubMed] [Google Scholar]
- 25.Coleman SL, Hoogendoorn B, Guy C, Smith SK, O’Donovan MC, Buckland PR. Streamlined approach to functional analysis of promoter-region polymorphisms. Biotechniques. 2002;33(2):412, 4,, 6 passim. [DOI] [PubMed] [Google Scholar]
- 26.Richardson DS, Allen PD, Kelsey SM, Newland AC. Effects of PARP inhibition on drug and Fas-induced apoptosis in leukaemic cells. Adv Exp Med Biol. 1999;457:267–79 [DOI] [PubMed] [Google Scholar]
- 27.Tanaka Y, Yoshihara K, Tohno Y, Kojima K, Kameoka M, Kamiya T. Inhibition and down-regulation of poly(ADP-ribose) polymerase results in a marked resistance of HL-60 cells to various apoptosis-inducers. Cell Mol Biol (Noisy-le-grand). 1995;41(6): 771–81 [PubMed] [Google Scholar]
- 28.Pottier N, Cheok MH, Yang W, Assem M, Tracey L, Obenauer JC, et al. Expression of SMARCB1 modulates steroid sensitivity in human lymphoblastoid cells: identification of a promoter SNP that alters PARP1 binding and SMARCB1 expression. Hum Mol Genet. 2007;16(19):2261–71 [DOI] [PubMed] [Google Scholar]
- 29.Ragusa M, Avola G, Angelica R, Barbagallo D, Guglielmino MR, Duro LR, et al. Expression profile and specific network features of the apoptotic machinery explain relapse of acute myeloid leukemia after chemotherapy. BMC Cancer. 2010;10:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomadaki H, Floros KV, Pavlovic S, Tosic N, Gourgiotis D, Colovic M, et al. Overexpression of the novel member of the BCL2 gene family, BCL2L12, is associated with the disease outcome in patients with acute myeloid leukemia. Clin Biochem. 2012;45(16–17):1362–7 [DOI] [PubMed] [Google Scholar]
- 31.Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S, et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses. Blood. 1998;91(9):3379–89 [PubMed] [Google Scholar]
- 32.Salomons GS, Smets LA, Verwijs-Janssen M, Hart AA, Haarman EG, Kaspers GJ, et al. Bcl-2 family members in childhood acute lymphoblastic leukemia: relationships with features at presentation, in vitro and in vivo drug response and long-term clinical outcome. Leukemia. 1999;13(10): 1574–80 [DOI] [PubMed] [Google Scholar]
- 33.Stoetzer OJ, Nussler V, Darsow M, Gullis E, Pelka-Fleischer R, Scheel U, et al. Association of bcl-2, bax, bcl-xL and interleukin-1 beta-converting enzyme expression with initial response to chemotherapy in acute myeloid leukemia. Leukemia. 1996;10 (Suppl 3):S18–S22 [PubMed] [Google Scholar]
- 34.Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res. 2000;6(5):1796–803 [PubMed] [Google Scholar]
- 35.Svingen PA, Karp JE, Krajewski S, Mesner PW, Jr, Gore SD, Burke PJ, et al. Evaluation of Apaf-1 and procaspases-2, -3, -7, -8, and -9 as potential prognostic markers in acute leukemia. Blood. 2000;96(12):3922–31 [PubMed] [Google Scholar]
- 36.Brunelle JK, Ryan J, Yecies D, Opferman JT, Letai A. MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. J Cell Biol. 2009;187(3):429–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100(1): 194–9 [DOI] [PubMed] [Google Scholar]
- 38.Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21(24):3232–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beverly LJ, Varmus HE. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene. 2009;28(9):1274–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moshynska O, Sankaran K, Pahwa P, Saxena A. Prognostic significance of a short sequence insertion in the MCL-1 promoter in chronic lymphocytic leukemia. J Natl Cancer Inst. 2004;96(9):673–82 [DOI] [PubMed] [Google Scholar]
- 41.Holleman A, den Boer ML, Kazemier KM, Janka-Schaub GE, Pieters R. Resistance to different classes of drugs is associated with impaired apoptosis in childhood acute lymphoblastic leukemia. Blood. 2003;102(13): 4541–6 [DOI] [PubMed] [Google Scholar]
- 42.Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam RW, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006;10(4):331–42 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.