

Abstract
Unusual N-acetylated sugars have been observed on the O-antigens of some Gram-negative bacteria and on the S-layers of both Gram-positive and Gram-negative bacteria. One such sugar is 3-acetamido-3,6-dideoxy-α-d-galactose or Fuc3NAc. The pathway for its production requires five enzymes with the first step involving the attachment of dTMP to glucose-1-phosphate. Here we report a structural and biochemical characterization of a bifunctional enzyme from Shewanella denitificans thought to be involved in the biosynthesis of dTDP-Fuc3NAc. On the basis of a bioinformatics analysis, the enzyme, hereafter referred to as FdtD, has been postulated to catalyze the third and fifth steps in the pathway, namely a 3,4-keto isomerization and an N-acetyltransferase reaction. For the X-ray analysis reported here, the enzyme was crystallized in the presence of dTDP and CoA. The crystal structure shows that FdtD adopts a hexameric quaternary structure with 322 symmetry. Each subunit of the hexamer folds into two distinct domains connected by a flexible loop. The N-terminal domain adopts a left-handed β-helix motif and is responsible for the N-acetylation reaction. The C-terminal domain folds into an antiparallel flattened β-barrel that harbors the active site responsible for the isomerization reaction. Biochemical assays verify the two proposed catalytic activities of the enzyme and reveal that the 3,4-keto isomerization event leads to inversion of configuration about the hexose C-4' carbon.
3-acetamido-3,6-dideoxy-α-d-galactose, hereafter referred to as Fuc3NAc, is an unusual dideoxysugar found in the O-antigens of various Gram-negative bacteria and in the S-layers glycans of some Gram-positive bacteria. The biosynthetic pathway for its production in Aneurinibacillus thermoaerophilus L420-91T was elucidated in 2003 and is shown in Scheme 1.1 The first step, catalyzed by a thymidylyltransferase (RmlA), involves the attachment of dTMP to glucose-1-phosphate. In the second step, the C-6' hydroxyl group is removed, and the C-4' hydroxyl group is converted to a keto moiety by the action of a 4,6-dehydratase (RmlB). These two steps are common to the biosynthesis of many di-, tri-, and tetradeoxysugars.2 The next three enzymes in the pathway are specific for the production of Fuc3NAc: FdtA which catalyzes a 3,4-ketoisomerization, FdtB which functions as a PLP-dependent aminotransferase, and FdtC which is responsible for the N-acetylation of the C-3' amino group.
Scheme 1.

Research from our laboratory has shown that the A. thermoaerophilus 3,4-ketoisomerase (FdtA) is a dimer and belongs to the well-characterized “cupin” superfamily.3 Members of this superfamily include both metal-dependent and metal-independent enzymes as well as seed storage and sugar-binding proteins.4–6 Although the molecular architectures of the aminotransferase (FdtB) and the N-acetyltransferase (FdtC) have not been reported thus far (Scheme 1), it can be postulated, based on amino acid sequences, that the former belongs to the aspartate aminotransferase superfamily and that the latter is a member of the left-handed β-helix superfamily. Proteins of the left-handed β-helix superfamily typically function as trimers.
In the case of A. thermoaerophilus, all of the enzymes required for the production of dTDP-Fuc3NAc exist on separate polypeptide chains. This is not the case for other organisms, however. Take for example, Shewanella denitrificans, which is a Gram-negative bacterium first isolated from the Baltic Sea.7 In this organism, a gene has been identified that apparently encodes for a bifunctional protein with both 3,4-ketoisomerase and N-acetyltransferase activities, corresponding to the third and fifth steps shown in Scheme 1. The quaternary structure of this hypothetical protein is intriguing since the sugar isomerases and the N-acetyltransferases are typically dimers and trimers, respectively. Given that the assignment of activity for this enzyme was based strictly on its predicted amino acid sequence, and curious as to its molecular architecture, we initiated a biochemical and structural analysis on this protein, hereafter referred to as FdtD. The research described herein represents the first detailed characterization of a bifunctional ketoisomerase/N-acetyltransferase.
Materials and Methods
Cloning of FdtD
The gene encoding FdtD was PCR-amplified from S. denitrificans, strain OS217 genomic DNA (ATCC BAA-1090), via standard procedures using primers such that the forward 5'-AAACATATGATTCATAAATTAGCAGATGTTCAATCTCAAAATATTGGTGACAATACC AAAGTCTGGC and the reverse 5'-AAACTCGAGGTTTTGTCTCATTTGTTTAAAAGTTGAATAATCACGAATGTAATCATCA GAATCATAGTAATGAG added Ndel and Xhol cloning sites, respectively. The purified PCR product was subsequently A-tailed and ligated into the pGEM-T vector (Promega), which was used to transform DH5-α Escherichia coli cells for subsequent screening and sequencing. A pGEM-T-fdtD vector construct of the correct sequence was then digested, and the fdtD gene ligated into the pET31 vector. DH5-α E. coli cells were transformed with the resulting plasmid and streaked onto LB agar plates supplemented with ampicillin. Multiple colonies were tested for the presence of the fdtD gene.
Protein Expression and Purification
The pET31-fdtD plasmid was used to transform Rosetta2(DE3) E. coli cells (Novagen). The cultures were grown in LB medium supplemented with ampicillin and chloramphenicol at 37°C with shaking until an optical density of 0.75 was reached at 600 nm. The flasks were cooled to room temperature, and the cells were induced with 1 mM IPTG and allowed to express protein at 23°C for 24 hours.
The cells were harvested by centrifugation and disrupted by sonication on ice. The lysate was cleared by centrifugation, and FdtD was purified utilizing Ni-NTA resin (Qiagen) according to the manufacturer’s instructions. The protein was dialyzed against 10 mM Tris-HCl (pH 8.0) and 200 mM NaCl and concentrated to 9.5 mg/mL based on an extinction coefficient of 0.96 (mg/mL)−1cm−1.
Selenomethionine-labeled protein was prepared as previously described.8 Cells were grown in minimal media. Prior to the addition of IPTG, methionine biosynthesis was suppressed by the addition of lysine, threonine, phenylalanine, leucine, isoleucine, valine, and selenomethionine. The selenomethionine-labeled protein was purified in the same manner as the wild-type enzyme and concentrated to 7.5 mg/mL.
Crystallization of FdtD
Crystallization conditions were initially surveyed by the hanging drop method of vapor diffusion using a laboratory-based sparse matrix screen. Single crystals of FdtD were subsequently grown via vapor diffusion against 100 mM HEPPS (pH 8.0), 7 −12% poly(ethylene glycol) 8000, and 100 mM MgCl2 using protein that had been incubated with 10 mM dTDP and 10 mM CoA. The crystals grew to maximum dimensions of ~0.1 × 0.3 × 0.4 mm in two weeks. They belonged to the P1 space group with unit cell dimensions of a = 85.3 Å, b = 109.4 Å, c = 127.8 Å, a = 79.2°, β = 80.0°, and γ = 84.9°. There were two hexamers in the asymmetric unit. The selenomethionine-labeled protein crystals were grown in a similar manner.
Structural Analysis of FdtD
Prior to X-ray data collection, crystals of either the wild-type enzyme or the selenomethionine-labeled protein were transferred to cryoprotectant solutions containing 26% poly(ethylene glycol) 8000, 15% ethylene glycol, 600 mM NaCl, 10 mM dTDP, 10 mM CoA, and 100 mM MgCl2 buffered at pH 8 with 100 mM HEPPS. X-ray data were collected at the Structural Biology Center Beamline 19-BM at a wavelength of 0.9794 Å from both the wild-type and the selenomethionine-labeled protein crystals (Advanced Photon Source). The X-ray data were processed and scaled with HKL3000.9 Relevant X-ray data collection statistics are listed in Table 1.
Table 1.
X-ray Data Collection Statistics
| Selenomethionine- labeled FdtD |
Wild-type FdtD | |
|---|---|---|
| resolution limits | 50.0–2.73 (2.84–2.73)b | 30.0–2.20 (2.28–2.20)b |
| number of independent reflections | 114101 (12651) | 205722 (19668) |
| completeness (%) | 98.5 (98.3) | 91.1 (87.0) |
| redundancy | 2.1 (2.1) | 3.0 (2.5) |
| avg I/avg σ(I) | 28.9 (6.6) | 27.0 (4.7) |
| Rsym (%)a | 6.2 (17.3) | 7.1 (22.3) |
Rsym = (∑|I - Ī |/ ∑ I) × 100.
Statistics for the highest resolution bin.
The structure of the protein was determined via single wavelength anomalous dispersion (SAD). Analysis of the X-ray data measured from the selenomethionine-labeled crystals with SHELXD revealed 54-selenium atoms.10 Visual inspection of the anomalous difference map led to the location of six additional sites. Protein phases were calculated using these 60 sites with the program SOLVE.11 The overall figure of merit was 0.12. Initial attempts at molecular averaging with the program RESOLVE yielded only a two-fold relationship between the selenium sites.12,13 In order to obtain better phasing information, the selenium sites were visualized in COOT, which allowed for the identification of 12 constellations of five selenium sites each.14 These constellations allowed for 12-fold molecular averaging, which greatly improved the quality of the electron density map. At this point in the analysis, the overall figure of merit increased to 0.25. An initial model was built and subsequently refined against the wild-type X-ray data. Iterative rounds of model building with COOT and refinement with REFMAC reduced the Rwork and Rfree to 20.4% and 27.0%, respectively, from 30 – 2.2 Å resolution.15 Model refinement statistics are listed in Table 2. A Ramachandran plot of all non-glycinyl residues is presented in Figure S1 of Supporting Information.
Table 2.
Refinement Statistics
| resolution limits (Å) | 30.0-2.20 |
| aR-factor (overall)%/no. reflections | 20.8/195488 |
| R-factor (working)%/no. reflections | 20.4/185204 |
| R-factor (free)%/no. reflections | 27.0/10284 |
| number of protein atoms | 26602 |
| number of heteroatoms | 962 |
| average B values | |
| protein atoms (Å2) | 35.4 |
| ligand (Å2) | 44.6 |
| solvent (Å2) | 32.8 |
| weighted RMS deviations from ideality | |
| bond lengths (Å) | 0.013 |
| bond angles (°) | 2.2 |
| planar groups (Å) | 0.010 |
| Ramachandran regions (%)b | |
| most favored | 85.8 |
| additionally allowed | 13.4 |
| generously allowed | 0.6 |
| disallowed | 0.1 |
R-factor = (Σ|Fo - Fc | / Σ|Fo |) × 100 where Fo is the observed structure-factor amplitude and Fc is the calculated structure-factor amplitude.
Distribution of Ramachandran angles according to PROCHECK.29
Measurement of Ketoisomerase and N-acetyltransferase Activities
The 3,4-ketoisomerase activity of S. denitrificans FdtD was verified according to Scheme 2. A 1 mL reaction containing 1 mM dTDP-glucose, 2 mg/mL 4,6-dehydratase (RmlB from E. coli), 2 mg/mL FdtD, 4 mg/mL aminotransferase (QdtB from Thermoanaerobacterium thermosaccharolyticum), 40 mM sodium glutamate, and 100 mM HEPPS (pH 8) was incubated at 37°C for two hours. Enzymes were removed, and the reaction products were evaluated via HPLC. The amino sugar product had the same retention time as that of a standard sample of dTDP-3-amino-6-deoxygalactose (dTDP-Fuc3N). The E. coli 4,6-dehydratase was cloned, over-expressed, and purified in the laboratory (unpublished procedures). The aminotransferase was purified as previously reported.16
Scheme 2.

The N-acetyltransferase activity of FdtD was verified under the following assay conditions. A 1 mL reaction containing 1 mM dTDP-Fuc3N, 1 mM acetyl-CoA, 2 mg/mL FdtD, and 100 mM HEPPS (pH 8) was incubated at 37°C for two hours. Enzymes were removed, and the reaction products were evaluated via HPLC. The acetylated sugar product had the same retention time as that of a standard sample of dTDP-Fuc3NAc. We were also curious as to whether FdtD could function on 3-amino-3,6-dideoxy-α-d-glucose as well (dTDP-Qui3N). The above reaction was replicated using dTDP-Qui3N in place of dTDP-Fuc3N. Both the dTDP-Fuc3N and dTDP-Qui3N substrates required for these assays were prepared according to previously published procedures.17 The assays did, indeed, demonstrate that FdtD can function on both sugars that differ in stereochemistry about the C-4' position.
Kinetic Analyses
The kinetic constants for the 3,4-ketoisomerase reaction were determined via a coupled assay using a Beckman Coulter DU-640 spectrophotometer according to Scheme 3. Specifically it was possible to measure the activity of FdtD by monitoring the decrease in absorbance at 340 nm as NADPH is oxidized to NADP+ due to the action of KijD10. Previous studies from the laboratory have verified that KijD10 from Actinomadura kijaniata functions as an NADPH-dependent C-3' ketoreductase.18 The reaction mixtures contained 50 mM HEPES (pH 7.5), 0.2 mM NADPH, 1 mg/mL KijD10, and dTDP-4-keto-6-deoxyglucose varying from 0.005 to 2.0 mM. The reactions were initiated by the addition of 0.03 mg/mL FdtD.
Scheme 3.
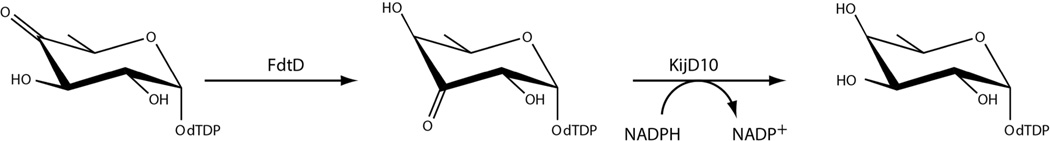
The kinetic constants for the N-acetyltransferase reaction were measured via a discontinuous assay using an ÄKTA HPLC. The reaction rates were determined by calculating the amount of dTDP-Fuc3NAc (or dTDP-Qui3NAc) produced on the basis of the peak area on the HPLC trace as measured at 267 nm. The area was correlated to concentration via a calibration curve created with standard samples that had been treated in the same manner as the reaction aliquots. Specifically, the Michaelis constants for dTDP-Fuc3N or dTDP-Qui3N were determined using 1 mL reactions containing 2 mM acetyl-CoA, 50 mM HEPPS (pH 8.5), and dTDP-Fuc3N varying from 0.005 mM to 1.0 mM or dTDP-Qui3N varying from 0.01 mM to 1.0 mM. The reactions were initiated by adding FdtD at a final concentration of 0.006 mg/ mL. Periodically, over two minutes, 250 µL samples were removed and quenched by the addition of 6 µL of 6 M HCl. To each quenched reaction, 200 µL aliquots of carbon tetrachloride were added, the samples were vigorously mixed, and then spun for two minutes at 14,000 × g. Samples of 200 µL were taken from the aqueous phase. These were diluted with 1 mL of water and loaded onto a 1 mL Resource-Q column for analysis. All HPLC analyses were performed using solutions at pH 4 and a gradient of 0 to 1.5 M ammonium acetate. The Michaelis constants for acetyl-CoA were determined in an analogous manner. In this case, the reactions contained 2 mM dTDP-Qui3N, 50 mM HEPPS (pH 8.5), and acetyl-CoA varying from 0.01 mM to 1.0 mM. The reactions were initiated by the addition of FdtD to a final concentration of 0.006 mg/ mL.
Plots of concentrations versus initial rates were analyzed with SigmaPlot8 and were fitted to the equation vo=(Vmax[S])/(KM+[S]). Kinetic parameters are listed in Table 3. Substrate saturation curves are presented in Figure S2 of Supporting Information.
Table 3.
Kinetic Parameters for FdtD
| dTDP-4-keto-6- deoxyglucosea |
dTDP-Fuc3Nb | dTDP-Qui3Nb | Acetyl-CoAb | |
|---|---|---|---|---|
| Km (µM) | 45 ± 3 | 45 ± 3 | 110 ± 20 | 19 ± 2 |
| kcat (s−1) | 4.4 ± 0.3 | 10.5 ± 0.5 | 7.9 ± 0.8 | 4.1 ± 0.3 |
| kcat/Km (M−1s−1) | 9.8 × 104 | 2.3 × 105 | 7.2 × 104 | 2.2 × 105 |
This is for the isomerase activity.
This is for the N-acetyltransferase activity.
Results and Discussion
Biochemical Characterization of S. denitrificans FdtD
On the basis of amino acid sequence analyses, we suspected that FdtD from S. denitrificans was involved in the production of dTDP-Fuc3NAc (Scheme 1). The carbohydrate composition of the S. denitrificans O-antigen has never been reported, however, and thus it was imperative to verify the catalytic activity of the enzyme. Indeed, a recent report on a similar bifunctional enzyme from Pasteurella multocida demonstrated that it plays a role in the biosynthesis of dTDP-3-acetamido-3,6-dideoxy-α-d-glucose (Qui3NAc) rather than Fuc3NAc. These two sugars differ simply in the stereochemistry about the hexose C-4' carbons with Qui3NAc and Fuc3NAc having the S- and R-configurations, respectively (Scheme 4). Whereas the general 3,4-ketoisomerase activity of the S. denitrificans FdtD was verified according to Scheme 2, the assay could not distinguish whether dTDP-Fuc3N or dTDP-Qui3N had been formed because, as we have previously shown, the aminotransferase (QdtB) employed in the experiments can function on either dTDP-3-keto-6-deoxygalactose or dTDP-3-keto-6-deoxyglucose.16 Thus it was possible that FdtD was producing dTDP-3-keto-6-deoxyglucose rather than dTDP-3-keto-6-deoxygalactose as indicated in Scheme 2.
Scheme 4.

NMR spectroscopy was subsequently employed to verify the stereochemistry of the product resulting from the 3,4-ketoisomerase activity of FdtD. Specifically, a 1H NMR spectrum was acquired on the aminated sugar product (Scheme 2) using a DMX-400 Mhz spectrometer at the Nuclear Magnetic Resonance Facility (NMRFAM), University of Wisconsin-Madison. It was not possible to follow the production of the dTDP-3-keto-6-deoxysugar directly via NMR due to its instability. Hence the aminated product was employed (Scheme 2). Analysis of the 1H NMR spectrum verified the stereochemistry about the C-4' carbon as that expected for dTDP-3-keto-6-deoxygalactose (Figure S3 of Supporting Information).
The kinetic parameters for both the 3,4-ketoisomerase and N-acetyltransferase activities of FdtD are listed in Table 3. The enzyme functions well as a 3,4-ketoisomerase with a catalytic efficiency of 9.8 × 104 s−1 M−1. Likewise, the catalytic efficiency of FdtD with respect to its N-acetyltransferase activity is good and comparable to that observed for similar enzymes from Thermoanaerobacterium thermosaccharolyticum,17 Caulobacter crescentus,19 and Campylobacter jejuni.20 Whereas FdtD can function as an N-acetyltransferase on either dTDP-Fuc3N or dTDP-Qui3N, its catalytic efficiency is ~3-fold higher with dTDP-Fuc3N as the substrate.
Overall Structure of S. denitrificans FdtD
Crystals of the S. denitrificans FdtD were obtained in the presence of dTDP and CoA. The asymmetric unit contained two complete hexamers, which corresponds to over 3500 amino acid residues. In all of the twelve subunits there were two regions where the electron density for the polypeptide chain backbone was disordered. One of these occurred in a loop between Lys 83 and Leu 95 and the other in a loop between Ala 152 and Asn 161. The α-carbons for the two hexamers in the asymmetric unit correspond with a root-mean-square deviation of ~0.38 Å. A superposition of the α-carbons for all twelve subunits in the asymmetric unit is shown in Figure S4 of Supporting Information. Their overall folds were virtually identical (non-crystallographic symmetry restraints were not imposed during the refinement procedures). Whereas all the subunits began at Met 1, they differed with respect to the number of residues visible in the electron density at the C-terminus (the shortest subunit ended at Lys 299 and the longest subunit terminated at Ala 310). The occupancies for the twelve CoA ligands were approximately the same. There was more variability in the electron densities for the dTDP ligands, however. In light of the similarity between the subunits, and for the sake of clarity, only the first hexamer in the X-ray coordinate file will be discussed.
Shown in Figure 1a is a ribbon representation of a single FdtD subunit. As can be seen, the polypeptide chain folds into two specific domains connected by the flexible loop (Ala 152 to Asn 161): an N-terminal β-helix motif delineated by Met 1 to Ala 152 and a cupin-fold formed by Asn 161 to the C-terminus. The β-helix motif consists of seven complete turns and harbors the active site responsible for N-acetylation. The cupin domain, which contains 12 β-strands and the C-terminal helix, is responsible for the 3,4-ketoisomerase activity of FdtD. Ten of the β-strands form an antiparallel flattened β-barrel that can be envisioned as two layers of four and six β-strands, respectively. Classical domain swapping occurs as the third and fourth β-strands, which adopt a β-hairpin motif, reach over from one subunit to the other in the dimer to complete the formation of a larger β-sheet.
Figure 1.

The structure of FdtD. A ribbon representation of one subunit of FdtD is shown in (a). The N-terminal and C-terminal domains, which contain the catalytic residues responsible for the N-acetyltransferase and 3,4-ketoisomerase activities of FdtD, are displayed in light blue and teal, respectively. A complete FdtD hexamer is depicted in (b). The dTDP and CoA ligands are shown in space-filling representations. The view in (b) is looking down a twofold rotational axis that relates one trimer to another within the hexamer. Thus the subunit colored in green in one trimer is related by a rotation of 180° to the subunit drawn in green in the second trimer. The FdtD hexamer displays 322 symmetry. The second twofold lies in the plane of the paper and is indicated by the horizontal arrow. The threefold axis also lies in the plane of the paper and is indicated by the vertical arrow. All figures were created using PyMOL.30
A ribbon representation of the complete FdtD hexamer is presented in Figure 1b. The molecule is quite elongated with overall dimensions of ~90 × 140 x 90 Å. The hexamer exhibits 322 symmetry with each subunit burying a surface area of ~3200 Å2. The six N-terminal β-helix domains of the hexamer form two “trimeric” stalks that are oriented on opposite sides of the protein. The cupin folds lie in the middle of the protein, wedged between the stalks.
The Cupin Domain
Given that the 3,4-keto isomerization step occurs prior to the amination and N-acetylation reactions in the biosynthesis of dTDP-Fuc3NAc (Scheme 1), the cupin domain will be discussed in detail first. It binds the dTDP ligand used in the crystallization experiments as shown in Figure 2a. A close-up view of the region surrounding the nucleotide is provided in Figure 2b. Tyr 286 forms a parallel stacking interaction with the thymine moiety of the nucleotide whereas the guanidinium group of Arg 205 lies within hydrogen bonding distance to O-4 of the ring. Both Arg 218 in subunit 1 and Arg 187 in subunit 2 of the dimer participate in electrostatic interactions with the pyrophosphoryl group of the nucleotide. Two ordered water molecules are observed: one that bridges the α- and β-phosphoryl groups of the nucleotide and a second that interacts with a β-phosphoryl oxygen. The backbone amide group of Gly 219 forms a hydrogen bond with one of the β-phosphoryl oxygens.
Figure 2.

The active site of the FdtD cupin domain. Electron density corresponding to the bound dTDP is presented in (a). The omit map, contoured at 2σ, was calculated at 2.2 Å resolution with coefficients of the form Fo-Fc, where Fo was the native structure factor amplitude and Fc was the calculated structure factor amplitude (the ligand was not included in the X-ray coordinate file so it did not contribute to the phasing). A close-up view of the region surrounding the dTDP ligand is shown in (b). The dashed lines indicate potential hydrogen bonds. Ordered water molecules are represented by small red spheres. Arg 187, highlighted in pink bonds, is contributed by the second subunit in the dimer as a result of domain swapping.
Shown in Figure 3a is a superposition of the FdtD cupin domain onto the 3,4-ketoisomerase subunit from A. thermoaerophilus, which was previously solved in the laboratory. Both enzymes catalyze the same reaction shown in Scheme 1. The α-carbons for these two proteins superimpose with a root-mean-square deviation of 1.0 Å. These two models represent the only sugar 3,4-ketoisomerases whose structures have been determined thus far by X-ray crystallography. Whereas the active sites are very similar between these two proteins (Figure 3b), it is noteworthy that Tyr 35 and His 95 in the A. thermoaerophilus protein have been replaced with a phenylalanine and a glycine residue, respectively, in the cupin domain of FdtD. Prior to this investigation, it was thought that Tyr 35 (or its equivalent) was conserved amongst the 3,4-ketoisomerase.21 The significance of these two substitutions in the cupin active site of FdtD will be described more fully below.
Figure 3.

Comparison of the cupin domain of FdtD and the A. thermoaerophilus 3,4-ketoisomerase (PDB code 2PA7). A superposition of the ribbon drawings for the A. thermoaerophilus 3,4-ketoisomerase (pink) and the FdtD cupin domain (light blue) is shown in (a). The bound dTDP ligands are drawn in stick representations. A close-up view of the two active sites, without the dTDP ligands, is displayed in (b). The A. thermoaerophilus 3,4-ketoisomerase and the FdtD cupin domain are colored in pink and light blue, respectively. The labels on the top refer to those amino acids found in the cupin domain of FdtD, whereas those on the bottom correspond to residues in the A. thermoaerophilus 3,4-ketoisomerase.
The sugar 3,4-ketoisomerases are an especially intriguing class of enzymes in that they do not require metal ions or cofactors for activity.1 All of them function on the same substrate, namely dTDP-4-keto-6-deoxyglucose (Scheme 1). Some retain the “glucose” configuration about C-4' in their products, whereas others invert the stereochemistry to yield the “galactose” configuration. Both the A. thermoaerophilus 3,4-ketoisomerase and the FdtD cupin domain invert the stereochemistry. Another 3,4-ketoisomerase from Streptomyce fradiae has been shown to retain the C-4' configuration.22 The three-dimensional structure of the S. fradiae enzyme is not yet known, however.
The overall mechanism of a sugar 3,4-ketoisomerase, whether it converts or retains the configuration about the hexose C-4' carbon, is thought to involve the initial abstraction of a proton from the pyranosyl C-3' carbon resulting in an enolate intermediate (Scheme 5). For the S. fradiae 3,4-ketoisomerase, which retains the “glucose” configuration about the C-4' carbon, it has been postulated that His 63 functions as the base to abstract the C-3' proton, that Arg 109 stabilizes the enolate intermediate, and that Tyr 49 functions as the active site acid that protonates the C-4' carbon on the opposite face of the sugar.21 The roles of His 63 and Tyr 49 in this “retaining” 3,4-ketoisomerase have been suggested through site-directed mutagenesis and model building experiments. We previously proposed for the A. thermoaerophilus 3,4-ketoisomerase, which inverts the configuration, that His 49 removes the proton from the sugar C-3' carbon and shuttles it to the sugar C-4' carbon on the same side of the pyranosyl moiety.3 This would result in the inversion of configuration about the C-4' carbon. We also suggested that His 51 functions in catalysis by shuttling protons between the C-3' and C-4' oxygens.
Scheme 5.

Thus far in our structural investigations of both the A. thermoaerophilus 3,4-ketoisomerase and FdtD, we have not trapped a nucleotide-linked sugar in the active site region. However, the structure of a C-3' epimerase from Streptomyces bikiniensis, another cupin family member, was recently solved in our laboratory in the presence of dTDP-6-deoxyglucose.23 The only difference between dTDP-6-deoxyglucose and dTDP-4-keto-6-deoxyglucose is the presence of a hydroxyl rather than keto group on the C-4' carbon. The S. bikiniensis C-3' epimerase functions in the biosynthesis of dTDP-6-deoxy-d-allose. As in the case of the 3,4-ketoisomerases, the mechanism for a C-3' epimerase is thought to involve the initial abstraction of a proton from the C-3' carbon. Importantly, the α-carbons for the cupin domain of FdtD and the S. bikiniensis C-3' epimerase superimpose with a root-mean-square deviation of 1.5 Å. In light of this structural correspondence, the position of dTDP-6-deoxyglucose in the C-3' epimerase structure provides an ideal template for modeling dTDP-4-keto-6-deoxyglucose into the cupin domain of FdtD.
Shown in Figure 4 is the FdtD substrate, dTDP-4-keto-6-deoxyglucose, positioned into the enzyme’s active site. The imidazole ring of His 221 is ideally located to remove the proton from the C-3' carbon and shuttle it to the C-4' carbon on the same side of the hexose ring, thereby inverting configuration about the C-4' carbon. Likewise, the imidazole ring of His 223 is in the proper location to function in proton transfer from the C-3' hydroxyl oxygen to the C-4' keto oxygen.
Figure 4.

Model of dTDP-4-keto-6-deoxyglucose bound in the FdtD cupin domain. The imidazole rings of His 221 and His 223 are located in positions ideally suited for proton transfer as indicated by the dashed lines. The carboxamide group of Gln 228 may function in stabilization of the enolate intermediate that is thought to occur during catalysis.
As noted above, the reactions for all sugar 3,4-ketoisomerases, whether they invert or retain the stereochemistry about the C-4' carbon, likely begin with the abstraction of a proton from the C-3' carbon to generate an enolate intermediate (Scheme 5). His 221 in the FdtD cupin domain is conserved in the A. thermoaerophilus 3,4-ketoisomerase as His 49 and in the S. fradiae 3,4-ketoisomerase as His 63. Although not tested by site-directed mutagenesis experiments, it was postulated that Arg 109 in the S. fradiae 3,4-ketoisomerase functions in stabilizing the enolate intermediate.21 Interestingly, the equivalent residue in the A. thermoaerophilus 3,4-ketoisomerase is a histidine, but more importantly in the FdtD cupin domain it is a glycine. This lack of conservation suggests that another amino acid is involved in stabilization of the enolate intermediate. With respect to the model shown in Figure 4, the carboxamide group of Gln 228 lies within hydrogen bonding distance of the sugar C-4' oxygen. Importantly, this residue is absolutely conserved amongst all of the 3,4-ketoisomerase amino acid sequences that have been reported thus far.
In the S. fradiae 3,4-ketoisomerase, which is a retaining enzyme, it was postulated that Tyr 49 lies on the opposite face of the pyranosyl ring of the substrate with respect to His 63, which initiates catalysis (Scheme 5). It was suggested to function, perhaps, by protonating the enolate intermediate. This would result in retention of stereochemistry about the C-4' carbon. In the FdtD cupin domain, the corresponding residue is a phenylalanine.
In light of this tyrosine to phenylalanine substitution, we subsequently conducted an amino acid sequence analysis of potential bifunctional 3,4-ketoisomerases/N-acetyltransferases (Figure S5 of Supporting Information). The alignments clearly reveal the presence of two different types of bifunctional enzymes, which will be referred to as Class A and B. In Class A, the cupin domain comes first in the amino acid sequence, followed by the β-helix motif. The P. multocida bifunctional enzyme, known to be a retaining 3,4-ketoisomerase, has this domain arrangement.24 All the enzymes in Class A contain a tyrosine in the same position as Tyr 49 in the S. fradiae 3,4-ketoisomerase, a retaining enzyme. The Class B bifunctional enzymes have the opposite arrangement of domains whereby the β-helix comes first in the amino acid sequence. In these enzymes, the tyrosine of the retaining enzymes has been replaced with a phenylalanine residue as in the FdtD cupin domain. Most likely these enzymes are involved in the production of dTDP-Fuc3NAc, rather than dTDP-Qui3NAc. It can thus be predicted that the O-antigens of those bacterial species having bifunctional enzymes belonging to Class B will contain Fuc3NAc rather than Qui3NAc. Indeed, the O-antigen of Pseudomonas syringae pv. tomato strain contains Fuc3NAc, and its bifunctional enzyme belongs to Class B (Figure S5 of Supporting Information).25
The β-Helix Motif
The last step in the biosynthesis of dTDP-Fuc3NAc is the N-acetylation of dTDP-Fuc3N (Scheme 1). In FdtD, this reaction is catalyzed by the β-helix domain, which lies at the N-terminus. As expected, electron density corresponding to CoA, utilized in the crystallization experiments, was observed binding to the β-helix domain as shown in Figure 5a. A close-up view of the binding pocket is presented in Figure 5b. The CoA adopts a decidedly curved conformation with an intra-molecular hydrogen bond formed between the hydroxyl group of the pantothenate unit and N-7 of the adenine ring. Both the β-mercaptoethylamine and pantothenate moieties interact with the protein via hydrogen bonding interactions through backbone amide and carbonyl groups (Asp 82, Ala 107, and Ala 125). Lys 83 bridges two of the pyrophosphoryl oxygens of the cofactor, whereas the side chains of Lys 131 and Arg 145 form salt bridges with the ribose phosphoryl group. An additional hydrogen bond occurs between the side chain of Thr 130 and the C-2 ribose hydroxyl group. Four water molecules lie within 3.2 Å of the CoA.
Figure 5.

Binding of CoA to the β-helix domain of FdtD. Electron density corresponding to bound CoA is shown in (a). The map was calculated as described in figure legend 2 and contoured at 2σ. A close-up view of the CoA binding pocket is displayed in (b). The CoA is highlighted in gray and orange bonds. Those residues belonging to subunits 1 and 2 are colored in blue and pink, respectively. Ordered water molecules are drawn as red spheres. The dashed lines indicate possible hydrogen bonding interactions between the cofactor and the protein.
In recent years, the X-ray structures of various N-acetyltransferases (or N-acyltransferases) that function on nucleotide-linked sugars have been reported.17,19,20,26–28 These enzymes all belong to the left-handed β-helix superfamily. From these studies it has become clear that the sugar N-acyltransferases have evolved into two separate protein classes that differ with respect to substrate binding orientations and reaction mechanisms. Class 1 enzymes employ a conserved active site histidine as a catalytic base to remove a proton from the amino group of the sugar substrate as it attacks the acetyl group of acetyl-CoA. Members of Class 1 include the N-acetyltransferases from C. jejuni and Caulobacter crescentus.19,20,26 Class 2 enzymes, such as those from T. thermosaccharolyticum, Bordetella petrii, and Bacillus cereus, have active sites devoid of enzymatic bases, and thus their reaction mechanisms are thought to proceed through substrate-assisted catalysis.17,27,28
Structurally, the β-helix domain of FdtD most closely matches the model for the B. petrii N-acetyltransferase as shown in Figure 6a. These two enzymes demonstrate an amino acid sequence identity and similarity of 37% and 53%, respectively and their α-carbons superimpose with a root-mean-square deviation of 1.3 Å (for 114 structurally equivalent α-carbons). As can be seen in Figure 6a, the ligands, CoA in FdtD and acetyl-CoA in the B. petrii N-acetyltransferase, align especially well. Both contain β- helices of seven turns, but the polypeptide chain of the B. petrii enzyme extends farther at the C-terminal end where it folds into a three-stranded anti-parallel β-sheet. In the B. petrii N-acetyltransferase, the regularity of the β-helix is interrupted by an extended loop between Asn 84 and Asp 100. In FdtD, this region is disordered.
Figure 6.

Comparison of the FdtD N-acetyltransferase domain with the N-acetyltransferases from B. petrii (PDB code 3MQH) and T. thermosaccharolyticum (3FSC). The structure of the β-helix domain of FdtD most closely resembles that of the enzyme from B. petrii. A superposition of the ribbon drawings for FdtD (blue) and the B. petrii enzyme (pink) is presented in (a). The B. petrii enzyme is larger with a globular domain following the β-helix motif. In both enzymes, the regularity of the β-helix is broken by an extended loop, which is ordered in the B. petrii enzyme, but disordered in the FdtD model. The conformation of the acetyl-CoA ligand, when bound to the B. petrii enzyme, matches that observed for CoA when complexed to FdtD. Shown in (b) is a superposition of the active site of the T. thermosaccharolyticum N-acetyltransferase with bound dTDP-Fuc3N (gray) onto the FdtD active site (blue). The dashed lines indicate hydrogen bonds that occur between the T. thermosaccharolyticum N-acetyltransferase and dTDP-Fuc3N. The residues in the T. thermosaccharolyticum enzyme responsible for anchoring the sugar into the active site include His 134, Glu 141, Asn 159, and Asp 160. These residues correspond to Lys 56, Asp 63, Asn 81, and Asp 82 in FdtD. Mutations of these residues in the T. thermosaccharolyticum enzyme did not result in loss of enzymatic activity.
Whereas these two enzymes demonstrate high structurally homology, the substrate for the B. petrii N-acetyltransferase, UDP-2-acetamido-3-amino-2,4-dideoxy-d-glucoronic acid, is not a good mimic for the FdtD substrate. The structure of the N-acetyltransferase from T. thermosaccharolyticum, on the other hand, has been solved in the presence of dTDP-Fuc3N, which is also the substrate for FdtD.17 Thus a superposition of the structures for the T. thermosaccharolyticum protein and FdtD provides significant insight as to the location of the sugar substrate in the FdtD active site (Figure 6b). Note that the CoA ligands bound to these two enzymes superimpose well. Additionally, in the FdtD structure, electron density was observed that corresponded to a thymine molecule at low occupancy, most likely a contamination of the dTDP solutions used in the crystallization experiments (Figure S6 in Supporting Information). The position of the thymine group matches well with the thymine moiety observed in the structural analysis of the T. thermosaccharolyticum N-acetyltransferase.
In the T. thermosaccharolyticum enzyme, Glu 141, Asn 159, Asp 160 from one subunit of the trimer, and His 134 from another subunit, are involved in hydrogen bonding to the dTDP-Fuc3N ligand (Figure 6b). Site-directed mutagenesis studies have demonstrated that none of these residues is required for its enzymatic activity. The corresponding residues in FdtD are Asp 63, Asn 81, Asp 82, and Lys 56 (Figure 6b). If the location of dTDP-Fuc3N in the T. thermosaccharolyticum enzyme active site is a good mimic for substrate positioning in FdtD, then the only residue that lies near the C-3' amino group is Asn 81, which cannot serve as an enzymatic base. It can thus be concluded that FdtD belongs to the Class 2 sugar N-acetyltransferases. Amino acid sequence analyses suggest that the reaction mechanisms for the N-acetyltransferase components of both the Class A and Class B bifunctional enzymes proceed through substrate-assisted catalysis whereby the sulfur of acetyl-CoA ultimately serves as the catalytic base by accepting a proton from the sugar amino group.
In summary, the investigation described herein represents the first detailed analysis of a bifunctional 3,4-ketoisomerase/N-acetyltransferase that functions on nucleotide-linked sugars. The enzyme adopts a hexameric quaternary structure with each subunit displaying a distinctly bilobal appearance. The N-acetyltransferase domain of the subunit assumes a left-handed β-helix motif whereas the isomerase domain adopts a cupin-type fold. Amino acid sequence alignments indicate that there are two types of bifunctional enzymes. The primary sequences of the Class A enzymes show that the cupin domain is followed by the left-handed β-helix motif. These enzymes catalyze the formation of dTDP-QuiNAc, which has the S-configuration (or glucose configuration) about the hexose C-4'. In these proteins, the tyrosine residue that is thought to act as an active site acid is conserved. The primary sequences of the Class B bifunctional enzymes demonstrate that the β-helix motif is followed by the cupin-domain as in the case of FtdD. These enzymes catalyze the formation of dTDP-FucNAc, which has the R-configuration about the hexose C-4', and the conserved tyrosine has been replaced with a phenylalanine residue.
Supplementary Material
Acknowledgments
We thank Professor Grover L. Waldrop for helpful discussions. A portion of the research described in this paper was performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source (U. S. Department of Energy, Office of Biological and Environmental Research, under Contract DE-AC02-06CH11357). We gratefully acknowledge Dr. Norma E. C. Duke for assistance during the X-ray data collection at Argonne and Christine N. Bradford for help with the NMR experiments.
Glossary
Abbreviations
- CoA
coenzyme A
- HEPES
N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid
- HEPPS
N-2-hydroxyethylpiperazine-N'-3-propanesulfonic acid
- HPLC
high-performance liquid chromatography
- IPTG
isopropyl-β-d-1-thiogalactopyranoside
- LB
lysogeny broth
- NADP+
nicotinamide adenine dinucleotide phosphate, oxidized
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced
- NMR
nuclear magnetic resonance
- NTA
nitrilotriacetic acid
- PCR
polymerase chain reaction
- TB
terrific broth
- dTDP
thymidine diphosphate
- Tris
tris-(hydroxymethyl)aminomethane
Footnotes
This research was supported in part by an NIH grant (DK47814 to H. M. H.)
The authors have no competing financial interests.
Associated Content
Supporting Information
Figures S1, S2, S3, S4, S5, and S6. This material is available free of charge via the Internet at http://pub.acs.org.
Coordinates have been deposited in the Protein Data Bank (entry 4MZU).
References
- 1.Pfoestl A, Hofinger A, Kosma P, Messner P. Biosynthesis of dTDP-3-acetamido-3,6-dideoxy-alpha-d-galactose in Aneurinibacillus thermoaerophilus L420-91T. J Biol Chem. 2003;278:26410–26417. doi: 10.1074/jbc.M300858200. [DOI] [PubMed] [Google Scholar]
- 2.Holden HM, Cook PD, Thoden JB. Biosynthetic enzymes of unusual microbial sugars. Curr Opin Struct Biol. 2010;20:543–550. doi: 10.1016/j.sbi.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis ML, Thoden JB, Holden HM. The X-ray structure of dTDP-4-keto-6-deoxy-d-glucose-3,4-ketoisomerase. J Biol Chem. 2007;282:19227–19236. doi: 10.1074/jbc.M702529200. [DOI] [PubMed] [Google Scholar]
- 4.Dunwell JM, Culham A, Carter CE, Sosa-Aguirre CR, Goodenough PW. Evolution of functional diversity in the cupin superfamily. Trends Biochem Sci. 2001;26:740–746. doi: 10.1016/s0968-0004(01)01981-8. [DOI] [PubMed] [Google Scholar]
- 5.Dunwell JM, Purvis A, Khuri S. Cupins: the most functionally diverse protein superfamily? Phytochemistry. 2004;65:7–17. doi: 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 6.Galperin MY, Koonin EV. Divergence and convergence in enzyme evolution. J Biol Chem. 2012;287:21–28. doi: 10.1074/jbc.R111.241976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brettar I, Christen R, Hofle MG. Shewanella denitrificans sp. nov., a vigorously denitrifying bacterium isolated from the oxic-anoxic interface of the Gotland Deep in the central Baltic Sea. International journal of systematic and evolutionary microbiology. 2002;52:2211–2217. doi: 10.1099/00207713-52-6-2211. [DOI] [PubMed] [Google Scholar]
- 8.Thoden JB, Holden HM. Molecular structure of galactokinase. J Biol Chem. 2003;278:33305–33311. doi: 10.1074/jbc.M304789200. [DOI] [PubMed] [Google Scholar]
- 9.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution-from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 10.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 11.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 4):849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 8):965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 15.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 16.Thoden JB, Schaffer C, Messner P, Holden HM. Structural analysis of QdtB, an aminotransferase required for the biosynthesis of dTDP-3-acetamido-3,6-dideoxy-alpha-d-glucose. Biochemistry. 2009;48:1553–1561. doi: 10.1021/bi8022015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thoden JB, Cook PD, Schaffer C, Messner P, Holden HM. Structural and functional studies of QdtC: an N-acetyltransferase required for the biosynthesis of dTDP-3-acetamido-3,6-dideoxy-alpha-d-glucose. Biochemistry. 2009;48:2699–2709. doi: 10.1021/bi802313n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubiak RL, Holden HM. Combined structural and functional investigation of a C-3"-ketoreductase involved in the biosynthesis of dTDP-L-digitoxose. Biochemistry. 2011;50:5905–5917. doi: 10.1021/bi200514b. [DOI] [PubMed] [Google Scholar]
- 19.Thoden JB, Reinhardt LA, Cook PD, Menden P, Cleland WW, Holden HM. Catalytic mechanism of perosamine N-acetyltransferase revealed by high-resolution X-ray crystallographic studies and kinetic analyses. Biochemistry. 2012;51:3433–3444. doi: 10.1021/bi300197h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olivier NB, Imperiali B. Crystal structure and catalytic mechanism of PglD from Campylobacter jejuni. J Biol Chem. 2008;283:27937–27946. doi: 10.1074/jbc.M801207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tello M, Rejzek M, Wilkinson B, Lawson DM, Field RA. Tyl1a, a TDP-6-deoxy-d-xylo-4-hexulose 3,4-isomerase from Streptomyces fradiae: structure prediction, mutagenesis and solvent isotope incorporation experiments to investigate reaction mechanism. Chembiochem. 2008;9:1295–1302. doi: 10.1002/cbic.200800021. [DOI] [PubMed] [Google Scholar]
- 22.Melancon CE, 3rd, Hong L, White JA, Liu YN, Liu HW. Characterization of TDP-4-Keto-6-deoxy-d-glucose-3,4-ketoisomerase from the D-Mycaminose Biosynthetic Pathway of Streptomyces fradiae: In Vitro Activity and Substrate Specificity Studies. Biochemistry. 2007;46:577–590. doi: 10.1021/bi061907y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kubiak RL, Phillips RK, Zmudka MW, Ahn MR, Maka EM, Pyeatt GL, Roggensack SJ, Holden HM. Structural and functional studies on a 3'-epimerase involved in the biosynthesis of dTDP-6-deoxy-d-allose. Biochemistry. 2012;51:9375–9383. doi: 10.1021/bi3012737. [DOI] [PubMed] [Google Scholar]
- 24.Harper M, St Michael F, Vinogradov E, John M, Boyce JD, Adler B, Cox AD. Characterization of the lipopolysaccharide from Pasteurella multocida Heddleston serovar 9: identification of a proposed bi-functional dTDP-3-acetamido-3,6-dideoxy-alpha-d-glucose biosynthesis enzyme. Glycobiology. 2012;22:332–344. doi: 10.1093/glycob/cwr147. [DOI] [PubMed] [Google Scholar]
- 25.Zdorovenko GM, Solyanik LP, Yakovleva LM, Paramonov NA. Characterization of O-antigens from different strains of Pseudomonas syringae pv. tabaci. Biochemistry. Biokhimiia. 1997;62:28–37. [PubMed] [Google Scholar]
- 26.Rangarajan ES, Ruane KM, Sulea T, Watson DC, Proteau A, Leclerc S, Cygler M, Matte A, Young NM. Structure and active site residues of PglD, an N-acetyltransferase from the bacillosamine synthetic pathway required for N-glycan synthesis in Campylobacter jejuni . Biochemistry. 2008;47:1827–1836. doi: 10.1021/bi702032r. [DOI] [PubMed] [Google Scholar]
- 27.Thoden JB, Holden HM. Molecular structure of WlbB, a bacterial N-acetyltransferase involved in the biosynthesis of 2,3-diacetamido-2,3-dideoxy-d-mannuronic acid. Biochemistry. 2010;49:4644–4653. doi: 10.1021/bi1005738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kubiak RL, Holden HM. Structural studies of AntD: an N-Acyltransferase involved in the biosynthesis of D-Anthrose. Biochemistry. 2012;51:867–878. doi: 10.1021/bi201650c. [DOI] [PubMed] [Google Scholar]
- 29.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 30.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific; 2002. The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.