

Abstract
Objectives
To review the contributions of cardiovascular disease to Alzheimer’s disease and Vascular Dementia.
Methods
Review of the literature
Results
Alzheimer’s disease and Vascular Dementia both share significant risk attributable to cardiovascular risk factors. Hypertension and hypercholesterolemia at midlife are significant risk factors for both subsequent dementia. Diabetes and obesity are also risk factors for dementia. Stressful medical procedures, such as coronary artery bypass and graft operations also appear to contribute to the risk of Alzheimer’s disease. Apolipoprotein E is the major risk factor for Alzheimer’s disease. Apolipoprotein E does not appear to contribute to Alzheimer’s disease by increasing serum cholesterol, but it might contribute to the disease through a mechanism involving both Aβ and an increase in neuronal vulnerability to stress.
Discussion
The strong association of cardiovascular risk factors with Alzheimer’s disease and Vascular dementia suggest that these diseases share some biological pathways in common. The contribution of cardiovascular disease to Alzheimer’s disease and Vascular Dementia suggest that cardiovascular therapies might prove useful in treating or preventing dementia. Anti-hypertensive medications appear to be beneficial in preventing vascular dementia. Statins might be beneficial in preventing the progression of dementia in subjects with Alzheimer’s disease.
Keywords: beta-amyloid, cholesterol, diabetes, hypertension, statins
Introduction
Heart disease is the major killer of people in the developed world. Death is frequently not the only outcome of disease processes, though. The vascular insufficiency associated with heart disease impairs the function of many organs, including the brain. Multiple studies indicate that cerebral blood flow declines with age and is exacerbated in individuals with AD. Cerebral blood flow can decrease due to systemic (e.g., cardiac insufficiency) or more localized effects (e.g., loss of elasticity associated with atherosclerotic buildup; direct blockage of cerebral blood flow by plaque formation in the cerebral vasculature). Reduced cerebral blood flow can directly impair cognitive function and leads to a syndrome termed vascular dementia (VaD), which is prevalent in the elderly. Declines in cerebral blood flow are also associated with Alzheimer’s disease (AD), the most common form of dementia in the elderly. The causes of the loss of cerebral blood flow in AD are poorly understood, but are likely to result both from cardiovascular disease and from the accumulation of β-amyloid in and around the cerebral vasculature. The vascular insufficiency that occurs in VaD and AD also renders the brain more susceptible to stroke, which can further impair cognitive function. The effects of the vasculature on the brain are more severe than on other organs because our cognitive function is what defines our identity. The body can tolerate a 50% loss of renal or hepatic function without serious consequences or fundamentally changing our interaction with world. A 50% loss of cognitive function, though, causes a devastating loss of our ability to socialize, to carry out activities of daily living and to function as a human being. All of these different outcomes emphasize the critical dependence of cognitive function on vascular function.
VaD and AD: Two different pathologies
The differing etiologies of VaD and AD are emphasized by the strikingly different pathologies of these two disorders. AD is associated with the accumulation of inclusions in the neuropil. The pathology of AD is defined by the accumulation of neuritic plaques in the extracellular neuropil and neurofibrillary tangles in neurons [1]. Neuritic plaques are composed of a small peptide, termed Aβ, while neurofibrillary tangles are composed of fibrils of tau protein. Aβ is derived from its parent protein, amyloid precursor. A small fraction of amyloid precursor is cleaved to yield a peptide that is 40 amino acids long, termed Aβ40 [2]. Approximately 5% of the Aβ is slightly longer, and is termed Aβ42; this longer form of Aβ has a much greater tendency to aggregate and tends to form the seed for larger accumulations of Aβ [3]. Plaques and tangles tend to accumulate in the cerebral cortex, and correspondingly, the cerebral cortex is the area of the brain that exhibits the greatest reduction in volume during the course of AD. Vascular pathology is frequently observed in cases of AD, but the pathology that is present tends to occur in the cortex [4]. Vascular dementia, by definition, is associated with vascular pathology. Subjects with VaD exhibit abundant white matter intensities and abundant small infarcts. The infarcts, though, are commonly subcortical. AD and VaD also share some features in common [4]. Both AD and VaD share the pathology of a cholinergic deficit. Heart disease is a risk factor for both AD and VaD, and the two diseases can occur concurrently [5]. Many individuals with dementia that cannot be accurately classified are either AD or VaD [6]. The overlap between the two diseases raises the possibility that the two illnesses share some aspects of their pathophysiology in common. The weakened state caused by reduced cerebral blood flow likely provokes neuronal dysfunction in VaD and exacerbates damage caused by oligomeric Aβ and tau in AD.
Heart Disease and AD
Multiple epidemiological studies demonstrate that patients with heart disease are at higher risk of AD. The incidence of AD in subjects over 65 years is 2.2%/yr, but this figure jumps to 3.4%/yr in subjects with hypertension and 5.7%/yr in those with peripheral artery disease [7]. Apolipoprotein E4 (apo E4), which is single most important genetic risk factor for AD (this is discussed in more detail below), exacerbates this effect. Subjects positive for the apo ε4 allele and cardiovascular disease perform worst on cognitive exams [8]. The more the number of cardiovascular risk factors the greater risk of AD. Subjects who have diabetes, hypertension, heart disease and smoke have a risk of incident AD that is elevated 3.4-fold, compared to subjects without cardiovascular risk factors [5]. Hypertension and CVD are intimately linked with cholesterol metabolism. Mutations in genes required for processing of cholesterol in the circulation, such as the low-density lipoprotein receptor, greatly increase levels of cholesterol and also greatly increase heart disease [9]. Cholesterol is also important to the general population. Increased levels of cholesterol are strongly associated with increased vascular and heart disease. Serum cholesterol levels also affect the incidence of AD, however the linkage between cholesterol and AD is not nearly as strong as is the linkage between cholesterol and heart disease [10, 11]. Elevated cholesterol is associated with AD, but factoring out the effects of apo E4, which increases cholesterol, eliminates effect observed in elderly subjects but leaves a strong signal for midlife [12, 13].
Analyses of midlife factors related to heart disease highlight the fact that AD is a disease that evolves slowly. Indices related heart disease, such as obesity, hypertension and cholesterol show a strong relationship between elevation during midlife and subsequent incidence of AD [12, 14]. For instance, obesity at midlife exhibits almost a 2-fold increase in risk of AD [15]. Hypertension at midlife shows over a 3-fold elevation of risk, and elevated cholesterol at midlife shows a 2.1-fold increase in risk [12]. The striking effect at midlife, particularly for cholesterol, contrasts with ambiguous data from late-life studies where obesity and cholesterol, in particular, are not associated with increased incidence of AD [16]. The midlife association might seem puzzling, because AD doesn’t appear until years after midlife. The best explanation appears to be that the stresses associated with vascular disease affect the pathophysiology of AD only a chronic stress. Acute stresses might injure the brain, but the type of injury associated with an acute stress might not lead to the accumulation of neuritic plaques and neurofibrillary tangles, which are required for the diagnosis of AD. These data are consistent with a model in which the neuritic plaques and neurofibrillary tangles that are associated with AD require many years to form. Cerebrovascular insufficiency associated with elevated cholesterol or hypertension might be insufficient to cause neurodegeneration on its own, but might be sufficient to augment production of Aβ, accumulation of Aβ or accumulation of fibrillar tau. This augmentation would increase the incidence of AD by accelerating the accumulation of pathology. In this model, cardiovascular disease might enhance this pathology only when present over many years, such as would occur in individuals with midlife elevation of blood pressure or cholesterol.
Diabetes and AD
Diabetes represents one of the strongest risk factors for AD. Subjects with diabetes at midlife show increased rates of cognitive decline [17, 18]. The presence of diabetes at midlife almost doubles the risk of incident AD later in life [19]. In addition, co-morbidity of diabetes and cardiovascular risk factors (hypertension, elevated cholesterol) increases the risk of AD in subjects over 3-fold [5]. Whether the pathophysiology of diabetes directly stimulates the pathophysiology of AD remains an open question. Insulin clearly modulates pathways that could directly affect Aβ deposition and neurodegeneration. Insulin crosses the blood brain barrier where it acts on neurons. Insulin stimulates glucose uptake and stimulates the signal transduction cascade mediated by the kinase Akt, which is associated with neuronal survival [20, 21]. Insulin is degraded by insulin degrading enzyme (IDE), and IDE levels are reduced in AD and in animal models of AD [22, 23]. IDE degrades Aβ in addition to degrading insulin [24]. The reduced levels of IDE in AD might increase the accumulation of Aβ. Reduced IDE in AD does not explain the link between diabetes and AD because IDE levels are increased in type II diabetes, particularly those on hypoglycemic medications. The alternative possibility is that diabetes increases the risk of AD by increasing cardiovascular disease. Cardiovascular disease is a frequent outcome of diabetes, and the discussion above presented the case suggesting that CVD increases the risk of AD.
Diabetes also frequently occurs in combination with obesity, hypercholesterolemia and hypertension. This syndrome is termed metabolic syndrome (Table I) [25]. Metabolic syndrome is rapidly becoming a major health issue, especially in developed countries, and the incidence of metabolic syndrome is expected to rise dramatically in the coming decades. The increasing rates of metabolic syndrome in an aging population will undoubtedly lead to increasing rates of AD because the factors that make of metabolic syndrome are also the risk factors for AD.
Table I.
Metabolic syndrome (ATPIII) criteria:
|
Cerebral Blood Flow
Severe cardiovascular disease is associated with reduced blood flow to end organs. Reduced cerebral blood flow has emerged as one of the most consistent observations among subjects with AD. Subjects with AD have a 7–10% reduction in blood flow [26, 27]. The reduction in cerebral blood flow correlates strongly with indices of cognition. Among subjects with mild cognitive impairment, those with the greatest cognitive decline had the largest decreases in cerebral blood flow [28]. Volumes of the hippocampal and amygdala also correlate with cerebral blood flow, with reduced cerebral blood flow being associated with smaller volumes. One of the critical questions is whether the reduction in cerebral blood flow is driven only by the cardiovascular disease or whether the process of AD also contributes to the reduction in cerebral blood flow. Studies of subjects with mild cognitive impairment indicate that reduced cerebral blood flow is present at this relatively early stage, and that the blood flow correlates with the cognitive status and future progression [28]. This result indicates that reductions in blood flow occur early in the course of AD and raise the possibility that the process of AD leads to reduced cerebral blood flow, although it does not rule out the possibility that vascular disease is driving the changes. A strong case can be made for the argument that decreased blood flow caused by cardiovascular disease would increase stress on the brain, which could speed the course of AD. The critical factor appears to be delivery of blood to sites distal from the heart. The type of CVD that is most tightly linked to cognitive decline and incidence of AD is peripheral vascular disease [7]. For example, Newman and colleagues showed that incident dementia is increased by about 50% in subjects with CVD without peripheral artery disease, but by 150% in subjects with CVD plus peripheral artery disease [7].
Coronary Artery Bypass and Graft Operations: A source of stress on the brain?
Another source of stress on the brain is iatrogenic. Major heart surgery, such as coronary artery bypass operations (CABG), is commonly associated with a short period in which cognitive function is depressed. The cognitive loss associated with major surgery is termed post-operative cognitive decline. This decline occurs in 20–50% of individuals within 12 weeks after a CABG [29, 30]. The decline is transient, and cognitive function typically recovers by 12 months after the operation. Whether the recovery is truly complete, though, remains an open question. Subjects at risk for AD might recover sufficiently from the stress of the operation to exhibit normal cognition, as judged by a simple cognitive test. This apparent recovery, though, might mask an underlying deficit that becomes apparent only the aging/neurodegenerative process proceeds. To test this, we examined the incidence of AD among patients receiving surgical treatment for the coronary artery disease [31]. We compared the incidence of AD in subjects who whose coronary artery disease was treated with a CABG operation to those whose coronary artery disease was treated with insertion of stents. After controlling for confounding factors, such as co-morbid illnesses, length of hospital stay, age and gender, we observed an increased incidence of AD that became statistically significant 5 years after the operation. By this time point, subjects receiving CABG operations exhibited a 50% increase in the incidence of AD [31]. Only one other study has followed cases this far past the CABG operation, and this study failed to observe an increase in the incidence of AD associated with CABG operations [30]. The difference between the two studies might lie in the nature of the population. Our study focused on the Veterans Affairs population, which is a population that generally has a larger number of co-morbid illnesses than the typical American. If other studies confirm that the incidence of AD is increased among patients receiving major operations, part of the future decision tree for treatment of coronary artery disease might include an assessment of the risk for incident AD, perhaps based on family history, apo E genotype and co-morbid illnesses.
Apo E, Cholesterol and AD: Distinguishing between neurobiology and vascular biology
The importance of apo E has been alluded to throughout this article. The apo ε4 genotype is overwhelmingly the single strongest genetic risk factor for late onset AD. Over 1200 studies have confirmed this observation, and factoring the effect of apo E4 is now standard for study examining risk factors for AD. These studies indicate that the apo ε4 allele is associated with an increased incidence of AD in a dose dependent manner [32]. The increased incidence appears to occur because the apo ε4 genotype reduces the age of onset of AD by about 10 years [33]. Individuals with apo E tend to get AD between age 55–65, instead of the typical age of onset, which is ages 65–75 [33]. Apo E functions as a lipid transport protein [34]. It acts with apo AI in forming lipoprotein particles in the peripheral circulation and is the major lipid transport protein for cholesterol in the central nervous system, where apo AII is absent. Apo E has three isoforms in human, termed apo E2, 3 and 4. Expression of the apo E4 protein in humans leads to hypercholesterolemia [35]. The function of apo E in the brain, however, is more complicated because apo E is important for synaptic maintenance and apo E also binds Aβ [36, 37]. Multiple studies have examined the relative importance of the function of apo E4 in transporting cholesterol compared to the other actions of apo E4, such as promoting Aβ oligomerization. In each of these studies, the higher incidence of AD correlates with the apo ε4 genotype rather than elevated levels of serum cholesterol [38]. The linkage of AD with the presence of apo E4, rather than with serum cholesterol levels, suggests that apo E4 is increasing risk of AD through a mechanism unrelated to serum cholesterol. Two types of mechanism could account for the tendency of apo E4 to reduce the age of onset of AD. One possibility is that apo E4 is directly harmful to neurons (e.g., by reducing axonal outgrowth), a second possibility is that apo E4 increases the accumulation of aggregated Aβ. In experimental models apo E4 reduces neurogenesis [39]. Apo E4 also appears to render humans more vulnerable to neurological disease. Apo E4 increases the risk of dementia after traumatic brain injury [40, 41]. Apo E4 also increases the risk of Parkinson disease [42]. However, apo E4 does not increase the risk of other neurodegenerative diseases, such as Huntington disease [43]. On the other hand, a large body of data consistently support the hypothesis that apo E4 reduces the age of onset of AD by promoting the accumulation of Aβ. Apo E binds Aβ, and apo E4 increases Aβ fibrillization and also reduces uptake of Aβ (and cholesterol). Transgenic mice that over-express apo E4 show increased accumulation of aggregated Aβ and more severe pathology. Data are clearly present for both mechanisms, and both mechanisms account for how apo E4 contributes to the pathophysiology of AD.
Vascular Dementia and AD
Vascular dementia (VaD) complements AD as one of the main causes of dementia in the elderly. The incidence of stroke increases dramatically with advancing age at roughly half the rate of that that seen for AD. The incidence of stroke doubles every decade beyond the age of 55, while the incidence of AD doubles every five years of age beyond 65 [44]. The prevalence of post-stroke dementia (PSD) is about 30%, with 7% occurring within the first year and up to 48% of stroke patients developing dementia after 25 years [45]. Dementia developing from strokes, though, is just one aspect of vascular disease. Micro-infarcts represent an important contribution to dementia, producing VaD. The occurrence of VaD is strongly linked to untreated hypertensive illness [4]. The pathology associated with VaD differs distinctly from that of AD because the infarcts tend to occur in sub-cortical regions such as the amygdala, thalamus or white matter tracts [4]. The high prevalence of stroke, VaD and AD, combined with shared cardiovascular risk factors, create a large degree of co-morbidity of these disorders in the aging population [6]. Subjects with mixed dementia respond to medications that benefit AD subjects, but the medications are less effective than with cases of pure AD. Given the aging of the population and improved survival post-stroke, it is likely that the incidence and prevalence of PSD will increase in the coming decades.
Treatment
The strong linkage between cardiovascular disease and dementia has important treatment implications (Table II). If hypertension and cholesterol had causal roles in dementia, one might expect that therapies reducing hypertension and lowering cholesterol would be beneficial. The clinical data generated to date, however, are surprisingly ambiguous for pharmacotherapy of AD, although epidemiological studies suggest that non-pharmacological approaches might also benefit AD. Multiple studies indicate that exercise is beneficial for the vascular system, and it appears that the brain is much like the vascular system. Exercise appears to be protective against AD [46], and the protection is particularly for individuals who carry the apo ε4 allele [47].
Table II.
Common Risk Factors for Cardiac Disease and AD
| Advancing age |
| ApoE4 allele |
| Elevated cholesterol and LDL |
| Hypertension |
| Diabetes (especially insulin-dependent) |
| Cigarette smoking |
| Elevated homocysteine or low folate levels |
Interest in the potential use of statins for therapy of AD stem in part from cross-sectional epidemiological studies indicating that subjects taking statins had a lower incidence and prevalence of AD [10, 48–50]. The case for the potential benefit of statins in treating AD is particularly interesting because of the multiple intersection lines of evidence suggesting that statins should be beneficial. As I will outline below, the clinical data are surprisingly ambiguous despite extensive preclinical data suggesting that statins should work. The role of apo E4 in AD necessarily sparked interest in the potential role of cholesterol in AD. This work lead to the discovery that production of Aβ is highly dependent on cellular cholesterol metabolism. Studies demonstrated that decreasing cellular cholesterol, either by treatment with statins or by treatment with β-cyclomethyldextran, also decreased secretion of Aβ. The exact mechanism depends on the mode of cholesterol reduction. Production of Aβ is also regulated by other sterol species linked to the cholesterol metabolic pathway. For instance, oxysterols, which are oxidative products of cholesterol, decrease Aβ production in neurons. Inhibiting the enzyme that produces cholesterol esters, decreases Aβ production. The reason for the linkage appears to be that Aβ modulates sphingomyelin metabolism and also modulates cholesterol production, though to a lesser degree. This work also extends to the in vivo setting. Statins decrease Aβ production in guinea pigs and mice, and inhibiting cholesterol production reduces the accumulation of neuritic plaques in mice genetically engineered to over-produce human Aβ.
The problem with the cholesterol-AD link appears to lie in the translation from the animal to the human context. Statins appear able to reduce cholesterol metabolism in the brain because subjects treated with statins (lovastatin, simvastatin or pravastatin) for 6 weeks all show a 20% reduction in the brain selective oxysterol, 24(S) hydroxycholesterol [51]. Whether the reduction of cholesterol metabolism translates to a reduction in Aβ levels is ambiguous. One study found a reduction in Aβ associated with statin treatment [52]. Two studies show changes but were insufficiently powered to determine the significance of the change [53, 54]. Two other studies have failed to observe decreases in Aβ associated with statin usage [55, 56]. Studies determining whether statins offer a the therapeutic benefit to subjects with AD or at risk for AD are similarly ambiguous. As mentioned above, the initial wave of epidemiological studies suggested that statin use is associated with a reduced risk of AD. However, recent studies have examined the issue using analyses based on multiple wave formats or prospective hazard curves [57–59]. These studies fail to observe any benefit of statins, when the data are analyzed using the prospective formats, although the data do show a reduced association of statins and AD in cross-sectional analyses of the data. These results suggest that the reduced association of statins with AD observed in the earlier studies was the result of uncorrected bias in the databases. Prospective clinical trials of statins and AD are also ambiguous. Two large studies failed to observe any reduction in the incidence of dementia among statin users [60, 61]. Incident dementia might be somewhat different than progression of dementia because studies of incident dementia examine subjects who do not have AD, while studies of the progression of dementia examine studies who have AD. One difference between the two paradigms might be the relative importance of inflammation. Statins are known to reduce inflammation [62]. Our own studies of AD subjects who were either on or off statin therapy indicated no difference in the amount of plaque pathology, but a significant reduction in the amount of inflammatory pathology [63]. Two small studies have observed delayed progression of some parameters of cognition in subjects with AD. Simons and colleagues noted that simvastatin reduces the progressive loss of cognitive function with one test (mini-mental status test), but not with a second test (ADAS-Cog) [53]. Sparks recently observed that AD subjects treated with Atorvastatin showed reduced cognitive loss for most measures of cognition, but the reduction only achieved statistical significance for the global depression scale [54]. These data raise the possibility that statins might not prevent AD, but might reduce the progression of AD.
The clinical data in favor of anti-hypertensive therapy for prevention of dementia appears to be stronger than that for statins. One reason might be that anti-hypertensive therapy has a clear benefit for subjects with VaD. Multiple studies show a clear reduction in incidence of dementia among users of anti-hypertensive therapies [64, 65]. The reduction, though, might be greatest for those with vascular dementia. Forette and colleagues observed that dementia was reduced among those on anti-hypertensive medications, but only in subjects who had evidence of strokes [66]. The reason for this difference might lie in the differing etiologies of AD compared to VaD or stroke. The pathologies of AD and VaD are strikingly different. Classic AD typically lacks micro-infarcts, while VaD is characterized by micro-infarcts. If anti-hypertensive therapy primarily protects against the occurrence of micro-infarcts, then one might expect anti-hypertensive therapy to benefit only subjects at risk for VaD. This distinction emphasizes the increasing importance of accurate clinical tests to identify dementia and distinguish between different types of dementia. If therapies have different outcomes depending on whether the patient has AD or VaD, then we must be able to distinguish between these two diseases accurately. Three strategies are being pursued to develop accurate clinical tests to differentiate AD from VaD. The most promising avenue of research lies with neuroimaging. AD is characterized by the accumulation of aggregated Aβ, which is a material that has strong β-pleated sheet structure. Klunk and colleagues have generated a brain permeable analog of thioflavine, which can be labeled for imaging by positron emission tomography and is a compound that binds β-pleated sheets. Preliminary evidence suggests that this compound, Pittsburgh compound-B, identifies subjects with AD [67]. A second approach is to use ELISA assays based on levels of tau and Aβ in cerebrospinal fluid [68]. This approach also holds promise, although it is unclear whether routine use of lumbar puncture would be accepted in the clinic. A final approach is to use high throughput technology, such as SELDI mass spectroscopy, to identify biomarkers in human serum. Many investigators are pursuing this approach, but no consensus has developed over a group of markers to use for clinical testing.
Summary
The pathology of AD differs greatly from that of VaD or stroke. AD exhibits cortical pathology with the accumulation of neuritic plaques and neurofibrillary tangles. VaD exhibits subcortical pathology characterized by abundant white matter intensities and abundant small infarcts. Despite these differing pathologies, both diseases share in common risk factors associated with cardiovascular disease (fig. 1). Hypertension, obesity and diabetes are all associated with increasing levels of AD and VaD. The association of hypertension and cholesterol with AD is only strongly apparent when examined at midlife, which suggests that only chronic vascular factors elicit pathology characteristic of AD. Therapies for cardiovascular disease appear to be quite beneficial for subjects at risk for VaD. The value of medicines for treating cardiovascular disease in treating AD is being examined in detail. Statins offer a possibility of reducing the progression, but not incidence, of AD. Anti-hypertensive therapy is less beneficial to cases of pure AD, but since many individuals have mixed dementia, this type of therapy might be quite valuable for many patients suffering from dementia.
Figure 1.
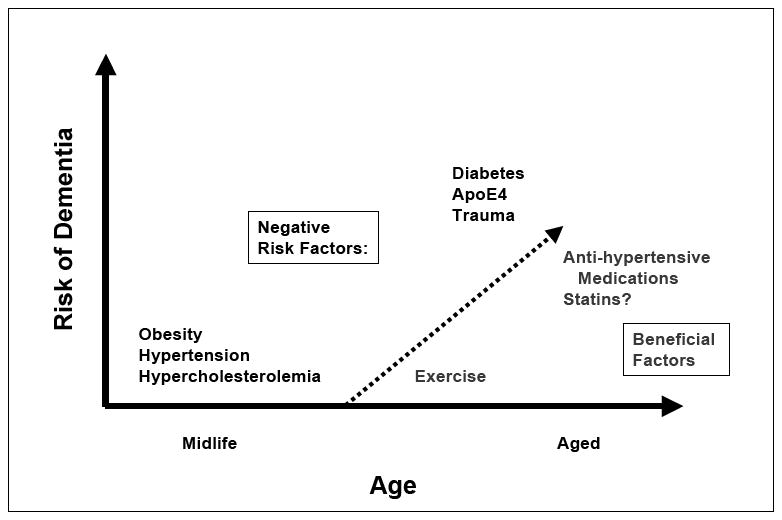
Summary of positive and negative factors influencing the incidence or progression of AD
Abbreviations
- AD
Alzheimer’s disease
- Apo E
Apolipoprotein E
- CVD
Cardiovascular disease
- CABG
Coronary Artery Bypass and Graft
- IDE
Insulin Degrading Enzyme
- PSD
Post Stroke Dementia
- VaD
Vascular Dementia
References
- 1.Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol. 1997;56:321–39. doi: 10.1097/00005072-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J. Amyloid, the Presenilins and Alzheimer’s disease. TINS. 1997;20:154–9. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki N, Cheung T, Cai X, et al. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (βAPP717) mutants. Science. 1994;264:1336–40. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 4.Andin U, Gustafson L, Passant U, et al. A clinico-pathological study of heart and brain lesions in vascular dementia. Dement Geriatr Cogn Disord. 2005;19:222–8. doi: 10.1159/000083801. [DOI] [PubMed] [Google Scholar]
- 5.Luchsinger JA, Reitz C, Honig LS, et al. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65:545–51. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langa KM, Foster NL, Larson EB. Mixed dementia: emerging concepts and therapeutic implications. Jama. 2004;292:2901–8. doi: 10.1001/jama.292.23.2901. [DOI] [PubMed] [Google Scholar]
- 7.Newman AB, Fitzpatrick AL, Lopez O, et al. Dementia and Alzheimer’s disease incidence in relationship to cardiovascular disease in the Cardiovascular Health Study cohort. J Am Geriatr Soc. 2005;53:1101–7. doi: 10.1111/j.1532-5415.2005.53360.x. [DOI] [PubMed] [Google Scholar]
- 8.Kang JH, Logroscino G, De Vivo I, et al. Apolipoprotein E, cardiovascular disease and cognitive function in aging women. Neurobiol Aging. 2005;26:475–84. doi: 10.1016/j.neurobiolaging.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 9.van Aalst-Cohen ES, Jansen AC, de Jongh S, et al. Clinical, diagnostic, and therapeutic aspects of familial hypercholesterolemia. Semin Vasc Med. 2004;4:31–41. doi: 10.1055/s-2004-822984. [DOI] [PubMed] [Google Scholar]
- 10.Yaffe K, Barrett-Connor E, Lin F, et al. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol. 2002;59:378–84. doi: 10.1001/archneur.59.3.378. [DOI] [PubMed] [Google Scholar]
- 11.Pappolla MA, Bryant-Thomas TK, Herbert D, et al. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology. 2003;61:199–205. doi: 10.1212/01.wnl.0000070182.02537.84. [DOI] [PubMed] [Google Scholar]
- 12.Kivipelto M, Helkala EL, Laakso MP, et al. Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. Brit Med Journal. 2001;322:1447–51. doi: 10.1136/bmj.322.7300.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kivipelto M, Helkala EL, Laakso MP, et al. Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med. 2002;137:149–55. doi: 10.7326/0003-4819-137-3-200208060-00006. [DOI] [PubMed] [Google Scholar]
- 14.Breteler MM. Early life circumstances and late life Alzheimer’s disease. Epidemiology. 2001;12:378–9. doi: 10.1097/00001648-200107000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Kivipelto M, Ngandu T, Fratiglioni L, et al. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol. 2005;62:1556–60. doi: 10.1001/archneur.62.10.1556. [DOI] [PubMed] [Google Scholar]
- 16.Notkola IL, Sulkava R, Pekkanen J, et al. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer’s disease. Neuroepidemiology. 1998;17:14–20. doi: 10.1159/000026149. [DOI] [PubMed] [Google Scholar]
- 17.Knopman D, Boland LL, Mosley T, et al. Cardiovascular risk factors and cognitive decline in middle-aged adults. Neurology. 2001;56:42–8. doi: 10.1212/wnl.56.1.42. [DOI] [PubMed] [Google Scholar]
- 18.Ott A, Stolk RP, Hofman A, et al. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–7. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- 19.Ott A, Stolk RP, van Harskamp F, et al. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–42. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 20.Kulik G, Klippel A, Weber MJ. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Molecular & Cellular Biology. 1997;17:1595–606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cross DA, Alessi DR, Cohen P, et al. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 22.Qiu WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: review and hypothesis. Neurobiol Aging. 2006;27:190–8. doi: 10.1016/j.neurobiolaging.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Zhao L, Teter B, Morihara T, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24:11120–6. doi: 10.1523/JNEUROSCI.2860-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–7. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yaffe K, Kanaya A, Lindquist K, et al. The metabolic syndrome, inflammation, and risk of cognitive decline. Jama. 2004;292:2237–42. doi: 10.1001/jama.292.18.2237. [DOI] [PubMed] [Google Scholar]
- 26.Encinas M, De Juan R, Marcos A, et al. Regional cerebral blood flow assessed with 99mTc-ECD SPET as a marker of progression of mild cognitive impairment to Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2003;30:1473–80. doi: 10.1007/s00259-003-1277-z. [DOI] [PubMed] [Google Scholar]
- 27.Bartenstein P, Minoshima S, Hirsch C, et al. Quantitative assessment of cerebral blood flow in patients with Alzheimer’s disease by SPECT. J Nucl Med. 1997;38:1095–101. [PubMed] [Google Scholar]
- 28.Ruitenberg A, den Heijer T, Bakker SL, et al. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol. 2005;57:789–94. doi: 10.1002/ana.20493. [DOI] [PubMed] [Google Scholar]
- 29.Newman MF, Kirchner JL, Phillips-Bute B, et al. Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. N Engl J Med. 2001;344:395–402. doi: 10.1056/NEJM200102083440601. [DOI] [PubMed] [Google Scholar]
- 30.Selnes OA, Royall RM, Grega MA, et al. Cognitive changes 5 years after coronary artery bypass grafting: is there evidence of late decline? Arch Neurol. 2001;58:598–604. doi: 10.1001/archneur.58.4.598. [DOI] [PubMed] [Google Scholar]
- 31.Lee TA, Wolozin B, Weiss KB, et al. Assessment of the emergence of Alzheimer’s disease following coronary artery bypass graft surgery or percutaneous transluminal coronary angioplasty. J Alzheimers Dis. 2005;7:319–24. doi: 10.3233/jad-2005-7408. [DOI] [PubMed] [Google Scholar]
- 32.Corder E, Saunders A, Strittmatter W, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 33.Khachaturian AS, Corcoran CD, Mayer LS, et al. Apolipoprotein E epsilon4 count affects age at onset of Alzheimer disease, but not lifetime susceptibility: The Cache County Study. Arch Gen Psychiatry. 2004;61:518–24. doi: 10.1001/archpsyc.61.5.518. [DOI] [PubMed] [Google Scholar]
- 34.Poirier J, Davignon J, Bouthillier D, et al. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342:697–9. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 35.Nathan B, Bellosta S, Sanan D, et al. Differential effects of apolipoprotein E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–2. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- 36.Strittmatter W, Saunders A, Schmechel D, et al. Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. PNAS. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strittmatter W, Weisgraber K, Huang D, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. PNAS. 1993;90:8098–102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prince M, Lovestone S, Cervilla J, et al. The association between APOE and dementia does not seem to be mediated by vascular factors. Neurology. 2000;54:397–402. doi: 10.1212/wnl.54.2.397. [DOI] [PubMed] [Google Scholar]
- 39.Jordan J, Galindo MF, Miller RJ, et al. Isoform-specific effect of apolipoprotein E on cell survival and beta-amyloid-induced toxicity in rat hippocampal pyramidal neuronal cultures. Journal of Neuroscience. 1998;18:195–204. doi: 10.1523/JNEUROSCI.18-01-00195.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith C, Graham DI, Murray LS, et al. Association of APOE e4 and cerebrovascular pathology in traumatic brain injury. J Neurol Neurosurg Psychiatry. 2006;77:363–6. doi: 10.1136/jnnp.2005.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diaz-Arrastia R, Gong Y, Fair S, et al. Increased risk of late posttraumatic seizures associated with inheritance of APOE epsilon4 allele. Arch Neurol. 2003;60:818–22. doi: 10.1001/archneur.60.6.818. [DOI] [PubMed] [Google Scholar]
- 42.Li YJ, Hauser MA, Scott WK, et al. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology. 2004;62:2005–9. doi: 10.1212/01.wnl.0000128089.53030.ac. [DOI] [PubMed] [Google Scholar]
- 43.Saft C, Andrich JE, Brune N, et al. Apolipoprotein E genotypes do not influence the age of onset in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2004;75:1692–6. doi: 10.1136/jnnp.2003.022756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seshadri S, Beiser A, Kelly-Hayes M, et al. The lifetime risk of stroke: estimates from the Framingham Study. Stroke. 2006;37:345–50. doi: 10.1161/01.STR.0000199613.38911.b2. [DOI] [PubMed] [Google Scholar]
- 45.Leys D, Henon H, Mackowiak-Cordoliani MA, et al. Poststroke dementia. Lancet Neurol. 2005;4:752–9. doi: 10.1016/S1474-4422(05)70221-0. [DOI] [PubMed] [Google Scholar]
- 46.Larson EB, Wang L, Bowen JD, et al. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med. 2006;144:73–81. doi: 10.7326/0003-4819-144-2-200601170-00004. [DOI] [PubMed] [Google Scholar]
- 47.Rovio S, Kareholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4:705–11. doi: 10.1016/S1474-4422(05)70198-8. [DOI] [PubMed] [Google Scholar]
- 48.Wolozin B, Kellman W, Ruosseau P, et al. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–43. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 49.Jick H, Zornberg GL, Jick SS, et al. Statins and the risk of dementia. Lancet. 2000;356:1627–31. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 50.Rockwood K, Kirkland S, Hogan DB, et al. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol. 2002;59:223–7. doi: 10.1001/archneur.59.2.223. [DOI] [PubMed] [Google Scholar]
- 51.Vega GL, Weiner MF, Lipton AM, et al. Reduction in levels of 24S-hydroxycholesterol by statin treatment in patients with Alzheimer disease. Arch Neurol. 2003;60:510–5. doi: 10.1001/archneur.60.4.510. [DOI] [PubMed] [Google Scholar]
- 52.Friedhoff LT, Cullen EI, Geoghagen NS, et al. Treatment with controlled-release lovastatin decreases serum concentrations of human beta-amyloid (Abeta) peptide. Int J Neuropsychopharmacol. 2001;4:127–30. doi: 10.1017/S1461145701002310. [DOI] [PubMed] [Google Scholar]
- 53.Simons M, Schwarzler F, Lutjohann D, et al. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double- blind trial. Ann Neurol. 2002;52:346–50. doi: 10.1002/ana.10292. [DOI] [PubMed] [Google Scholar]
- 54.Sparks DL, Sabbagh MN, Connor DJ, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol. 2005;62:753–7. doi: 10.1001/archneur.62.5.753. [DOI] [PubMed] [Google Scholar]
- 55.Sjogren M, Gustafsson K, Syversen S, et al. Treatment with simvastatin in patients with Alzheimer’s disease lowers both alpha- and beta-cleaved amyloid precursor protein. Dement Geriatr Cogn Disord. 2003;16:25–30. doi: 10.1159/000069989. [DOI] [PubMed] [Google Scholar]
- 56.Hoglund K, Wiklund O, Vanderstichele H, et al. Plasma Levels of Beta-Amyloid(1-40), Beta-Amyloid(1-42), and Total Beta-Amyloid Remain Unaffected in Adult Patients With Hypercholesterolemia After Treatment With Statins. Arch Neurol. 2004;61:333–337. doi: 10.1001/archneur.61.3.333. [DOI] [PubMed] [Google Scholar]
- 57.Zandi P, Sparks L, Khachaturian A, et al. Do Statins Reduce Risk of Incident Dementia and AD? The Cache County Study. Arch Gen Psychiatry. 2004 doi: 10.1001/archpsyc.62.2.217. in press. [DOI] [PubMed] [Google Scholar]
- 58.Li G, Higdon R, Kukull WA, et al. Statin therapy and risk of dementia in the elderly: a community-based prospective cohort study. Neurology. 2004;63:1624–8. doi: 10.1212/01.wnl.0000142963.90204.58. [DOI] [PubMed] [Google Scholar]
- 59.Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol. 2005;62:1047–51. doi: 10.1001/archneur.62.7.1047. [DOI] [PubMed] [Google Scholar]
- 60.Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–30. doi: 10.1016/s0140-6736(02)11600-x. [DOI] [PubMed] [Google Scholar]
- 61.Group HPSC. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22. doi: 10.1016/S0140-6736(02)09327-3. [DOI] [PubMed] [Google Scholar]
- 62.Marz W, Koenig W. HMG-CoA reductase inhibition: anti-inflammatory effects beyond lipid lowering? J Cardiovasc Risk. 2003;10:169–79. doi: 10.1097/01.hjr.0000073686.78271.6d. [DOI] [PubMed] [Google Scholar]
- 63.Wolozin B, Mange J, Bryant R, et al. Cholesterol, Alzheimer’s disease and Statins: Re-assessing the relationship between cholesterol, statins and Alzheimer’s disease. Acta Neuropathol Scandanavia. 2006 doi: 10.1111/j.1600-0404.2006.00687.x. (In Press) [DOI] [PubMed] [Google Scholar]
- 64.in’t Veld BA, Ruitenberg A, Hofman A, et al. Antihypertensive drugs and incidence of dementia: the Rotterdam Study. Neurobiol Aging. 2001;22:407–12. doi: 10.1016/s0197-4580(00)00241-4. [DOI] [PubMed] [Google Scholar]
- 65.Skoog I, Lithell H, Hansson L, et al. Effect of baseline cognitive function and antihypertensive treatment on cognitive and cardiovascular outcomes: Study on COgnition and Prognosis in the Elderly (SCOPE) Am J Hypertens. 2005;18:1052–9. doi: 10.1016/j.amjhyper.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 66.Forette F, Seux ML, Staessen JA, et al. The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Arch Intern Med. 2002;162:2046–52. doi: 10.1001/archinte.162.18.2046. [DOI] [PubMed] [Google Scholar]
- 67.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 68.Olsson A, Vanderstichele H, Andreasen N, et al. Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem. 2005;51:336–45. doi: 10.1373/clinchem.2004.039347. [DOI] [PubMed] [Google Scholar]