

Abstract
Activation of complement stimulates inflammation and provides an initial vigorous defense against infection. Insertion of the membrane attack complex in cell membranes of vascular endothelial cells induces changes in cell differentiation that promote coagulation, thrombosis, inflammation, and immunity. These changes are mediated by production of interleukin (IL)-1α by endothelial cells, which acts locally on endothelial cells to contain infection and promote healing of the affected site. In healthy tissues, however, promoting coagulation and inflammation would be dysphysiologic. Accordingly, endothelial cell activation by the membrane attack complex depends on both transcriptional regulation of IL-1α and availability of that cytokine to broadly modify endothelial cell physiology. Here, we report that the IL-1α gene contains a suppressor sequence that cooperates with histone modification to regulate production of IL-1α by endothelial cells. The suppressor sequence binds C/EBP (CCAAT enhancer–binding protein) family DNA-binding proteins isolated from the nucleus of quiescent endothelial cells. These results suggest constitutive suppression of IL-1α maintains quiescence of endothelium and that terminal complement complexes remove that suppression, allowing IL-1α transcription and, ultimately, activation of endothelium to proceed.
Keywords: calcineurin, complement, endothelium, histone deacetylase, inflammation, interleukin-1
Quiescent endothelial cells actively maintain the form and function of blood vessels. For example, quiescent endothelial cells promote angiogenesis by synthesizing growth factors such as vascular endothelial growth factor and platelet-derived growth factor1 and control angiogenesis by producing inhibitors of cellular growth and migration, such as heparan sulfate2 and nitric oxide3 (reviewed elsewhere4,5). Endothelial cells preserve blood fluidity and prevent thrombosis in conditions of health by presenting a nonadhesive and anticoagulant surface6,7 and relaxing vascular smooth muscle.8 This antiinflammatory, anticoagulant, prosuffusing posture maintains blood and tissue homeostasis.
Endothelial cells also sense injury and infection. Thus, endothelial cells express receptors for endotoxin,9 thrombin,10 and inflammatory cytokines such as interleukin (IL)-1.11–13 Stimulation of these receptors by corresponding agonists causes changes in endothelial cells that promote inflammation,14–16 coagulation,14,17,18 and diminished blood flow.19 This response has been called endothelial cell activation.20,21
Ideally, endothelial cells should coordinate responses to local and systemic inflammatory mediators for which they bear receptors. For example, endothelial cells respond to endotoxin via Toll-like receptor 422 and to cytokines by activating latent cytoplasmic nuclear factor (NF)-κB transcription factors, thus initiating a global nuclear factor NF-κB–driven proinflammatory gene transcription program. In this way, receptor-mediated activation of endothelial cells begins an inflammatory paradigm that includes transcription of more than 100 genes involved in acute and chronic inflammation,23 including inflammatory mediators such as adhesion molecules and chemokines,14–16 procoagulant molecules such as plasminogen activator inhibitor-1,18 vasoconstrictors such as thromboxane A2,19 and production of IL-1α.13,20,22,24–26
Activation of complement on endothelial cells can change endothelial cell physiology in ways similar to receptor-mediated activation.17 Activation of complement may occur spontaneously27 or in the context of disease or tissue trauma.28–30 The fragments of C3 and C5 (C3a and C5a) activate G protein–coupled receptors (C3aR, C5aR) and promote fusion of Weibel–Palade bodies with cell membranes.31 Externalization of Weibel–Palade bodies promotes adhesion of leukocytes but does not activate endothelial cells, as commonly understood.32 Insertion of C5b678 complexes in endothelial cell membranes promotes formation of polymeric C9, the membrane attack complex, which can activate endothelial cells fully. However, by itself, the membrane attack complex does not stimulate the broad range of changes described above but, rather, stimulates only the production of IL-1α, which may act on endothelium in an autocrine manner, evoking the broader manifestations of receptor-mediated activation including production of procoagulant, vasoactive, and inflammatory mediators typical of activated endothelium.17,33 Consistent with this concept, inhibiting the function of IL-1α prevents activation of endothelial cells by the membrane attack complex.34 On the other hand, if IL-1α is carried away by free-flowing blood, the broader range of changes may not occur.33
How the membrane attack complex selectively activates IL-1α expression in endothelial cells is incompletely understood. The membrane attack complex acts via calcineurin to activate NK-κB–dependent transcription of the IL-1α gene, whereas cytokines and other agonists function independently of calcineurin.35 Still unclear is how transcription of IL-1α is differentially controlled. Because acetylation of histones controls transcription of other inflammatory cytokines,36–38 we asked whether histone acetylation may regulate transcription of IL-1α. We report here that the IL-1α gene contains a suppressor sequence that cooperates with histone modification to regulate production of IL-1α and thus governs endothelial cell response to complement.
Materials and Methods
Materials
Human serum was used as a source of complement.17,34 Recombinant tumor necrosis factor (TNF)α and antibodies against IL-1α were from R&D Systems (Minneapolis, Minn). FK506 and trichostatin A were from Calbiochem (La Jolla, Calif). Antibodies to CCAAT enhancer–binding proteins (C/EBPs) were from Santa Cruz Biotechnology (Santa Cruz, Calif).
Endothelial Cell Culture and Analysis of IL-1α mRNA
Porcine aortic endothelial cells (passages 4 to 8) were cultured as described.34 Human aortic endothelial cells were from Cambrex Bio-Science Walkersville Inc (Santa Rosa, Calif) and cultured as suggested by the supplier. Total RNA from confluent cultures of endothelial cells was isolated by the guanidinium thiocyanate method. RT-PCR was performed as described previously.34 The identities of the PCR products were confirmed by sequencing (data not shown). To compare relative levels of various mRNA to GAPDH mRNA, a semiquantitative RT-PCR strategy was used.17 Sequences of oligonucleotides for PCR were as follows:
Human IL-1α: CCAGGCGTAGGTCTGGAGTCTCACTTGTCT and TGTTGCGGCAGGAAGGCTTAGGTATTATTC
Human GAPDH: CATGCCATCACTGCCACCCAGAAGACTGTG and GAAATGAGCTTGACAAAGTGGTCGTTGAGG
Porcine IL-1α: CGGGAAGATTCTGAAGAAGAGACGGTTGAG and TGGGCGGCTGATTTGAAGTAGTCCATATTG
Porcine GAPDH: CCGCGTCCCTGAGACACGATGGTGAAGGTC and TTCAAGTGAGCCCCAGCCTTCTCCATGGTC
The amplified products were fractionated on a 1.2% agarose–Tris/boric acid/EDTA (TBE) gel and visualized by staining with ethidium bromide. The resulting bands were quantitated using the Gel Doc 2000 and Quantity One software (Bio-Rad, Hercules, Calif).
Construction of IL-1α Luciferase Reporter Plasmids, Transfection, and Luciferase Assay
The porcine IL-1α promoter region was cloned from liver genomic library prepared in Lambda FIX II vector (Stratagene, La Jolla, Calif). The 5′ noncoding region of IL-1α encompassing a 3.25-kb DNA fragment was sequenced. The human IL-1α promoter containing 156 bp upstream of the IL-1α transcription start site and the repressor region was isolated from genomic DNA from human aortic endothelial cells. The DNA region was amplified using AGTTTTAGCCAGTATCGAGTTGAATGAACATAGAA and AAGCCTGAGTCAGTCTTCTTCGCCTTTTGTAATTG or AGTTTTAGCCAGTATCGAGTTGAATGAACATAGAA and AACTAGTTTCCTATGAGGGCATGGGTGAATACAACTGA oligonucleotides (GenBank accession no. X03833.1). The PCR product was isolated and sequenced.
Human and porcine IL-1α promoter regions, point mutants, and deletions were ligated to pGL3 basic luciferase reporter vector (Promega). Transient transfections of endothelial cells on 24-well plates at 40% to 50% confluence were performed using 0.5 μg of reporter plasmid and 0.1 μg of Renilla luciferase control reporter plasmid (pRL-TK; Promega, Madison, Wis) and Superfect transfection reagent (Qiagen, Valencia, Calif). Luciferase activity was assayed using the Dual-Luciferase Reporter Assay system (Promega) and a TD-20/20 Luminometer (Turner Designs, Sunnyvale, Calif).
Electrophoretic Mobility-Shift Assays
Proteins were extracted from cell nuclei of confluent cultures of porcine or human endothelial cells as described.39 Nuclear protein content was measured using Bio-Rad Protein Assay (Bio-Rad).
All oligonucleotides were from the Mayo Clinic Molecular Biology Core Facility. Probes were prepared by end-labeling with [γ32P]ATP (Amersham Bioscience) and T4 polynucleotide kinase (New England Biolabs Inc) and purified using G-25 Sepahdex Quik Spin Column (Roche Diagnostics, Indianapolis, Ind).
In a typical electrophoretic mobility-shift assay (EMSA) reaction, nuclear extract (4 to 5 μg) and labeled probes (0.25 pmol per reaction) were incubated on ice for 15 minutes, as described.39 For antibody supershift experiments, 1 μL of various antibodies was included. Protein–DNA complexes were resolved on 4% nondena-turing polyacrylamide gels. The gels were dried and subjected to autoradiography.
Results
Complement Activates IL-1α Gene Transcription
We previously found that the membrane attack complex of complement, and other pore-forming proteins, activate endothelial cells by calcium- and calcineurin-dependent production and secretion of IL-1α.17,34,35 Generation of the membrane attack complex of complement is specifically required because serum depleted of complement component C8, or serum that has been heated to inactivate complement, does not activate endothelial cells.17 Thus, as Figure 1A shows, activation of complement on cultured porcine aortic endothelial cells stimulates expression of IL-1α mRNA. Cells treated with the calcineurin inhibitor FK506 express significantly less IL-1α mRNA in response to complement than cells not treated with the drug, suggesting calcineurin-dependent signals stimulate IL-1α transcription.35
Figure 1.
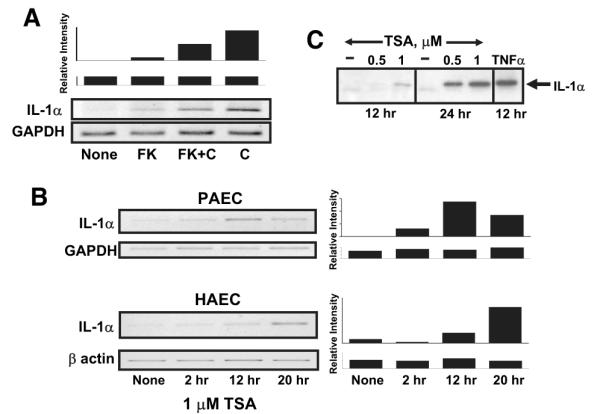
IL-1α expression is activated by complement and regulated by histone deacetylase in endothelial cells. Expression of IL-1α was evaluated in confluent cultures of porcine (PAEC) or human (HAEC) aortic endothelial cells. A, Complement-stimulated IL-1α expression requires calcineurin. Porcine aortic endothelial cells were left untreated (none) or were treated with 100 nmol/L FK506 (FK) for 2 hours to inhibit calcineurin. The cells were then were exposed to 12% human serum for 4 hours to activate complement (indicated as C). Total cellular RNA was prepared and analyzed for IL-1α mRNA using quantitative RT-PCR. The samples were also analyzed for GAPDH as a control for equal mRNA loading and quality. The representative gels shown at the bottom are representative of 3 separate experiments. The densitometry scan bar graph at the top reflect IL-1α/GAPDH mRNA levels. B, IL-1α expression is regulated by histone deacetylase. Confluent cultures of porcine or human aortic endothelial cells were treated with 1 μmol/L trichostatin A (TSA) for various periods of time, as shown, to inhibit histone deacetylase. Total cellular RNA was prepared, and expression of IL-1α mRNA or control GAPDH or β-actin mRNA was evaluated by quantitative RT-PCR as described above. C, Porcine aortic endothelial cells were treated with 0.5 or 1.0 μmol/L trichostatin A or with the drug vehicle alone for 12 or 24 hours. A positive control sample was stimulated with 10 nmol/L TNFα. Secretion of IL-1α into cell culture supernatants was analyzed from equal volumes of supernatant by immunoblot using anti-porcine IL-1α antibodies. The results show that complement or an inhibitor of histone deacetylase induces IL-1α gene expression in endothelial cells.
Histone Acetylation Regulates IL-1α Gene Expression
Epigenetic modification of chromosome structure regulates transcription of the IL-1α gene in CD4+ T cells40 and promotes activation of inflammatory genes such as IL-6 and TNFα in human blood monocytes36 and mouse microglia.37 Because epigenetic modifications that regulate gene expression also operate in endothelium,41 we asked whether acetylation of histones regulates transcription of IL-1α in endothelial cells. We investigated porcine endothelial cells because we determined previously that these cells, like human endothelial cells, respond to complement and other pore-forming proteins by regulating the availability of IL-1α.17,34,35,42,43 We cultured porcine aortic endothelial cells with trichostatin A, an inhibitor of histone deacetylase, and then tested for expression of IL-1α by RT-PCR. Endothelial cells cultured with trichostatin A had increased expression of IL-1α mRNA within 2 hours of adding the inhibitor (Figure 1B) and reached levels 40-fold higher than untreated cells in 20 hours. IL-1α protein was secreted by the endothelial cells treated with trichostatin A, as detected by immunoblot analysis of cell culture supernatants using anti-porcine IL-1α–specific antibodies (Figure 1C).
Identification of a Novel IL-1α Gene Suppressor Element
Cytokines, chemokines, and complement stimulate IL-1α transcription by activating nuclear transport of NF-κB.23,35 Because inhibition of histone deacetylase stimulated IL-1α production without addition of an NF-κB agonist, we reasoned that changes in chromatin structure imparted by histone acetylation may diminish the activity of a constitutive suppressor of IL-1α gene transcription. To determine whether a cis-acting element suppresses the IL-1α gene, we cloned the porcine IL-1α gene promoter, along with the contiguous 5′ and 3′ regions, into luciferase reporter plasmids and tested for suppressor function. Reporter plasmids containing the firefly luciferase gene regulated by various regions of the IL-1α gene promoter were transfected into porcine aortic endothelial cells, and the production of luciferase was assayed 36 hours later. Figure 2A shows a schematic representation of the promoter region and the first 2 exons of the porcine IL-1α gene.44,45 (See Figure I in the online data supplement [http://circres.ahajournals.org] for the complete nucleotide sequence of this region of the porcine IL-1α gene [NM 214029].) The 5′ end of this sequence, immediately preceding exon 1, contains the IL-1α minimal gene promoter, and this region strongly induces expression of the IL-1α luciferase reporter in porcine aortic endothelial cells (Figure 2B). However, luciferase production was abolished when the promoter was combined with 700 bp of the contiguous 3′ sequence (Figure 2B), suggesting the latter includes a suppressor.
Figure 2.

Identification of a repressor element in the porcine and human IL-1α gene. A, Schematic representation of the porcine IL-1α gene promoter and first 2 exons. The nucleotide sequence of a repressor element (boxed) is shown. Because the repressor element is located “downstream” or 3′ to the IL-1α gene promoter, between exon 1 and exon 2, we named it repressor downstream, or RepD (see below). B, The IL-1α gene promoter or the promoter plus 700 bp of contiguous 3′ sequence (downstream) encoding the first 2 exons of IL-1α was cloned into a luciferase reporter plasmid, and the reporter plasmids were transfected into porcine aortic endothelial cells. Production of the luciferase reporter measured 36 hours later revealed that the 700-bp sequence abolished luciferase reporter gene production. The bar graph reflects the mean±SEM of triplicate samples and is representative of 3 experiments. Deletions within the 700-bp repressor region sequence showed that the 163-bp region located ≈100 bp 3′ of the IL-1α gene promoter contained the repressor.
To determine which region(s) within the 700-bp sequence downstream of the promoter region suppresses IL-1α transcription, we generated nested deletion mutants of the region in luciferase reporter plasmids and tested them for suppression of reporter production. Thus, porcine aortic endothelial cells were transfected with the mutated reporter plasmids and tested for suppression of IL-1α gene promoter activity. This analysis revealed that the sequence responsible for suppression of the IL-1α gene promoter resides within a region located 169 bp 3′ of the IL-1α transcriptional start site (Figure 2A and 2B). Because this region of the IL-1α gene repressed gene expression and is located “downstream” of the gene promoter and transcriptional start site, and within the first intron of the gene, we call it “repressor downstream” or RepD.
To identify a specific nucleotide sequence within RepD that acts as a suppressor, we mutated specific sequences within suppressor region and tested the mutants for suppression of the IL-1α gene promoter. A 60-bp sequence suppressed luciferase reporter gene expression in porcine endothelial cells (Figure 3). Mutations or deletions within regions we call RepD-A or RepD-B abolished much RepD repressor function and mutations or deletions in both regions fully disabled the repressor (Figure 3). RepD specifically controls the IL-1α promoter, as it has no repressor activity when placed 5′ or 3′ of the SV40 promoter (data not shown).
Figure 3.
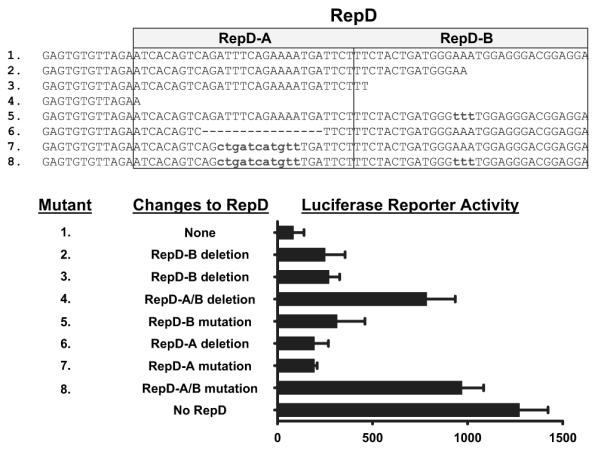
Resolution and characterization of the IL-1α gene repressor RepD sequence. Luciferase reporter plasmids containing the IL-1α gene promoter and RepD or nucleotide deletions or substitutions within the 60-bp RepD sequence reveal that RepD consists of 2 functional domains (shown as RepD-A and RepD-B). The bar graph reflects the mean±SEM for triplicate samples and is representative of 3 experiments.
A RepD Sequence in the Human IL-1α Gene
We have previously shown that pore-forming proteins induce production of IL-1α in both swine and human endothelial cells.34 Accordingly, we asked whether RepD also regulates transcription of human IL-1α. We first tested whether IL-1α expression by human aortic endothelial cells was regulated by epigenetic modification by treating the cells with an inhibitor of histone deacetylase. Like porcine cells, human aortic endothelial cells treated with trichostatin A had increased expression of IL-1α mRNA detected by RT-PCR (Figure 1B). To test whether the human IL-1α gene is regulated by RepD, we cloned the human IL-1α gene promoter along with the contiguous 5′ and 3′ sequences into luciferase reporter vectors and tested for suppressor function by transfection of human aortic endothelial cells. The human IL-1α gene contained a RepD-A sequence (Figure 3) approximately 270 bp 3′ of the gene promoter region (Figure 4A; see supplemental Figure II for the complete nucleotide sequence of this region of the human IL-1α gene). The human IL-1α RepD-A contains a 12-bp sequence identical to the porcine RepD-A, and luciferase production in human aortic endothelial cells transfected with reporter plasmids containing human IL-1α promoter and RepD-A region was suppressed by ≈50% (Figure 4B). Suppression by porcine RepD was ≈90% (Figure 3). Greater suppression of the reporter gene with porcine RepD may reflect the presence of the RepD-B sequence that is lacking in human RepD. This difference in reporter gene suppression between species should not be taken to predict the degree of suppression in vivo, which likely also depends on cytokine balance, complement levels, flora, etc.
Figure 4.

Identification of RepD in the human IL-1α gene. A, Schematic representation of the human IL-1α gene promoter and contiguous 270 bp of 3′ sequence. The nucleotide sequence of the RepD-A sequence is shown. B, Luciferase reporter plasmids regulated by the human IL-1α gene promoter plus the contiguous 270 bp of 3′ sequence, or by the IL-1α promoter alone, were transfected into human aortic endothelial cells. Production of the luciferase reporter gene measured 36 hours later revealed that the additional 270 bp containing the RepD-A sequence suppressed luciferase reporter gene activation. The bar graph reflects the mean±SEM for triplicate samples and is representative of 3 experiments.
RepD-Binding Proteins
As a first step toward identifying the protein(s) that regulate the IL-1α gene in endothelial cells, we extracted proteins from the nuclei of quiescent porcine aortic endothelial cells, in which transcription of IL-1α is suppressed, and performed EMSAs using the nuclear proteins and 32P-labeled oligonucleotide probes derived from RepD. Nuclear proteins from the endothelial cells formed 2 separate complexes with the full RepD sequence probe (Figure 5). To distinguish the 2 complexes formed with RepD, we tested oligonucleotides representing the 5′ half (RepD-A) or the 3′ half (RepD-B) of RepD as cold competitors in EMSAs. The oligonucleotide derived from RepD-A inhibited formation of the slower-migrating complex but not the faster-migrating complex formed between porcine endothelial cell nuclear protein(s) and the RepD probe (Figure 5). In contrast, oligonucleotides containing poly-thymidine or a poly-adenosine mutations that disrupt RepD repressor activity (Figure 3) did not compete with the labeled RepD probe, suggesting the protein(s) that forms the slow migrating complex binds specifically to sequences within RepD-A. The oligonucleotide derived from RepD-B partially inhibited formation of the faster-migrating complex but not the slower-migrating complex. As would be expected, RepD oligonucleotides with a poly-adenosine mutation of RepD-B prevented competition (Figure 5). The slower-migrating complex bound to RepD-A and the faster-migrating complex to RepD-B because a slower-migrating complex formed with the radiolabeled RepD-A probe and a faster-migrating complex formed on the radiolabeled RepD-B probe (Figure 5), consistent with the 2 complexes that formed on the full-length RepD probe. These results suggest that 2 distinct protein complexes synergistically regulate IL-1α gene transcription by binding to RepD.
Figure 5.

Protein factors in endothelial cell nuclei bind RepD sequences. Nuclear proteins were extracted from porcine aortic endothelial cells and incubated with 32P-labeled oligonucleotide probes derived from the RepD sequence as shown (top). A control reaction contained labeled probe without protein (first lane). The protein–DNA complexes were resolved by nondenaturing polyacrylamide gel electrophoresis and detected on autoradiography film. The experiment revealed that 2 protein–DNA complexes form simultaneously on the radio-labeled RepD sequence. The slower-migrating complex is diminished when cold competitor oligonucleotide probes from intact RepD or from RepD-A are included but not when cold RepD-A sequences include the specific mutations shown. The faster-migrating complex is diminished by cold RepD or RepD-B oligonucleotides but not when RepD-B contains the mutation shown. Protein–DNA complexes formed with 32P-labeled oligonucleotide probes derived from RepD-A or RepD-B confirm that the slower-migrating complex forms specifically with the RepD-A sequence and that the faster-migrating complex forms specifically with the RepD-B sequence (right). Data are representative of 3 experiments.
RepD Binds C/EBP From Endothelial Cells
The RepD sequence contains a site potentially recognized by C/EBP. The promoter regions of TNFα, IL-8, and G-CSF genes also have such sequences.46 To investigate whether the DNA-binding complexes from endothelial cells contain C/EBP, we performed EMSAs using 32P-labeled oligonucleotide probes with native RepD-A sequences or RepD-A mutated within the putative C/EBP-binding region (Figure 6). RepD-A oligonucleotides with changes within the C/EBP-binding sequence did not form complexes with nuclear proteins extracted from porcine aortic endothelial cells (Figure 6A), suggesting protein(s) with the C/EBP DNA-binding specificity may regulate IL-1α via RepD.
Figure 6.

The IL-1α gene repressor RepD binds to a C/EBP. Nuclear proteins were extracted from porcine (PAEC) or human (HAEC) aortic endothelial cells and incubated with 32P-labeled oligonucleotide probes derived from the RepD-A sequence, as shown in A. The protein–DNA complexes were resolved by nondenaturing polyacrylamide gel electrophoresis and detected by autoradiography of dried gels. A, Nuclear proteins from porcine or human aortic endothelial cells form complexes with RepD-A oligonucleotides but not with RepD-A oligonucleotides containing mutations in the CEBP-β–binding sequence. B, Antibodies to C/EBP-β (lanes marked β) react with protein–RepD-A oligonucleotide complexes and retard migration of the migration through gels. Antibodies against other proteins including C/EBP-α (lanes marked α) or NFATc (data not shown) were without effect. Shown at left side are several examples of experiments using porcine or human porcine aortic endothelial cells probed with the RepD-A probe and anti C/EBP or control (-) antibodies. Antibodies against C/EBP-α or C/EBP-β do not react with the faster-moving complex that forms with RepD-B (right).
To determine whether C/EBP family members may regulate IL-1α transcription, we performed electrophoretic mobility “supershift” analysis using antibodies to C/EBP family members or to other transcription factors. Antibodies directed against C/EBPβ retarded mobility of the RepD-A complex to a small but reproducible extent (Figure 6B), whereas antibodies specific for C/EBPα (Figure 6B) or NFATc (data not shown) did not. None of the antibodies reacted with the component(s) of the RepD-B–protein complex (Figure 6B). These results suggest that suppression of IL-1α gene is mediated by RepD interaction with C/EBPβ, although other nuclear proteins may play a role by binding to the RepD-B sequence.
Discussion
Activation of complement is a seminal event in inflammation and immunity.33 Complement is the first system to be activated by infectious microorganisms, and it initiates an immediate and potent defense. Besides directly killing micro-organisms, the membrane attack complex promotes procoagulant posture of endothelial cells14,17 and recruits platelets to vessel walls, both of which circumscribe sites of infection and produce a barrier that isolates the pathogen.33 Although complement clearly defends the host, it may also damage healthy endothelium and tissues.47 For this reason, presumably, endothelial cells resist injury from complement and rapidly repair such injury that may occur.28,33 This resistance reflects both control of production and responsiveness to IL-1α.34 Here, we report that a transcriptional repressor controls IL-1α production by quiescent endothelial cells.
The physiological changes in endothelial cells induced by the membrane attack complex are predominantly mediated by production of IL-1α and autocrine or paracrine action of that cytokine.33 Because the membrane attack complex is produced constitutively,33 the quiescent condition of endothelial cells depends on control of IL-1α transcription. We show here that such control is exerted, at least in part, by repression of IL-1α transcription enforced by epigenetic modification of DNA40 and/or histones (Figure 1) and sequence-specific binding of repressor proteins. Toward the latter, we report a DNA sequence within the IL-1α gene that we call RepD and that functions as a transcriptional repressor. RepD differs from a repressor-binding site reported by McDowell et al48 located between −448 and −435 nucleotides with respect to the human IL-1α transcriptional start site. Although McDowell et al did not report which physiological stimuli disable the upstream repressor, we found that histone deacetylase inhibitors activate IL-1α transcription independently of other agonists. Thus, cellular signaling pathways that regulate histone deacetylase may regulate IL-1α by disabling the function of a repressor(s). Given our previous finding that the membrane attack complex activates calcineurin-dependent IL-1α transcription in endothelial cells by targeting NF-κB to the IL-1α gene,35 and the report by Zhang et al49 that calcineurin promotes export of histone deacetylase(s) from the nucleus,49 leaving unopposed histone acetylases that acetylate histones in chromatin and thereby “open” certain genes for activation by NF-κB50 or other transcriptional activators, we speculate the membrane attack complex may selectively target transcription of the IL-1α gene by calcineurin-dependent removal of histone deacetylase activity from the nucleus.
Suppression of IL-1α transcription in endothelial cells by RepD and derepression by calcineurin-dependent signals and histone deacetylase may provide a switch by which IL-1α, via action on endothelium, participates in host defense in one context and protects endothelium from harm in another. After the initial encounter with a microorganism, complement activation increases production of membrane attack complexes and promotes small vessel permeability by disrupting endothelial integrity.28 These changes promote generation of thrombin, activation of platelets, and formation of fibrin clots and platelet plugs, contributing to reduced blood flow and sequestration of infectious agents. When blood flow is reduced, IL-1α accumulates and stimulates production of chemokines, influx of leukocytes, acute inflammation,14,16 and subsequent initiation of adaptive immune responses.51,52 However, in normal tissues, the proinflammatory action of IL-1α would be detrimental and is prevented by multilayered mechanisms. Thus, under physiological conditions, transcription of the IL-1α gene is constitutively suppressed, as we show here, and endothelial cells remain inured to small amounts of complement spontaneously activated even in healthy tissues. Moreover, even if transcriptional suppression fails, small amounts of IL-1α produced are dissipated in the rapid blood flow. Blood vessels adjacent to injured or infected sites may experience increased exposure to IL-1α, but endothelial cell responses to IL-1α are antagonized by a secreted IL-1α receptor antagonist,53 synthesis of a nonsignaling IL-1 receptor,54 constitutive suppression of IL-1α transcription, and feedback inhibition of IL-1α synthesis by IL-1 receptor signaling [S.S. and J.L.P., unpublished results, 2006]. Thus, constitutive suppression of IL-1α gene transcription and removal of that suppression by histone modifications allow proinflammatory production of IL-1α to predominate in appropriate circumstances and potentially enable complement, via highly regulated production of IL-1α, to serve as a master regulator of endothelial function in health and disease.
Supplementary Material
Acknowledgments
Sources of Funding This work supported by NIH grants HL52297 and AI53733.
Footnotes
Disclosures None.
References
- 1.Yonekura H, Sakurai S, Liu X, Migita H, Wang H, Yamagishi S, Nomura M, Abedin MJ, Unoki H, Yamamoto Y, Yamamoto H. Placenta growth factor and vascular endothelial growth factor B and C expression in microvascular endothelial cells and pericytes. Implication in autocrine and paracrine regulation of angiogenesis. J Biol Chem. 1999;274:35172–35178. doi: 10.1074/jbc.274.49.35172. [DOI] [PubMed] [Google Scholar]
- 2.Bingley JA, Hayward IP, Campbell JH, Campbell GR. Arterial heparan sulfate proteoglycans inhibit vascular smooth muscle cell proliferation and phenotype change in vitro and neointimal formation in vivo. J Vasc Surg. 1998;28:308–318. doi: 10.1016/s0741-5214(98)70167-3. [DOI] [PubMed] [Google Scholar]
- 3.Cornwell TL, Arnold E, Boerth NJ, Lincoln TM. Inhibition of smooth muscle cell growth by nitric oxide and activation of cAMP-dependent protein kinase by cGMP. Am J Physiol. 1994;267:C1405–C1413. doi: 10.1152/ajpcell.1994.267.5.C1405. [DOI] [PubMed] [Google Scholar]
- 4.Nelson RM, Venot A, Bevilacqua MP, Linhardt RJ, Stamenkovic I. Carbohydrate-protein interactions in vascular biology. Annu Rev Cell Dev Biol. 1995;11:601–631. doi: 10.1146/annurev.cb.11.110195.003125. [DOI] [PubMed] [Google Scholar]
- 5.Rossant J, Howard L. Signaling pathways in vascular development. Annu Rev Cell Dev Biol. 2002;18:541–573. doi: 10.1146/annurev.cellbio.18.012502.105825. [DOI] [PubMed] [Google Scholar]
- 6.Esmon CT. The roles of protein c and thrombomodulin in the regulation of blood coagulation. J Biol Chem. 1989;264:4743–4746. [PubMed] [Google Scholar]
- 7.Broze GS., Jr. Tissue factor pathway inhibitor. Thromb Haemost. 1995;74:90–93. [PubMed] [Google Scholar]
- 8.Palmer RMJ, Ferridge AG, Moneada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 9.Faure E, Equils O, Sieling PA, Thomas L, Zhang FX, Kirschning CJ, Polentarutti N, Muzio M, Arditi M. Bacterial lipopolysaccharide activates NF-kappaB through toll-like receptor 4 (TLR-4) in cultured human dermal endothelial cells. Differential expression of TLR-4 and TLR-2 in endothelial cells. J Biol Chem. 2000;275:11058–11063. doi: 10.1074/jbc.275.15.11058. [DOI] [PubMed] [Google Scholar]
- 10.Coughlin SR. Protease-activated receptors in vascular biology. Thromb Haemost. 2001;86:298–307. [PubMed] [Google Scholar]
- 11.Nawroth PP, Stern DM. Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med. 1986;163:740–745. doi: 10.1084/jem.163.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pober JS, LaPierre LA, Stolpen AH, Brock TA, Springer TA, Fiers W, Bevilacqua MP, Mendrick DL, Gimbrone MA., Jr. Activation of cultured human endothelial cells by recombinant lymphotoxin: comparison with tumor necrosis factor and interleukin 1 species. J Immunol. 1987;138:3319–3324. [PubMed] [Google Scholar]
- 13.Strieter RM, Kunkel SL, Showell HJ, Remick DG, Phan SH, Ward PA, Marks RM. Endothelial cell gene expression of a neutrophil chemotactic factor by TNF-alpha, LPS, and IL-1 beta. Science. 1989;243:1467–1469. doi: 10.1126/science.2648570. [DOI] [PubMed] [Google Scholar]
- 14.Tedesco F, Pausa M, Nardon E, Introna M, Mantovani A, Dobrina A. Cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J Exp Med. 1997;185:1619–1627. doi: 10.1084/jem.185.9.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kishimoto T. The biology of interleukin-6. Blood. 1989;74:1–10. [PubMed] [Google Scholar]
- 16.Huber AR, Kunkel SL, Todd RF, III, Weiss SJ. Regulation of transendothelial neutrophil migration by endogenous interleukin-8. Science. 1991;254:99–102. doi: 10.1126/science.1718038. [DOI] [PubMed] [Google Scholar]
- 17.Saadi S, Holzknecht RA, Patte CP, Stern DM, Platt JL. Complement-mediated regulation of tissue factor activity in endothelium. J Exp Med. 1995;182:1807–1814. doi: 10.1084/jem.182.6.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalady MF, Lawson JH, Sorrell RD, Platt JL. Decreased fibrinolytic activity in porcine-to-primate cardiac xenotransplantation. Mol Med. 1998;4:629–637. [PMC free article] [PubMed] [Google Scholar]
- 19.Bustos M, Coffman TM, Saadi S, Platt JL. Modulation of eicosanoid metabolism in endothelial cells in a xenograft model: role of cyclooxygenase-2. J Clin Invest. 1997;100:1150–1158. doi: 10.1172/JCI119626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stern D, Nawroth P, Handley D, Kisiel W. An endothelial cell-dependent pathway of coagulation. Proc Natl Acad Sci U S A. 1985;82:2523–2527. doi: 10.1073/pnas.82.8.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA., Jr. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J Clin Invest. 1985;76:2003–2011. doi: 10.1172/JCI112200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 23.Kempe S, Kestler H, Lasar A, Wirth T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005;33:5308–5319. doi: 10.1093/nar/gki836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nawroth PP, Handley DA, Esmon CT, Stern DM. Interleukin 1 induces endothelial cell procoagulant while suppressing cell-surface anticoagulant activity. Proc Natl Acad Sci U S A. 1986;83:3460–3464. doi: 10.1073/pnas.83.10.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pober JS. Cytokine-mediated activation of vascular endothelium. Am J Pathol. 1988;133:426–433. [PMC free article] [PubMed] [Google Scholar]
- 26.Warner SJC, Auger KP, Libby P. Interleukin 1 induces interleukin 1. II. Recombinant human interleukin 1 induces interleukin 1 production by adult human vascular endothelial cells. J Immunol. 1987;139:1911–1917. [PubMed] [Google Scholar]
- 27.Matsuo S, Ichida S, Takizawa H, Okada N, Baranyi L, Iguchi A, Morgan BP, Okada H. In vivo effects of monoclonal antibodies that functionally inhibit complement regulatory proteins in rats. J Exp Med. 1994;180:1619–1627. doi: 10.1084/jem.180.5.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saadi S, Platt JL. Transient perturbation of endothelial integrity induced by natural antibodies and complement. J Exp Med. 1995;181:21–31. doi: 10.1084/jem.181.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mollnes T, Lea T, Mellbye O, Pahle J, Grand O, Harboe M. Complement activation in synovial fluid and tissue from patients with juvenile rheumatoid arthritis. Arthritis Rheum. 1986;29:715–721. doi: 10.1002/art.1780290603. [DOI] [PubMed] [Google Scholar]
- 30.Corvetta A, Pomponio G, Rinaldi N, Luchetti MM, Di Loreto C, Stramazzotti D. Terminal complement complex in synovial tissue from patients affected by rheumatoid arthritis, osteoarthritis and acute joint trauma. Clin Exp Rheumatol. 1992;10:433–438. [PubMed] [Google Scholar]
- 31.Pinsky DJ, Naka Y, Liao H, Oz MC, Wagner DD, Mayadas TN, Johnson RC, Hynes RO, Heath M, Lawson CA, Stern DM. Hypoxia-induced exocytosis of endothelial cell Weibel-Palade bodies. J Clin Invest. 1996;97:493–500. doi: 10.1172/JCI118440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reidy MA, Chopek M, Chao S, McDonald T, Schwartz SM. Injury induces increase of von Willebrand factor in rat endothelial cells. Am J Pathol. 1989;134:857–864. [PMC free article] [PubMed] [Google Scholar]
- 33.Saadi S, Wrenshall LE, Platt JL. Regional manifestations and control of the immune system. FASEB J. 2002;16:849–856. doi: 10.1096/fj.01-0690hyp. [DOI] [PubMed] [Google Scholar]
- 34.Saadi S, Holzknecht RA, Patte CP, Platt JL. Endothelial cell activation by pore forming structures: pivotal role for IL-1a. Circulation. 2000;101:1867–1873. doi: 10.1161/01.cir.101.15.1867. [DOI] [PubMed] [Google Scholar]
- 35.Brunn GJ, Saadi S, Platt JL. Differential regulation of endothelial cell activation by complement and interleukin 1alpha. Circ Res. 2006;98:793–800. doi: 10.1161/01.RES.0000216071.87981.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miao F, Gonzalo IG, Lanting L, Natarajan R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem. 2004;279:18091–18097. doi: 10.1074/jbc.M311786200. [DOI] [PubMed] [Google Scholar]
- 37.Suuronen T, Huuskonen J, Pihlaja R, Kyrylenko S, Salminen A. Regulation of microglial inflammatory response by histone deacetylase inhibitors. J Neurochem. 2003;87:407–416. doi: 10.1046/j.1471-4159.2003.02004.x. [DOI] [PubMed] [Google Scholar]
- 38.Huang N, Katz JP, Martin DR, Wu GD. Inhibition of IL-8 gene expression in Caco-2 cells by compounds which induce histone hyper-acetylation. Cytokine. 1997;9:27–36. doi: 10.1006/cyto.1996.0132. [DOI] [PubMed] [Google Scholar]
- 39.Brunn GJ, Falls EL, Nilson AE, Abraham RT. Protein-tyrosine kinase-dependent activation of STAT transcription factors in interleukin-2- or interleukin-4-stimulated T lymphocytes. J Biol Chem. 1995;270:11628–11635. doi: 10.1074/jbc.270.19.11628. [DOI] [PubMed] [Google Scholar]
- 40.van Rietschoten JG, Verzijlbergen KF, Gringhuis SI, van der Pouw Kraan TC, Bayley JP, Wierenga EA, Jones PA, Kooter JM, Verweij CL. Differentially methylated alleles in a distinct region of the human interleukin-1alpha promoter are associated with allele-specific expression of IL-1alpha in CD4+ T cells. Blood. 2006;108:2143–2149. doi: 10.1182/blood-2006-01-021147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fish JE, Matouk CC, Rachlis A, Lin S, Tai SC, D’Abreo C, Marsden PA. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem. 2005;280:24824–24838. doi: 10.1074/jbc.M502115200. [DOI] [PubMed] [Google Scholar]
- 42.Saadi S, Platt JL. Endothelial cell responses to complement activation. In: Volanakis JE, Frank MM, editors. The Human Complement System in Health and Disease. Marcel Dekker Inc; New York: 1998. pp. 335–353. [Google Scholar]
- 43.Ihrcke NS, Wrenshall LE, Lindman BJ, Platt JL. Role of heparan sulfate in immune system-blood vessel interactions. Immunol Today. 1993;14:500–505. doi: 10.1016/0167-5699(93)90265-M. [DOI] [PubMed] [Google Scholar]
- 44.Maliszewski CR, Renshaw BR, Schoenborn MA, Urban JFJ, Baker PE. Porcine IL-1 alpha cDNA nucleotide sequence. Nucleic Acids Res. 1990;18:4282. doi: 10.1093/nar/18.14.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Straubinger AF, Viveiros MM, Straubinger RK. Identification of two transcripts of canine, feline, and porcine interleukin-1 alpha. Gene. 1999;236:273–280. doi: 10.1016/s0378-1119(99)00274-7. [DOI] [PubMed] [Google Scholar]
- 46.Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, Nakajima T, Hirano T, Kishimoto T. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 1990;9:1897–1906. doi: 10.1002/j.1460-2075.1990.tb08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turesson C, Englund P, Jacobsson LT, Sturfelt G, Truedsson L, Nennesmo I, Lundberg IE. Increased endothelial expression of HLA-DQ and interleukin 1alpha in extra-articular rheumatoid arthritis. Results from immunohistochemical studies of skeletal muscle. Rheumatology (Oxford) 2001;40:1346–1354. doi: 10.1093/rheumatology/40.12.1346. [DOI] [PubMed] [Google Scholar]
- 48.McDowell TL, Symons JA, Duff GW. Human interleukin-1 alpha gene expression is regulated by Sp1 and a transcriptional repressor. Cytokine. 2005;30:141–153. doi: 10.1016/j.cyto.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 49.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kilgore KS, Flory CM, Miller BF, Evans VM, Warren JS. Membrane attack complex of complement induces interleukin-8 and monocyte chemoattractant protein-1 secretion from human umbilical vein endothelial cells. Am J Pathol. 1996;149:953–961. [PMC free article] [PubMed] [Google Scholar]
- 52.Selvan RS, Kapadia HB, Platt JL. Complement-induced expression of chemokine genes in endothelium: regulation by IL-1-dependent and -independent mechanisms. J Immunol. 1998;161:4388–4395. [PubMed] [Google Scholar]
- 53.Shiraishi M, Csete M, Yasunaga C, McDiarmid SV, Vannice JL, Busuttil RW, Shaked A. The inhibitor cytokine interleukin-1 receptor antagonist synergistically augments cyclosporine immunosuppression in a rat cardiac allograft model. J Surg Res. 1995;58:465–470. doi: 10.1006/jsre.1995.1073. [DOI] [PubMed] [Google Scholar]
- 54.Colotta F, Orlando S, Fadlon EJ, Sozzani S, Matteucci C, Mantovani A. Chemoattractants induce rapid release of the interleukin 1 type II decoy receptor in human polymorphonuclear cells. J Exp Med. 1995;181:2181–2186. doi: 10.1084/jem.181.6.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.