

Abstract
The microbial production, isolation and structure elucidation of four new napyradiomycin congeners (1–4) is reported. The structures of these compounds, which are new additions to the marine-derived meroterpenoids, were defined by comprehensive spectroscopic analysis and by X-ray crystallography. Using fluorescence-activated cell sorting (FACS) analysis, napyradiomycins 1–4 were observed to induce apoptosis in the colon adenocarcinoma cell line, HCT-116, indicating the possibility of a specific biochemical target for this class of cytotoxins.
Marine microbial metabolites are receiving increased attention as a new resource for the discovery of new therapeutic agents. Successes in the clinical development of salinosporamide A (aka marizomib) and the halimide derivative NPI-2358 (aka plinabulin) have validated marine microbes as a valuable source for novel therapeutic agents.1 In the course of exploring marine microorganisms, we have focused on bacterial strains producing metabolites with novel carbon skeletons and potentially useful anticancer properties. A largely marine actinomycete taxon, tentatively designated MAR4 (Family Streptomycetaceae), was found to produce a host of meroterpenoids of the napyradiomycin class.2–4 The napyradiomycins were first discovered from cultures of the actinomycete Chainia rubra isolated in Japan in 1986.5,6 In subsequent work, several other strains have also been found to produce napyradiomycins.7–9 The napyradiomycins were initially characterized for their antimicrobial activity, but have since been found to inhibit gastric (H+-K+) ATPases, and to behave as estrogen receptor antagonists.8,9 An examination of the biological potential of these molecules in the treatment of cancer, however, has not been reported, and specific information defining their interactions with targets in cancer cells is unknown. Using HCT-116 colon carcinoma cytotoxicity as a guide, we have repeatedly observed significant growth inhibition from compounds in the napyradiomycin class, and these observations have now been supplemented by assessing the induction of apoptosis, a mechanism that includes numerous pathways of significant utility in the treatment of cancer. In this paper, we report the isolation, structure elucidation and biological evaluation of four cytotoxic napyradiomycin meroterpenoids (1–4), and demonstrate the induction of cell death by the activation of apoptosis in four napyradiomycin congeners.
RESULTS AND DISCUSSION
Actinomycete strain CNQ525, previously identified as a member of the MAR4 clade (Streptomycetaceae) by phylogenetic analysis,2 was cultured under saline conditions at 30 °C for seven days and solid-phase extracted using Amberlite XAD-7 resin for 2 h. We had previously examined this strain and found that it produced a complex variety of napyradiomycins and that the metabolites produced were dependent upon culture conditions.2 On re-cultivation under a variety of conditions, the organic extracts that were obtained were fractionated first by silica flash chromatography and subsequently by C18 reversed-phase HPLC to yield the new napyradiomycins 1–4. Not wishing to add to the complex nomenclature of the napyradiomycins, we further defined these metabolites as shown using their strain number and molecular weights consistent with an earlier paper.2
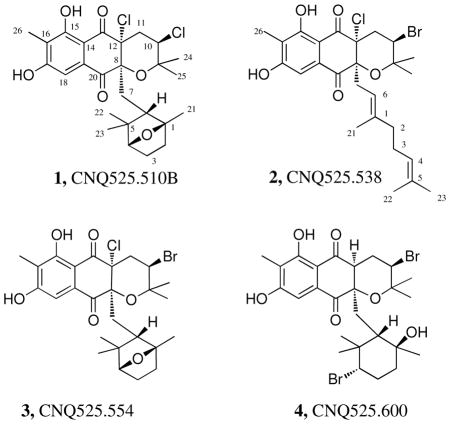
Napyradiomycin CNQ525.510B (1), was analyzed by LC-MS to have the same mass as compound CNQ525.510A reported by Soria-Mercado et al.2 (Figure 1), but with a slightly shorter elution time by HPLC analysis using a C18 column and an H2O/CH3CN gradient. Analysis by HR-TOF mass spectrometry showed a quasimolecular ion ([M+H]+ m/z 511.1652) corresponding to the molecular formula C26H3235Cl2O6, confirming that the two metabolites were isomeric. Initial examination of the NMR spectroscopic data revealed that 1 lacked the double bond at C-11 – C-12 that was present in compound CNQ525.510A2 (For CNQ525.510A – δH 4.56, d, J = 1.9, H-11; For 1 – δHa 2.37, m, H-11a; δHb 2.54, dd, J = 3.8, 14.0, H-11b). Analysis of HMBC, HSQC, and 1H NMR spectroscopic data for 1 revealed the presence of one aromatic methyl group (δC 8.6, C-26, δH 2.21, s, H-26), five other methyl groups (δC 21.1, C-21, δH 1.48, s, H-21; δC 21.4, C-22, δH 0.29, s, H-22; δC 31.7, C-23, δH 0.89, s, H-23; δC 22.8, C-24, δH 1.31, s, H-24; δC 29.5, C-25, δH 1.49, s, H-25) attached to sp3 carbons, as well as one aromatic proton (δH 7.26, s, H-18) (Table 1). The molecular formula for 1, and the lack of an olefin in comparison to napyradiomycin CNQ525.510A, suggested that an additional ring was present in this derivative. It was envisioned that the new ring was a cyclic ether formally produced by transannular displacement of chloride in the cyclohexane ring. Unfortunately, this could not be unambiguously confirmed by NMR methods due to difficulty of detecting HMBC correlations through the C-1 quaternary carbon and the ether linkage. Consequently, 1 was crystallized by slow evaporation from a 50:50 hexane/EtOAc solution and the poorly formed crystals subjected to X-ray analysis. The X-ray experiment succeeded in providing the absolute stereostructure shown of 1 as shown in Figure 2. In napyradiomycin CNQ525.510A (1) it is apparent that the bicyclic ether was indeed conceptually formed by transannular displacement of chloride. The structure of 1 was further supported by comprehensive 2D NMR analysis (Table 1).
Figure 1.

Structures of related napyradiomycin meroterpenoids used as structural comparisons.
Table 1.
| Carbon # | δC, type | δH, mult (J in Hz) | COSY | HMBCc |
|---|---|---|---|---|
| 1 | 90.6, C | |||
| 2 | 30.0, CH2 | 1.59, m | H-3a, H-3b | 1, 3, 6 |
| 1.23, m | H-3a, H-3b | 1, 3, 6 | ||
| 3 | 26.9, CH2 | 1.59, m | 1, 2, 4, 5 | |
| 1.67, m | 1, 2, 4, 5 | |||
| 4 | 87.2, CH | 3.82, d (4.6) | H-3a, H-3b | 1, 2, 3, 5, 6 |
| 5 | 41.3, C | |||
| 6 | 49.0, CH | 1.81, bs | 1, 2, 5, 7, 21, 22, 23 | |
| 7 | 37.0, CH2 | 1.47, d (15.4) | H6 | 1, 5, 6, 8, 12, 20 |
| 2.07, dd (3.4, 15.4) | H6 | 1, 5, 6, 8, 12, 20 | ||
| 8 | 83.9, C | |||
| 9 | 79.3, C | |||
| 10 | 59.0, CH | 4.50, dd (3.8, 12.0) | 9, 11, 12, 24, 25 | |
| 11 | 42.6, CH2 | 2.37, m | H10 | 8, 9, 10, 12, 13, |
| 2.54, dd (3.8, 14.0) | H10 | 8, 9, 10, 12, 13, | ||
| 12 | 81.7, C | |||
| 13 | 194.1, C | |||
| 14 | 107.7, C | |||
| 15 | 163.6, C | |||
| 16 | 120.2, C | |||
| 17 | 163.4, C | |||
| 18 | 107.7, CH | 7.26, s | 14, 15, 16, 17, 18, 19, 20, 26 | |
| 19 | 131.8, C | |||
| 20 | 192.9, C | |||
| 21 | 21.1, CH3 | 1.48, s | 1, 2, 6 | |
| 22 | 21.4, CH3 | 0.29, s | 4, 5, 22 | |
| 23 | 31.7, CH3 | 0.89, s | 4, 5, 23 | |
| 24 | 22.8, CH3 | 1.31, s | 9, 10, 24 | |
| 25 | 29.5, CH3 | 1.49, s | 9, 10, 25 | |
| 26 | 8.6, CH3 | 2.21, s | 14, 15, 16, 17, 18, 19 | |
| OH (15) | 12.39, s | 14, 15, 16 |
1H spectra were recorded in CDCl3 at 500 MHz,
Assignments were made based on interpretation of COSY, HMBC and HSQC experiments.
HMBC correlations are from the proton(s) stated to the indicated carbons.
Figure 2.

Computer-generated structure drawing of napyradiomycin CNQ525.510B (1) depicting the compound’s absolute configuration.
Bromine substitution on the monoterpenoid cyclohexane ring at C-4 has been previously reported,4 yet bromine substitution at C-12 or C-10, the other reported sites of chlorination, has only recently been observed.10 The novel chloronium ion based biosynthetic mechanism of terpene cyclization demonstrated by Winter et al.11 for this series of metabolites encouraged us to examine whether the vanadium dependent chloroperoxidases that chlorinate C-10 and the FADH2-dependent halogenase that chlorinates C-12 could also use bromine in the cyclization. To explore this, strain CNQ525 was cultivated with a nutrient medium supplemented with sodium bromide. Examination of the organic extract from the bromide enriched cultivation yielded a new congener, napyradiomycin CNQ525.538 (2), that contained two halogens. The mass spectrometric isotope ionization pattern observed suggested the presence of one chlorine and one bromine substituent. High resolution TOF negative ion mass spectrometric data illustrated a quasi-molecular ion ([M-H]− m/z 537.1042) corresponding to the molecular formula C26H3279Br35ClO5. Analysis of the HSQC, HMBC and 1H NMR spectra revealed the presence of one aromatic methyl group (δC 8.1, C-26, δH 2.22, s, H-26), and two aliphatic methyl groups (δC 16.2, C-21, δH 1.28, s, H-21; δC 25.5, C-22, δH 1.58, s, H-22; and three olefinic methyls (δC 17.4, C-23, δH 1.44, s, H-23; δC 29.1, C-24, δH 1.49, s, H-24; δC 23.2, C-25, δH 1.23, s, H-25). The presence of three olefinic methyl groups suggested that the geranyl group in this molecule had not been cyclized. The recognition of an aromatic methyl and a linear terpenoid substituent suggested that this compound was similar to the published napyradiomycin SF2415B3 (Figure 1),12,13 but with substitution of bromine for one of the chlorine atoms (numbering of carbons in 2 is reflective of previous numbering published for SF2415B3.) Comparison of the published spectroscopic data for compound SF2415B3 with those obtained for 2 showed the molecules were almost identical. Reflecting the addition of bromine at C-10, there were variations in both the proton and carbon shifts at C-10 (SF2415B3 – δC 58.8, C-10, δH 4.43, dd, J = 4.9, 11.3, H-10; for 2 – δC 50.7, C-10, δH 4.52, dd, J = 6.3, 9.6, H-10). Napyradiomycin CNQ525.538 (2) is a rare example of a napyradiomycin that contains bromine at a position other than C-4. The overall structure was subsequently confirmed by comprehensive 2D NMR analysis (Table 2).
Table 2.
| Carbon # | δC, type | δH, mult. (J in Hz) | COSY | HMBCc |
|---|---|---|---|---|
| 1 | 142.7, C | |||
| 2 | 39.6, CH2 | 1.55, m | ||
| 3 | 25.7, CH2 | 1.54, m | ||
| 4 | 123.6, CH | 4.84 bs | H-3 | 3, 22, 23 |
| 5 | 131.6, C | |||
| 6 | 115.1, CH | 4.67, t (8.1) | H-7 | 21, 2, 7 |
| 7 | 41.3, CH2 | 2.67, d (8.1) | 1, 6, 8, 12,-20 | |
| 8 | 83.4, C | |||
| 9 | 78.4, C | |||
| 10 | 50.7, CH | 4.52, dd (6.3, 9.6) | H-11a, H-11b | 9, 11, 24, 25 |
| 11 | 43.7, CH2 | 2.55, m | 8, 9, 10, 12, 13 | |
| 2.57, m | ||||
| 12 | 78.6, C | |||
| 13 | 193.2, C | |||
| 14 | 109.7, C | |||
| 15 | 162.6, C | |||
| 16 | 120.6, C | |||
| 17 | 161.7, C | |||
| 18 | 107.7, CH | 7.18, s | 13, 14, 16, 17, 19, 20 | |
| 19 | 131.8, C | |||
| 20 | 196.4, C | |||
| 21 | 16.2, CH3 | 1.28, s | 1, 2, 6, 7 | |
| 22 | 25.5, CH3 | 1.58, s | 4, 5, 23 | |
| 23 | 17.4, CH3 | 1.44, s | 4, 5, 22 | |
| 24 | 29.1, CH3 | 1.49, s | 9, 10, 25 | |
| 25 | 23.2, CH3 | 1.23, s | 9, 10, 24 | |
| 26 | 8.1, CH3 | 2.22, s | 14, 15, 16, 18, 19 | |
| OH (15) | 12.11, s | 14, 15, 16 |
1H Spectra were recorded in CDCl3 at 600 MHz,
Assignments were made based on analysis of COSY, HMBC and HSQC data.
HMBC correlations are from the proton(s) stated to the indicated carbons.
Further fractionation of the extract yielded another compound that had a similar mass as the known napyradiomycin A80915B, but clearly lacked the UV profile of a napyradiomycin with the diazo moiety attached to the dihydronapthoquinone core.7 Examination of the isotope ionization pattern of the new compound suggested that it was not tri-chlorinated as in napyradiomycin A80915B, but instead contained one chlorine and one bromine substituent. This new congener, napyradiomycin CNQ525.554 (3), was analyzed by HRFTMS, which illustrated a quasi-molecular ion ([M+H]+ m/z 555.1150) that corresponded to the molecular formula C26H3279Br35ClO6. Compound 3 had a similar molecular formula to 1 but contained bromine rather than chlorine. The 1H NMR spectroscopic data for 1 and 3 were virtually identical, however analysis of the HSQC NMR spectrum showed a significant upfield shift for C-10 (for 2 = δC 59.0, C-10; for 3 = δC 51.0, C-10) suggesting C-10 as the site of bromination. Subsequent comprehensive analysis of 1D and 2D NMR data (Table 3) demonstrated that 3 was indeed the brominated analog of 1 with the bromine positioned at C-10.
Table 3.
| Carbon # | δC, type | δH, mult (J in Hz) | COSY | HMBCc |
|---|---|---|---|---|
| 1 | 90.6, C | |||
| 2 | 29.4, CH2 | 1.54, m | 1, 3, 6 | |
| 1.20, m | 1, 3, 6 | |||
| 3 | 26.7, CH2 | 1.55, m | 1, 2, 4, 5 | |
| 1.62, m | 1, 2, 4, 5 | |||
| 4 | 87.2, CH | 3.77, d (4.8) | H-3a, H-3b | 1, 2, 3, 5, 6 |
| 5 | 41.0, C | |||
| 6 | 48.6, CH | 1.78, bs | 1, 2, 7, 8, 21, 22, 23 | |
| 7 | 36.7, CH2 | 1.41, dd (5.1, 15.5) | H6 | 1, 5, 6, 8, 20 |
| 2.03, dd (3.4, 15.5) | H6 | 1, 5, 6, 8, 20 | ||
| 8 | 83.6, C | |||
| 9 | 78.3, C | |||
| 10 | 51.0, CH | 4.58, dd (3.8, 12.2) | H11a, H-11b | 9, 11, 12, 24, 25 |
| 11 | 43.6, CH2 | 2.49, dd (12.2, 14.0) | 8, 9, 10, 12, 13, | |
| 2.60, dd (3.8, 14.0) | 8, 9, 10, 12, 13, | |||
| 12 | 81.8, C | |||
| 13 | 193.9, C | |||
| 14 | 107.5, C | |||
| 15 | 163.4, C | |||
| 16 | 119.9, C | |||
| 17 | 163.1, C | |||
| 18 | 107.4, CH | 7.22, s | 14, 16, 17, 19, 20 | |
| 19 | 131.6, C | |||
| 20 | 192.9, C | |||
| 21 | 20.9, CH3 | 1.45, s | 1, 2, 6 | |
| 22 | 31.6, CH3 | 0.85, s | 4, 5, 23 | |
| 23 | 21.2, CH3 | 0.23, s | 4, 5, 22 | |
| 24 | 29.9, CH3 | 1.46, s | 9, 10, 25 | |
| 25 | 23.9, CH3 | 1.36, s | 9, 10, 24 | |
| 26 | 8.5, CH3 | 2.16, s | 14, 15, 16, 17, 18 | |
| OH (15) | 12.36, s | 14, 15, 16 |
1H spectra were recorded in CDCl3 at 600 MHz.
Assignments were made based on COSY, HMBC and HSQC experiments.
HMBC correlations are from the proton(s) stated to the indicated carbons.
Until now only napyradiomycins that were mono-brominated had been isolated. Careful inspection of the bromide enriched cultivation extract illustrated the presence of another new metabolite assigned as napyradiomycin CNQ525.600 (4). Negative ion HR-MALDI TOF MS analysis of 4 showed a quasi-molecular ion ([M−H]− m/z 599.0649) that corresponded to the molecular formula C26H3479Br2O6. NMR spectroscopic data for 4 showed five aliphatic methyl groups (δC 24.4, C-21, δH 1.21, s, H-21; δC 17.5, C-22, δH 0.72, s, H-22; δC 30.0, C-23, δH 0.41, s, H-23; δC 21.0, C-24, δH 1.50, s, H-24; δC 21.0, C-25, δH 1.36, s, H-25), one aromatic methyl group (δC 8.1, C-26, δH 2.18, s, H-26), one aromatic proton (δH 7.52, s, H-18), and the characteristic proton resonance from the hydrogen bonded –OH at C-15 (δH 12.47). The monoterpenoid cyclohexane ring in 4 was found to contain a hydroxy group at C-1 and a bromine at C-4. This is similar to the monoterpenoid ring seen in napyradiomycin A80915C, which has a chlorine atom attached at C-4.7 Comparison of the 1H NMR spectrum of 4 with the published data from napyradiomycin A80915C showed a downfield chemical shift for the C-4 proton (A80915C = δH 3.47, dd, J =3.7, 12.1, H-4; for 4 = δH 3.71, dd, J =3.9, 12.5, H-4). This is the first example of a napyradiomycin with a hydroxy group and a bromine atom on the cyclohexane ring. Comprehensive analysis of COSY, HMBC and HSQC NMR spectroscopic data revealed that 4 had a hydrogen atom at C-12. Only one other napyradiomycin, compound CNQ525.512,2 contains a hydrogen atom at C-12 with all other napyradiomycins containing either a chlorine or a hydroxy functional group at C-12 or a double bond at the C-11 and C-12 positions. Comparison of the 1H NMR spectroscopic data from compound 4 with that from CNQ525.512 showed a downfield chemical shift for the C-10 proton (CNQ525.512 = δH 3.92, dd, J = 3.9, 12.3, H-10; for 4 = δH 4.02, dd, J = 3.8, 12.5, H-10) suggesting the presence of the second bromine at C-10. The hydroxy and bromine containing cyclohexane ring, the presence of a bromine atom at C-10, and the presence of a hydrogen atom at C-12 makes 4 a significant new compound to be added to the napyradiomycin compound library. The structure of 4 was confidently assigned by comprehensive 2D NMR analysis (Table 4).
Table 4.
| Carbon # | δC, type | δH, mult (J in Hz) | COSY | HMBCc |
|---|---|---|---|---|
| 1 | 71.6, C | |||
| 2 | 42.2, CH2 | 1.44, dd (3.5, 13.8) | 1, 3, 21 | |
| 1.86, m | ||||
| 3 | 31.8, CH2 | 1.89, m | ||
| 2.04, dd (3.9, 13.8) | 1, 2, 5, 6 | |||
| 4 | 66.3, CH | 3.71, dd (3.9, 12.5) | H-3a, H-3b | 2, 3, 5, 22, 23 |
| 5 | 40.4, C | |||
| 6 | 51.0, CH | 1.52, d (7.7) | 1, 2, 3, 4, 5, 21 | |
| 7 | 41.1, CH2 | 2.02, m | ||
| 1.90, m | ||||
| 8 | 81.2, C | |||
| 9 | 80.5, C | |||
| 10 | 53.0, CH | 4.02, dd (3.8, 12.5) | C-11a, C-11b | 9, 11, 12, 24, 25 |
| 11 | 35.0, CH2 | 2.24, m | ||
| 2.56, dt (3.8, 13.5) | 8, 10, 12, 13 | |||
| 12 | 58.8, CH | 3.12, dd (4.2, 13.5) | C-11a, C-11b | 7, 8, 10, 11, 13, 14, 20 |
| 13 | 199.3, C | |||
| 14 | 108.9, C | |||
| 15 | 163.0, C | |||
| 16 | 119.5, C | |||
| 17 | 162.9, C | |||
| 18 | 108.1, CH | 7.52, s | 16, 17, 19, 20, 26 | |
| 19 | 132.6, C | |||
| 20 | 193.6, C | |||
| 21 | 24.4, CH3 | 1.21, s | 1, 2, 6 | |
| 22 | 17.5, CH3 | 0.72, s | 4, 5, 23 | |
| 23 | 30.0, CH3 | 0.41, s | 4, 5, 22 | |
| 24 | 30.0, CH3 | 1.50, s | 9, 10, 25 | |
| 25 | 21.0, CH3 | 1.36, s | 9, 10, 24 | |
| 26 | 8.1, CH3 | 2.18, s | 14, 15, 16, 17, 18 | |
| OH (15) | 12.47, s | 14, 15, 16 |
1H spectra were recorded in CDCl3 at 600 MHz.
Assignments were made based on interpretation of COSY, HMBC and HSQC experiments.
HMBC correlations are from the proton(s) stated to the indicated carbons.
The relative configurations of the napyradiomycins 1–4 were determined using NOESY correlations and coupling constant analyses where possible (Tables 1–4), except for 1 the absolute configuration of which was defined by X-ray crystallographic analysis. For 2–4, the absolute configurations were assumed to be identical to 1 based on their stereospecific biosyntheses.
In the course of isolation of these four new molecules we were also able to isolate a significant number of previously identified napyradiomycin congeners. This allowed for a more detailed examination of the specific mechanism of action for this class. Previously, this family had been shown to be antibacterial, but also several cytotoxic napyradiomycins have been reported. Due to the high level of conjugation in the molecules, it seemed possible that the biological actions of these molecules could be the result of nucleophilic addition (acting as Michael acceptors), thus causing broad, non-specific cellular damage. On the other hand, if the mechanism of action was binding to a specific protein target then structural variations in the molecules should have a significant effect on the potency of the molecules. In total, 14 napyradiomycins were screened against HCT-116 colon carcinoma at concentrations from 100 μM to 0.78 μM by incubating for three days. A large variation in the cytotoxicity of the compounds was noted despite relatively minor changes in structure (Table 5).
Table 5.
In vitro cytotoxicity (IC50 values) of the napyradiomycins 1–4 and related napyradiomycin congeners against HCT-116 colon carcinoma. The 95% confidence interval data are shown to the right. For several compounds the 95th percent confidence interval could not be established due to excessively high or low IC50 values - these are marked NA(not accessable).
| Compound | IC50 values | 95% CI |
|---|---|---|
| Napyradiomycin CNQ525.510B (1) | 17 μM | 10.6 – 26.0 μM |
| Napyradiomycin CNQ525.538 (2) | 6 μM | 3.5 – 10.5 μM |
| Napyradiomycin CNQ525.554 (3) | >100 μM | NA |
| Napyradiomycin CNQ525.600 (4) | 49 μM | 16.3 – 90.9 μM |
| Napyradiomycin B1 | 2 μM | 0.8 – 7.4 μM |
| Napyradiomycin B3 | 3 μM | 1.0 – 5.0 μM |
| Napyradiomycin B4 | 10 μM | 2.8 – 32.6 μM |
| A80915A | 3 μM | 1.0 – 9.5 μM |
| A80915B | <1 μM | NA |
| A80915C | 15 μM | 7.4 – 28.8 μM |
| A80915D | <1 μM | NA |
| CNQ525.510A | >100 μM | NA |
| CNQ525.512 | >100 μM | NA |
| etoposide control | 1 μM | 0.7 – 2.2 μM |
We then examined the effects of four napyradiomycins on the cell cycle by Fluorescence- Activated Cell Sorting methods (FACS) using HCT-116 colon carcinoma cells. Of interest was whether these compounds induced apoptosis, a regulated cell death, or necrosis, a non-regulated cell death. In healthy, multicellular organisms, a key part of controlled cell growth is apoptosis.14 Cancer cells often become resistant to apoptotic signals and therefore continue to grow under conditions in which normal cells would undergo regulated cell death. Anticancer drug leads that induce apoptosis have a greater potential to become therapeutic agents than non-specific cytotoxins, which are more likely to cause significant and undesirable side effects. The four napyradiomycins selected for FACS analysis were napyradiomycin CNQ525.510B (1), napyradiomycin A80915A, napyradiomycin A80915C, and the diazo-containing analog napyradiomycin A80915B. These napyradiomycins were incubated with HCT-116 cells, then detached with trypsin, washed, and stained with propidium iodide and Yo-pro apoptosis detecting dye. Samples were analyzed by FACS, with gating, using a camptothecin positive control. All compounds were found to induce apoptosis in a dose dependent manner (Figure 3 and Supporting Information). Due to the cancellation of cell particles during the flow cytometry analysis, this assay is not able to assess total cell death but can only analyze the state of the intact cells that are still present. Napyradiomycin CNQ525.510B (1) began to induce apoptosis at 4 μM, which is similar to compound A80915A. However, napyradiomycin A8015C did not begin to induce significant apoptosis at less than 8 μM. That all four of these diverse napyradiomycins induce apoptosis suggests that molecules in this class, perhaps possessing the common dihydronaphthoquinone functionality, may target a specific biochemical pathway, albeit at differing concentrations. Further, this suggests that there is likely a specific protein that these molecules are interacting with. The interesting bimodal effect seen in the diazo-containing compound (Figure 3C) may be due to two mechanisms taking place. At 1 μM and 2 μM there is significant cell death and apoptosis, which is lower at 4 μM and then is present again at 8 μM and 16 μM. We were not able to obtain the 50,000 events necessary in our analysis for the A808915B sample at 32 μM due to complete cell death. While unknown, the initial activity may be due to activity associated with the diazo group, while the secondary activity seen at higher concentrations may be due to the napyradiomycin acting at a core target.
Figure 3.

FACS analysis using HCT-116 cells illustrating the induction of apoptosis by treatment with four napyradiomycin derivatives. A) HCT-116 cells were incubated for 24 h with napyradiomycin CNQ525.510B (1); B) A80915 A; C) A80915C; and D) A80915B at various concentrations. Controls: E) are shown 1- no treatment of cells as negative control, 2- DMSO only as negative control, and 3- cell incubated with 10 μM camptothecin as a positive control. Samples were subsequently stained with propidium iodide, a marker of cell death, and Yo-Pro-1, a marker of apoptosis.
In summary, we have described the isolation and structure elucidation of four new napyradiomycin congeners, 1–4. By comparing structurally similar napyradiomycins in a FACS-based cytotoxicity bioassay, we have provided evidence of the induction of apoptosis. From these initial studies it is not yet clear if the mechanism of action of these molecules is unique, however these results suggest further examination.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a JASCO P-2000 polarimeter. UV spectra were measured on a Beckman Coulter DU800 spectrophotometer. IR spectra were recorded on a Perkin-Elmer 1600 FTIR spectrometer. NMR spectra were obtained on Varion Inova 500 and 300 MHz spectrometers, and a Bruker 600 MHz DRX-600 equipped with a 1.7 mm cryoprobe and Avance III console. High-resolution mass spectra were obtained on an Agilent ESI-TOF at the Scripps Center for Mass Spectrometry. Low-resolution LC-MS data were acquired using a Hewlett-Packard series 1100 system equipped with a reversed-phase C18 column (Phenomenex Luna, 4.6 × 100 mm, 5 μm) at a flow rate of 0.7 mL/min. Preparative HPLC separations were performed using a Waters 600E system controller and pumps with a Model 480 spectrophotometer. Purifications were achieved using Phenomenex Luna semi-preparative C18 (250 × 10 mm, 5 μm) and Phenomenex Luna semi-preparative silica (250 × 10 mm, 5 μm) columns at flow rates of 5.0 and 3.0 mL/min, respectively.
Isolation of Strain CNQ525
The actinomycete strain CNQ525 was isolated from a marine sediment sample collected at a depth of 152 m off the coast of La Jolla, CA as previously described.2 This strain was subjected to 16S rRNA sequence analysis (Genbank accession number EF177816) and found to be part of the MAR4 group that clades within the family Streptomycetaceae.2,3
Isolation of Napyradiomycins 1–4
For the isolation of compound 1, strain CNQ525 was cultured in 20 x 1L scale using A1bFe + C media (10 g starch, 4 g yeast extract, 2 g peptone, 1 g CaCO3, 5 mL of a 2% (w/v) KBr stock solution, and 5 mL of a 0.8% (w/v) Fe2(SO4)•4H2O stock solution in 1 L seawater). Cultures were shaken at 200 rpm on a rotary shaker, at 30 °C for 9 days, and then extracted by stirring for 6 h with Amberlite XAD-7 resin (20–30 g/L). The resin was filtered and extracted with acetone, and the filtered extraction solvent removed under vacuum to generate the organic extract (7.5 g). The organic extract was then fractionated by silica flash chromatography using various ratios of hexane/EtOAc. Napyradiomycin CNQ525.510B (1, 5.6 mg) was subsequently isolated from the 65% hexane/35% EtOAc flash column fraction using semi-preparative C8 HPLC performed isocratically with 80% CH3CN/20% H2O. For the isolation of 2–4, a 20 x 1L fermentation using an A1bFe + C high bromide medium was prepared by addition of NaBr to an artificial seawater medium (20 g NaBr, 0.6 g KBr, 9.3 g MgCl2•6H2O, 1.3 g CaCl2•2H2O, 3.4 g Na2SO4, 1.7 g NaHCO3, 0.002 g Na2HPO4, 1 liter distilled H2O). Cultures were shaken at 200 rpm on a rotary shaker, at 30 °C for 9 days, and then extracted by stirring for 6 h with Amberlite XAD-7 resin (20–30 g/L). The resin was filtered and extracted with acetone, and the filtered extraction solvent removed under vacuum to generate the organic extract. The extract (4.0 g) was then fractionated by silica flash chromatography using various rations of EtOAc/isooctane to produce ten fractions of differing polarity. These fractions were then analyzed by LC-MS to assess the presence of members of the napyradiomycin family. Napyradiomycin CNQ525.538 (2, 5.9 mg) was isolated from the 85% hexane/15% EtOAc flash column fraction using semi-preparative C8 RP HPLC performed isocratically with 80% CH3CN/20% H2O. Napyradiomycin CNQ525.554 (3, 10.4 mg) was isolated from the 65% hexane/35% EtOAc flash column fraction by semi-preparative C8 RP HPLC using a 70:30 to 100:0 CH3CN/H2O gradient over 40 min, and napyradiomycin CNQ525.600 (4, 1.0 mg) was isolated from the 65% hexane/35% EtOAc flash column fraction by semi-preparative C8 RP HPLC using a 70:30 to 100:0 CH3CN/H2O gradient.
Napyradiomycin CNQ525.510B (1): amorphous solid, [α]D −3.7 (c 0.5, CHCl3); UV λmax (log ε) 216 (4.15), 324 (3.70), 352 (3.65) nm; IR (film) νmax 2922, 2854, 1602, 1451, 1346, 1281, 1114, 1072 cm−1; NMR see Table 1; HRESITOFMS m/z 511.1652 [M+H]+ (calcd for C26H3335Cl2O6, 511.1654)
Napyradiomycin CNQ525.538 (2): colorless oil, [α]D −9 (c 0.5, CHCl3); UV λmax (log ε) 264 (4.11), 319 (3.69), 356 (3.65) nm; IR (film) νmax 3399, 2920, 2851, 1711, 1608, 1457, 1286, 1163, 1079, 1012, 909 cm−1; NMR see Table 2; HRESITOFMS m/z 537.1042 [M−H]− (calcd for C26H3179Br35ClO5, 537.1043)
Napyradiomycin CNQ525.554 (3): a light red oil, [α]D −42 (c 0.25, CHCl3); UV λmax (log ε) 257 (3.76), 303 (3.32), 355 (3.24) nm; IR (film) νmax 2922, 1701, 1604, 1454, 1285, 1197, 1118, 1081, 1007 cm−1; NMR see Table 3; HRESIFTMS m/z 555.1150 [M+H]+ (calcd for C26H3379Br35ClO6, 555.1149).
Napyradiomycin CNQ525.600 (4): a light red-orange oil, [α]D −20 (c 0.25, CHCl3); UV λmax (log ε) 262 (4.22), 322 (3.78), 355 (3.74) nm; IR (film) νmax 2921, 1697, 1622, 1455, 1309, 1115, 1055 cm−1; NMR see Table 5; HRMALDITOFMS m/z 599.0649 [M−H]− (calcd for C26H3379Br2O6, 599.0644).
X-Ray Analysis of Napyradiomycin CNQ525.510B (1)
A colorless block 0.15 × 0.15 × 0.15 mm in size (but coated with a gummy substance), obtained from a 50:50 hexane/EtOAc solution, was mounted on a Cryoloop with Paratone oil. The crystals obtained were very poor, however X-ray data were successfully collected in a nitrogen gas stream at 100(2) °K using phi and omega scans. Crystal-to-detector distance was 60 mm and exposure time was 5 seconds per frame using a scan width of 0.5°. Data collection was 98.2% complete to 67.00° in θ. A total of 7887 reflections were collected covering the indices, −11<=h<=12, −18<=k<=18, −12<=l<=12; 4187 reflections were found to be symmetry independent, with an Rint of 0.0191. Indexing and unit cell refinement indicated a primitive, monoclinic lattice. The space group was found to be P2(1) (No. 4). The data were integrated using the Bruker SAINT software program and scaled using the SADABS software program. Solution by direct methods (SIR-2004) produced a complete heavy-atom phasing model consistent with the proposed structure of napyradiomycin CNQ525.510B (1). All non-hydrogen atoms were refined anisotropically by full-matrix least-squares (SHELXL-97). All hydrogen atoms were placed using a riding model. Their positions were constrained relative to their parent atom using the appropriate HFIX command in SHELXL-97. The X-ray experiment defined the absolute configuration of 1 (Figure 2. See the Supporting Information for complete X-ray experimental details and crystal data). Crystallographic data for napyradiomycin 1 have been deposited with the Cambridge Crystallographic Data Centre under the deposition number CCDC 944922. Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Antiproliferative Bioassay
Aliquot samples of HCT-116 human colon adenocarcinoma cells were transferred to 96-well plates and incubated overnight at 37 °C in 5% CO2/air. Test compounds were dissolved in DMSO at 10 mg/mL and added to the plates in DMSO at an initial concentration of 100 μM and serially diluted. The plates were then further incubated for another 72 h, and at the end of this period, a CellTiter 96 aqueous non-radioactive cell proliferation assay (Promega) was used to assess cell viability. Inhibition concentration (IC50) values were deduced from the bioreduction of MTS/PMS by living cells into a formazan product. MTS/PMS was first applied to the sample wells, followed by incubation for 3 h. Etoposide (Sigma; IC50 = 1.5–4.9 μM) and DMSO (solvent) were used as the positive and negative controls in this assay. The quantity of the formazan product (in proportion to the number of living cells) in each well was determined by the Molecular Devices Emax microplate reader set to 490 nm wavelength. IC50 values were calculated using the analysis program Prism.
Flow Cytometry (Apoptosis) Assay
Flow cytometry (FACS) analysis was performed using Yo-Pro-1 (Invitrogen) as a marker of apoptotic cells and propidium iodide as a marker of necrotic cells. HCT116 cells were plated on to 6 well plates and allowed to grow in McCoy’s media with 10% FBS at 37 °C in 5% CO2/air until 50% confluent. Compounds at desired concentration were added to microtiter plates and incubated for 24 h. Cells were harvested after the incubation period using trypsin, and washed in cold phosphate-buffered saline (PBS) and adjust the cell density to ~1 × 106 cells/mL in PBS. For each assay, a 1 mL assay volume was used. Yo-Pro-1 stock solution (1 μL) was added (Component A) and 1 μL PI stock solution (Component B) were added to each 1 mL of cell suspension. HCT - 116 cells were incubated on ice at a final concentration of 0.1 μM Yo-Pro-1 and 3 μg/mL propidium iodide for 30 min prior to flow analysis. Cells were immediately analyzed on a Becton-Dickinson instrument, using 488 nm excitation with green fluorescence emission for Yo-Pro-1 and red fluorescence emission for propidium iodide. Cell gating was performed by analysis of both a negative control (DMSO) and a positive control (24 hours of 10 μM camptothecin). Cells were gated to exclude debris.
Supplementary Material
Acknowledgments
This work was the result of financial support from the NIH, National Cancer Institute, under grant R37 CA044848.
Footnotes
Copies of NMR and other spectra for napyradiomycins 1–4, as well as complete X-ray data for 1 and raw FACS data are available online at http://pubs.acs.org.
References
- 1.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 2.Soria-Mercado IE, Prieto-Davó A, Jensen PR, Fenical W. J Nat Prod. 2005;68:904–910. doi: 10.1021/np058011z. [DOI] [PubMed] [Google Scholar]
- 3.Gallagher K, Fenical W, Jensen PR. Curr Opin Biotechnol. 2010;21:794–780. doi: 10.1016/j.copbio.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Cheng YB, Jensen PR, Fenical W. Euro J Org Chem. 2013;18:3751–3757. doi: 10.1002/ejoc.201300349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiomi K, Iinuma H, Hamada M, Naganawa H, Manabe M, Matsuki C, Takeuchi T, Umezawa H. J Antibiot. 1986;39:487–493. doi: 10.7164/antibiotics.39.487. [DOI] [PubMed] [Google Scholar]
- 6.Shiomi K, Nakamura H, Iinuma H, Naganawa H, Takeuchi T, Umezawa H, Iitaka Y. J Antibiot. 1987;40:1213–1219. doi: 10.7164/antibiotics.40.1213. [DOI] [PubMed] [Google Scholar]
- 7.Fukuda DS, Mynderse JS, Baker PJ, Berry DM, Boeck LD, Yao RC, Mertz FP, Nakatsukasa WM, Mabe J, Ott J, Counter FT, Ensminger PW, Allen NE, Alborn W, Hobbs JN. J Antibiot. 1990;43:623–633. doi: 10.7164/antibiotics.43.623. [DOI] [PubMed] [Google Scholar]
- 8.Dantzig AH, Minor PL, Garrigus JL, Fukuda DS, Mynderse JS. Biochem Pharmacol. 1991;42:2019–26. doi: 10.1016/0006-2952(91)90603-3. [DOI] [PubMed] [Google Scholar]
- 9.Hori Y, Abe Y, Shigematsu N, Goto T, Okuhara M, Kohsaka M. J Antibiot. 1993;46:1890–1893. doi: 10.7164/antibiotics.46.1890. [DOI] [PubMed] [Google Scholar]
- 10.Wu Z, Li S, Li J, Chen Y, Saurav K, Zhang Q, Zhang H, Zhang W, Zhang S, Zhang C. Marine Drugs. 2013;11:2113–2125. doi: 10.3390/md11062113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winter JM, Moffitt MC, Zazopoulos E, McAlpine JB, Dorrestein PC, Moore BS. J Biol Chem. 2007;282:16362–16368. doi: 10.1074/jbc.M611046200. [DOI] [PubMed] [Google Scholar]
- 12.Gomi S, Ohuchi S, Sasaki T, Itoh J, Sezaki M. J Antibiot. 1987;40:740–749. doi: 10.7164/antibiotics.40.740. [DOI] [PubMed] [Google Scholar]
- 13.Shomura T, Gomi S, Ito M, Yoshida J, Tanaka E, Amano S, Watabe H, Ohuchi S, Itoh J, Sezaki M, Takebe H, Uotani K. J Antibiot. 1987;40:732–739. doi: 10.7164/antibiotics.40.732. [DOI] [PubMed] [Google Scholar]
- 14.Meier P, Vousden KH. Mol Cell. 2007;28:746–754. doi: 10.1016/j.molcel.2007.11.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.