

Abstract
Background
Activation of polymorphonuclear neutrophils (PMN) is thought to contribute to traumatic brain injury (TBI). Because hypertonic fluids can inhibit PMN activation, we studied whether hypertonic fluid resuscitation can reduce PMN activation in TBI patients.
Methods
Trauma patients with severe TBI were resuscitated with 250 ml of either 7.5% hypertonic saline (HS; n=22), HS + 6% dextran-70 (HSD; n=22), or 0.9% normal saline (NS; n=39) and blood samples were collected on hospital admission and 12 and 24 h after resuscitation. PMN activation (CD11b, CD62L, CD64) and degranulation (CD63, CD66b, CD35) markers and oxidative-burst activity as well as spontaneous PMN apoptosis were measured by flow cytometry.
Results
Relative to healthy controls, TBI patients showed increased PMN activation and decreased apoptosis of PMN. In the HS group, but not in the HSD group, markers of PMN adhesion (CD11b, CD64) and degranulation (CD35, CD66b) were significantly lower than in the NS group. These effects were particularly pronounced 12 h after resuscitation. Treatment with HS and HSD inhibited PMN oxidative burst responses compared to NS-treated patients. HS alone partially restored apoptosis. Despite these differences, the groups did not differ in clinical outcome parameters such as mortality and Extended Glasgow Outcome Scale.
Conclusions
This study demonstrates that pre-hospital resuscitation with HS can partially restore normal PMN activity and the apoptotic behavior or PMNs, while resuscitation with HSD was largely ineffective. Although the results are intriguing, additional research will be required to translate these effects of HS into treatment strategies that improve clinical outcome in TBI patients.
Keywords: Hypertonic saline, resuscitation fluids, hemorrhage, neutrophils, adhesion molecules, inflammation, multi-organ failure, oxidative burst, apoptosis
Introduction
Traumatic brain injury (TBI) is a leading cause of mortality and long-term neurological disability worldwide [1,2]. Functional outcome after severe TBI depends on the extent of the initial mechanical injury to the head as well as subsequent pathophysiological processes and corresponding treatments aimed at reducing brain damage [3]. These pathophysiological processes associated with TBI are complex and require multifactorial treatment approaches to reduce secondary injury after TBI. The primary mechanical insult that causes the disruption of the brain parenchyma, blood vessels, and diffuse axonal damage at the moment of impact is irreversible and can only be averted through primary prevention strategies [4]. However, subsequent secondary injury processes are amenable to therapeutic intervention with neuroprotective agents [5]. Secondary injury is mediated through several pathophysiological mechanisms, including the formation of post-traumatic brain edema, raised intracranial pressure, disruption of the blood-brain barrier, reduced cerebral perfusion pressure, and ischemia, all of which lead to irreversible brain damage [6,7]. A cascade of neurochemical and immunological events contribute to secondary brain damage through hypoxia, oxidative stress, release of inflammatory mediators, mitochondrial dysfunction, cellular infiltration, and apoptotic and necrotic cell death of neurons and glia cells [8].
Since it was first described nearly 100 years ago [9], hyperosmotic fluid therapy has been widely used in the care for TBI patients to reduce brain swelling that follows head injuries and triggers many of the subsequent pathological processes that cause permanent tissue damage. Numerous studies have implicated a dysfunctional innate inflammatory response with excessive polymorphonuclear neutrophil (PMN) activation in the processes contributing to secondary brain injury [10-12]. Delayed PMN apoptosis after critical injury is another mechanism that could increase PMN-induced tissue damage. Thus, inhibition of the activation of PMN and their defective apoptotic behavior may be a potential strategy for TBI management. Hypertonic crystalloids and colloids rapidly restore blood pressure by expanding intravascular volume while simultaneously reducing intracranial hypertension and cerebral edema in TBI patients [13,14].
We have previously shown that specific hypertonic saline resuscitation strategies can inhibit early post-traumatic PMN activation and secondary organ damage after hemorrhagic shock [15-18]. In the current study, we investigated whether pre-hospital hypertonic fluid resuscitation using hypertonic saline with or without dextran can help to reduce trauma-induced alterations of PMN responses in TBI patients. In particular, we characterized PMN activation markers, including a panel of cell-surface adhesion and degranulation molecules, oxidative burst capacity, and spontaneous PMN apoptosis.
Materials and Methods
Study design, setting, and participants
The study protocol was approved by the US Food and Drug Administration, the Canadian Institutes of Health Research, and all participating Institutional Review Boards as previously described [19]. This study was an a priori subgroup analysis of a larger randomized, placebo-controlled, double-blinded trial carried out by the Resuscitation Outcomes Consortium (ROC) — a multicenter clinical trials network designed to conduct interventional studies in the pre-hospital setting following life-threatening trauma [20,21]. For the current study, we included 83 severe TBI patients with head trauma and a Glasgow Coma Scale (GCS) ≤8 who were admitted to the study sites in Seattle or Toronto, where laboratory personnel was standing by around-the-clock to process serial blood samples for the immunological studies described below. The patients described in the current study included only those TBI patients without signs of hemorrhagic shock. Patients with hemorrhagic shock were assigned to another sub-study [21]. The enrollment criteria for patient selections were described previously [20]. Briefly, patients were excluded if they were <15 years of age, pregnant, or if they received intravenous fluid therapy in the field with >1,000 ml of isotonic crystalloid fluids, any colloids, or any blood products prior to treatment with study fluids, or if >4 h had passed after injury. Other exclusion criteria were pre-hospital cardiopulmonary resuscitation, severe hypothermia (body core temperature <28°C), drowning or asphyxia due to hanging, burns of >20% of the total body surface area, isolated penetrating head injury, inability to obtain intravenous access, or if a potential subject was known to be a prisoner. A group of 20 asymptomatic adult blood donors served as a healthy control group.
Interventions
The randomized, placebo-controlled, double-blinded, three-armed parent trial was described previously [20,21]. All study fluids were purchased from BioPhausia Inc., Stockholm, Sweden and provided in identical 250-ml infusion bags that contained either 7.5% NaCl + 6% dextran-70 (HSD; RescueFlow), 7.5% NaCl without dextran (HS), or 0.9% NaCl (normal saline, NS). These intravenous bags were distributed among the 11 different geographic regions participating in the parent trial of the ROC. For the current substudy, paramedics in Toronto and Seattle administered the fluids in a blinded fashion via intravenous access as the initial resuscitation fluid given within 4 h of the incident. Once the study fluid had been administered, additional fluids could be given as per local emergency medical service guidelines as previously described [21]. Clinical data collected upon hospital admission included age, gender, mechanism of injury, GCS, and Injury Severity Score (ISS). The severity of illness was quantified using the Glasgow Coma Scale (GCS) at study entry and the Multiple Organ Dysfunction Score (MODS) at the time of admission to the intensive care unit (ICU). The primary outcome measure for TBI patients was the neurological outcome at 6 months based on the Extended Glasgow Outcome Scale (GOS-E). Additional clinical outcome parameters collected were the 28-day survival rate, fluid and blood transfusion requirements, physiologic parameters, and evidence of infections.
Blood samples
In two of the eleven regional centers (Toronto and Seattle) participating in the parent ROC trial, study personnel was on stand-by to collect serial blood samples from TBI patients in order to assess cellular immune responses after HS, HSD, or NS treatment. Serial heparinized whole-blood samples of venous blood were collected at the time of admittance to the emergency department (≤ 3 hours of resuscitation) and 12 and 24 h after admission and immediately processed to assess PMN activation and cell-surface, adhesion, and degranulation markers. Separate blood samples were used to assess routine clinical laboratory values, including plasma sodium concentrations and leukocyte differential counts. Healthy control blood samples were obtained by venipuncture of 20 age-matched healthy volunteers.
Flow cytometric determination of neutrophil cell surface receptors
Whole blood samples were used to analyze the expression of specific surface molecules that indicate various states of PMN activation. PMN adhesion was assessed with antibodies that recognize CD62L (L-selectin), CD11b, and CD64 that are shed from (L-selectin) or increase (CD11b and CD64) in activated cells. We also assessed markers of degranulation using antibodies that recognize CD35, CD66b, and CD63. These degranulation markers are present in secretory vesicles (CD35), specific granules (CD66b), and azurophilic granules (CD63) and emerge on the cell membrane as a result of exocytosis of these granules. Freshly collected whole blood samples (100 μl) were placed in 12×75-mm polystyrene Falcon tubes and incubated for 20 min at room temperature in the dark with saturating concentrations of CD66b-FITC, CD63-PE, CD14-PerCP, CD11b-APC, CD64-FITC, CD35-PE, CD62L-APC antibody conjugates (BD Biosciences, San José, CA). CD14 was assessed to facilitate separation of gated cell populations. Appropriate isotype-matched antibodies were used in separate tubes to control for autofluorescence and non-specific binding. After the incubation period, erythrocytes were removed by adding 2 ml of FACS™ Lysing Solution (BD Biosciences) for 10 min followed by centrifugation at 500×g for 5 min at room temperature. Cells were washed once with CellWASH™ (BD Biosciences) and resuspended in 400 μl of a 1% paraformaldehyde solution. Stained cell suspensions were acquired on a dual-laser FACSCalibur flow cytometer (BD Biosciences) calibrated for four-color analysis using CaliBRITE™ beads (BD Biosciences). For each sample, 104 PMNs were acquired with CellQuest® software (BD Biosciences) using a live-gate setting to distinguish the PMN population from other cells and debris according to the CD14-PerCP fluorescence versus side-scatter (SSC) light characteristics. Data analysis was performed with FlowJo software v.8.7 (Tree Star Inc., Ashland, OR). Electronic analysis gates and quadrant markers were set to define positive and negative populations using fluorescence histogram data according to the non-specific staining of isotype-matched negative controls. Results were recorded as percentage (%)-positive cells and mean channel fluorescence intensity (MFI; in arbitrary units, a.u.). Absolute cell counts were obtained by multiplying the corresponding percentages of cells derived from FACS analysis by total leukocyte counts obtained with a hematology analyzer (Coulter Electronics, Hialeah, FL).
Assessment of spontaneous neutrophil apoptosis with Annexin V staining
The proportion of apoptotic PMNs was determined by PE-conjugated Annexin V (An-V) binding of externalized phosphatidylserine, in conjunction with the vital dye 7-amino-actinomycin D (7-AAD) obtained from BD Biosciences (Mississauga, ON), as previously described [22]. Briefly, fresh heparinized whole-blood samples were cultured in a 95% humidified atmosphere with 5% CO2 at 37°C. After 20 h, blood suspensions were drawn from the culture, washed in ice-cold 1× An-V Binding Buffer, centrifuged, and the pellet resuspended in 100 μl of binding buffer in the presence of 5 μl of PE-conjugated An-V and 5 μl of 7-AAD, along with saturating concentrations of anti-CD14-PerCP. The mixtures were incubated for 15 min at room temperature in the dark. After red cell lysis, samples were resuspended in 400 μl of An-V Binding Buffer and analyzed on a FACSCalibur flow cytometer with a live-gate set to count 10,000 PMN events per samples. Data were analyzed in FlowJo™ software and results were expressed as the total % of An-V-positive PMNs.
Oxidative burst activity
The intracellular oxidative burst capacity (i.e., generation of superoxide and secondary reactive oxygen species) of whole blood PMNs was measured with flow cytometry, using a commercially available fluorometric assay kit (PHAGOBURST® kit, Orpegen Pharma, Heidelberg, Germany). This test provides a quantitative assessment of burst activity by measuring the percentage and fluorescence intensity (MFI, in the FL1 530-nm emission channel) of cells that oxidize the fluorogenic substrate dihydrorhodamine (DHR)-123 to green fluorescent rhodamine (Rho)-123 in the presence of hydrogen peroxide]. Briefly, three 100-μl aliquots of each heparinized whole blood sample were placed in separate test tubes and combined with 20 μl of either wash buffer (as an unstimulated control), N-formyl-methionyl-leucyl-phenylalanine (fMLP), or phorbol 12-myristate 13-acetate (PMA) at final concentrations of 5 and 8.1 μM, respectively. Simultaneously, 20 μl of DHR-123 substrate solution was added to each tube and then samples were incubated in a water bath at 37°C for 20 min. In all cases, reactions were stopped by placing the test tubes into an ice bath. Erythrocytes were lysed by addition of 2 ml of BurstTest Lysing Solution for 10 min. After centrifugation at 250 × g for 5 min, the lysates were discarded and the remaining white cells resuspended and incubated with anti-CD14-APC at room temperature for 15 min. The cells were then washed with 3 ml of Wash Solution and centrifuged at 250 × g. Finally, 200 μl of and propidium iodide (PI) solution in PBS (200 μg/ml) was added to each sample on ice. Samples were analyzed using a FACSCanto (BD Biosciences) flow cytometer within 30 min. PMN populations were gated using a CD14-APC/SSC-defined gate; 10,000 PMN events were recorded for each sample. Data were analyzed in FlowJo™ software v.8.7 (Tree Star) and the results presented as the % Rho-123-positive PMNs and as Rho-123 MFI expressed as a percentage of the unstimulated control sample.
Statistical analyses
Baseline demographic data and clinical outcome scores are expressed as mean ± standard deviation (SD) and as median scores with interquartile ranges (IQR), respectively. All biomarker values were treated as normally distributed continuous variables and expressed as mean ± standard error of the mean (SEM). For statistical analyses, Student's t test was used for continuous variables and χ2 test was used for categorical predictor variables. The non-parametric Mann-Whitney U test was used for continuous variables that were not normally distributed. Serial comparisons (time × treatment) of biomarkers between treatment groups and control group were made using repeated measures ANOVA with post-hoc Bonferroni/Dunn testing. All analyses were two-tailed and p-values < 0.05 were considered statistically significant.
Results
Hypertonic fluid resuscitation did not improve clinical outcome
In the current study, we enrolled a total of 83 patients with severe head injuries and 20 healthy controls. All patients received intravenous fluid therapy in the field using equal volumes (250 ml) of either HS, HSD, or NS. After infusion of a bolus of these study fluids, the volume of further resuscitation fluid administered en route to the emergency department was kept at <1,000 ml. Table 1 shows detailed demographic, laboratory, and clinical information about these three patient groups. Gender, the average age, Glasgow Coma Scales (GCS), Multi-Organ Dysfunction Scores (MODS), and Injury Severity Scores (ISS) were similar in each treatment group. On admission, the first blood sample was drawn to determine PMN activation markers and to assess standard laboratory values. As expected, patients treated with hypertonic fluids showed peak plasma sodium and chloride levels that were ∼10 mM higher than the average values in patients treated with NS. There were no significant differences in outcome parameters among the treatment groups which is consistent with the results of the parent ROC study [20]
Table 1. Demographic, Laboratory and Clinical Characteristics of Severe TBI Patients.
| Resuscitation Group | ||||
|---|---|---|---|---|
|
|
||||
| Variables | All Patients | HS | HSD | NS |
| Demographics | ||||
| No. of patients | 83 | 22 | 22 | 39 |
| Sex, n (% male) | 61 (73.5%) | 19 (86.4%) | 13 (59.1%) | 29 (74.4%) |
| Age, mean (SD), years | 39.8 (19.5) | 39.1 (17.7) | 37.2 (21.0) | 36.2 (19.1) |
| Vital Signs | ||||
| ED SBP, mean (SD), mmHg | 140.4 (28.4) | 144.0 (25.0) | 148.8 (25.2)*,a | 133.8 (30.8) |
| GCS at entry, median (IQR) | 5.0 (3-7) | 5.0 (3-7) | 5.5 (3-7) | 5.0 (3-7) |
| Worst MODS, mean (SD) | 8.7 (9.0) | 7.5 (8.3) | 9.3 (8.9) | 8.6 (9.0) |
| ISS, median (IQR) | 27.0 (15-36) | 25.5 (17.5-35) | 29 (17-38) | 29 (16-35) |
| Injuries | ||||
| Type of injury, n (%) | ||||
| Blunt | 81 (97.6%) | 21 (95.5%) | 21 (95.5%) | 39 (100%) |
| Penetrating | 2 (2.4%) | 1 (4.5%) | 1 (4.5%) | 0 (0%) |
| Head AIS | 3.9 (1.1) | 4.1 (1.0) | 4.0 (1.2) | 3.8 (1.1) |
| Laboratory Values | ||||
| Sodium, mean (SD), mM | 143.0 (5.8) | 147.2 (3.9)*,a | 148.1 (6.6)*,a | 139.8 (2.7) |
| Chloride, mean (SD), mM | 113.0 (10.1) | 116.6 (4.8) | 120.0 (15.1) | 107.2 (3.9) |
| Osmolality, mean (SD), mOsm/l | 321.5 (25.8) | 323.5 (21.8) | 330.1 (25.3) | 315.6 (27.1) |
| Hemoglobin, mean (SD), g/l | 113.4 (24.7) | 121.5 (19.0)*,a | 111.3 (25.2) | 109.9 (27.0) |
| Hematocrit, mean (SD), l/l | 0.33 (0.07) | 0.35 (0.06) | 0.31 (0.08) | 0.32 (0.08) |
| Fluids | ||||
| Total pre-hospital fluid, mean (SD), l | 0.89 (0.63) | 0.82 (0.51) | 0.87 (0.56) | 0.78 (0.61) |
| Total fluids first 24 h, mean (SD), l | 5.9 (4.4) | 5.1 (2.9) | 5.8 (5.1) | 6.4 (4.7) |
| Infections | ||||
| One or more infections, n (%) | 26 (31.3%) | 8 (36.4%) | 7 (31.8%) | 10 (25.6%) |
| Outcomes | ||||
| Length of stay, mean (SD), d | 32.9 (50.0) | 30.8 (22.8) | 21.9 (27.8) | 28.7 (29.3) |
| Overall mortality, n (%) | 23 (27.7%) | 5 (22.7%) | 7 (31.8%) | 11 (28.2%) |
| ARDS free survival to 28 d, n (%) | 61 (73.5%) | 18 (81.8%) | 15 (68.2%) | 28 (71.8%) |
| GOS-E, 6-month, median (IQR) | 4.0 (1-6) | 3.5 (2-6) | 3.5 (1-6) | 4.0 (1-6) |
| DRS, 6-month, median (IQR) | 5 (2-6) | 5.5 (3-6) | 6 (2-6) | 5 (3-6) |
Abbreviations: NS, normal saline; HSD, hypertonic saline plus dextran; ED, emergency department admission; SBP, systolic blood pressure; GCS, Glasgow Coma Scale; ISS, Injury Severity Score; worst MODS, highest Multiple Organ Dysfunction Score during entire observation period; SD, standard deviation; IQR, interquartile range; d, days; GCS, Glasgow Coma Score; head AIS, Abbreviated Injury Score for the head; ISS, Injury Severity Score; ARDS, acute respiratory distress syndrome; GOS-E, Extended Glasgow Outcome Score; DRS, Disability Rating Score. Statistical differences with p < 0.05 between the HS or HSD groups and the NS group are indicated by astersiks followed by the type of test used (a, Student's t-test; b, Mann-Whitney U test; c, Chi-square test).
Neutrophil counts were elevated in all treatment groups
All TBI patients had significantly increased PMN counts on admission, which were up to 3-times higher than those of healthy controls and remained elevated above control values for the first 24 h after injury (Fig. 1). PMNs accounted for ∼ 80% of circulating white blood cells in TBI patients, compared to ∼60% in normal controls. Although the HS group appeared to have slightly lower circulating PMN counts than the other two treatment groups, there were no significant differences among the three groups. These findings suggest that the type of resuscitation fluid has little effect on the leukocyte counts and the mobilization of PMNs in response to TBI.
Fig. 1. TBI induces sustained neutrophilia.

Peripheral blood samples were collected in EDTA vacutainers from healthy controls (n=20) and trauma patients resuscitated with NS (n=39), HSD (n=22), or HS (n=22) at the time of emergency department (ED) admission (≤3 h post-resuscitation) and 12 and 24 h post-resuscitation. Both the percentage (square symbols) of neutrophils among all white blood cells and the concentration (circles) of neutrophils (PMNs) in the peripheral blood were elevated relative to levels in healthy controls (triangles) throughout the sampling period, as determined by a Beckman Coulter Hematology Analyzer. Statistics: ap < 0.05 vs. age-matched healthy controls by ANOVA.
HS attenuated neutrophil activation and adhesion markers
Next we studied whether hypertonic resuscitation fluids can attenuate PMN activation in TBI patients measuring PMN activation markers as previously reported [23]. Multi-color flow cytometry was used to quantify PMN expression of the cell-surface activation and adhesion markers CD11b (an integrin), CD62L (L-selectin), and CD64 (Fcγ receptor I). Upon admission to the emergency department (ED≤3 h), CD11b expression on the cell surface of PMNs was not significantly elevated in either treatment group when compared to healthy controls (Fig. 2A). However, after 12 h, the CD11b expression levels in patients treated with NS or HSD were significantly higher than those in healthy controls. In the HS group, however, CD11b levels remained near control levels. Shedding of CD62L (L-selectin) from the surface of PMNs is another marker of cell activation. On admission, CD62L shedding was only apparent in the HSD group but not in the HS or NS groups. After 12 h, all patient groups showed evidence of CD62L shedding. However, 24 h after admission, CD62L expression levels had returned to normal control values in all treatment groups (Fig. 2B). In addition to CD11b and CD62L, we measured the expression of CD64 (Fcγ receptor I) on the cell surface of PMNs as a marker of cell activation (Fig. 2C). On admission, none of the patient groups showed significantly elevated levels of this activation marker, when compared to healthy controls. At the 12-h time point, CD64 expression levels in the HSD and NS groups were significantly increased compared to the control and the HS groups. After 24 h, CD64 expression levels returned to control values in all but the NS group. The percentage of CD64 positive PMNs remained elevated above control values in all treatment groups.
Fig. 2. HS, HSD, and NS differentially modulate neutrophil activation and adhesion molecule expression after TBI.
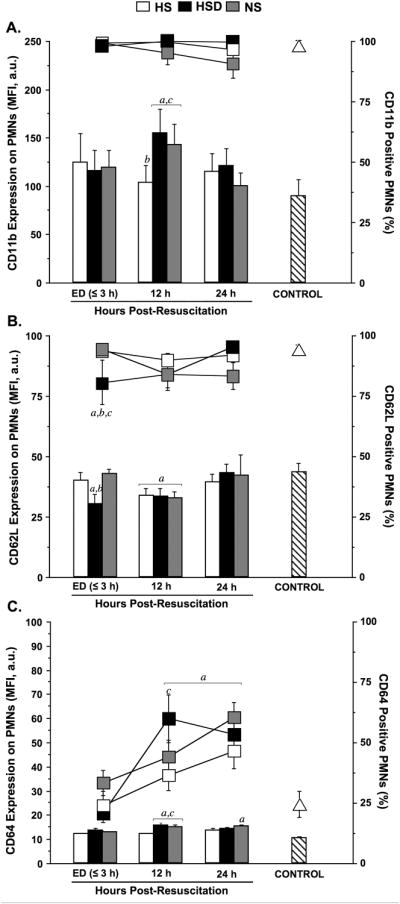
Cell-surface expression of CD11b (A), CD62L (B), and CD64 (C), were assessed by multiparameter flow cytometry using freshly drawn heparinized whole blood samples. Results were expressed as both the mean fluorescence intensity (MFI, bars) in arbitrary units (a.u.) and percentage antigen-positive (%, lines) neutrophils (PMNs). Blood was sampled serially from patients resuscitated with normal saline (NS; n=39), hypertonic saline-dextran (HSD; n=22), or hypertonic saline (HS; n=22) upon hospital admission (ED ≤3 h) and 12 and 24 h after resuscitation. Blood samples from age and gender matched healthy volunteers served as control (triangles; n=20). Statistical analyses: ap < 0.05 vs. age-matched healthy controls; bp < 0.05 vs. time-matched NS-treated patients; cp < 0.05 vs. time-matched HS-treated patients, by ANOVA.
HS reduced neutrophil degranulation markers
CD35, CD63, and CD66b are found in the membranes of various PMN granules and we measured the presence of these molecules on the cell surface as indicators of exocytosis of the contents of these vesicles as previously described [23]. On admission, CD35 expression, a marker of secretory vesicles degranulation, was similar in all patients groups and did not differ from healthy controls (Fig. 3A). At later time points, CD35 expression was only slightly different from controls with the exception of the HSD group, where CD35 expression levels were significantly higher than in healthy controls and in the NS group. Throughout the observation period, the expression of CD63, a marker of azurophilic granules, was significantly lower in all patient groups when compared to controls (Fig. 3B). These findings suggest that, PMNs expressing CD63 may be eliminated from the circulation of TBI patients or that CD63 expression may be lower in fresh PMNs recruited from the bone marrow of TBI patients compared to the PMNs in healthy controls. While they differed from healthy controls, CD63 expression levels were similar in all TBI patient groups. CD66b is a marker of specific granules. In the initial phase after injury, CD66b expression was elevated in patients treated with HSD or NS, but not in patients who received HS (Fig. 3C). After 24 h, CD66b expression had returned to control levels in all patient groups. Taken together, these results suggest that HS may reduce degranulation of PMNs, while HSD has either no effect or promotes degranulation.
Fig. 3. Neutrophil degranulation marker expression is differentially affected by HS and HSD.
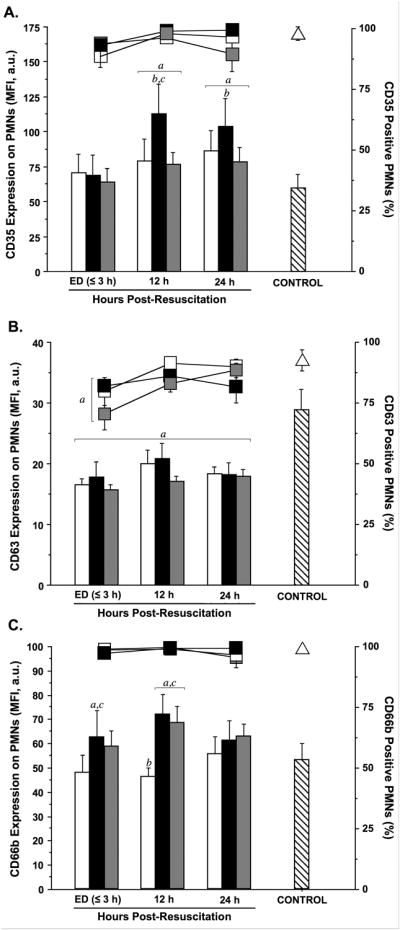
Cell-surface expression of CD35 (A), CD63 (B), and CD66b (C) were assessed by whole-blood multiparameter flow cytometry. Results are expressed as both the mean fluorescence intensity (MFI, bars) in arbitrary units (a.u.) and percentage antigen-positive (%, lines) neutrophils (PMNs). Blood was sampled serially from patients resuscitated with normal saline (NS; n=39), hypertonic saline-dextran (HSD; n=22), or hypertonic saline (HS; n=22) upon hospital admission (ED ≤3 h) and 12 and 24 h after resuscitation. Blood samples from matched healthy volunteers served as controls (triangles; n=20). Statistical analyses: ap < 0.05 vs. age-matched healthy controls; bp < 0.05 vs. time-matched NS-treated patients; cp < 0.05 vs. time-matched HS-treated patients, by ANOVA.
Hypertonic resuscitation decreased neutrophil oxidative burst activity
The oxidative burst of PMNs in response to fMLP stimulation was significantly higher in patients treated with HSD or NS than in patients who received HS (Fig. 4). However, while it was still lower than in the HSD and NS groups, oxidative burst in the HS group was also elevated above control values 24 h after resuscitation with these fluids. We obtained similar results when oxidative burst was stimulated with PMA instead of fMLP. However, under these circumstances, oxidative burst in the HS group, but not in the HSD or NS groups, was similar or lower than that in healthy controls. At the 24-h time point, values in all patient groups were significantly lower than in controls. Taken together, these findings suggest that hypertonic fluids, and particularly HS, can reduce oxidative burst of PMNs in response to the bacterial product fMLP and in response to other stimuli.
Fig. 4. Hypertonic fluid treatment reduces oxidative burst activity of neutrophils after TBI.

Neutrophil (PMN) oxidative burst activity was measured by assessing intracellular rhodamine (Rho)123 mean fluorescence intensity (MFI ± SEM) in arbitrary units (a.u.) of neutrophils in whole-blood samples from healthy controls (n=20) and trauma patients resuscitated with NS (n=39), HSD (n=22), or HS (n=22) at the time of emergency department (ED) admission (≤3 h post-resuscitation) and 12 and 24 h after resuscitation. Samples were incubated at 37°C with N-formyl-methionine-leucine-phenylalanine (fMLP, 5 μM) as a moderate stimulus or with phorbol 12-myristate 13-acetate (PMA, 8.1 μM) as a strong stimulus. Rho-123 MFI values were expressed as a percentage of unstimulated control samples. Statistical analyses: ap < 0.05 vs. age-matched healthy controls; bp < 0.05 vs. time-matched NS-treated patients; cp < 0.05 vs. time-matched HS-treated patients, by ANOVA.
HS partially normalized spontaneous neutrophil apoptosis after TBI
Reduced PMN apoptosis is thought to contribute to secondary organ damage after injury [24-26]. We studied PMN apoptosis using Annexin V (AnV) staining as described previously [22]. In healthy controls, ∼30% of PMNs underwent apoptosis after an overnight incubation period (Fig. 5). In all patient groups, the percentage of AnV-positive PMNs was significantly lower than in healthy controls suggesting that PMN apoptosis is attenuated by TBI. While average apoptosis levels in the HSD and NS groups remained at or below 10% throughout the observation period, we found significantly higher levels in the HS group. These findings indicate that HS, but neither HSD nor NS, can prevent the decrease in PMN apoptosis after TBI.
Fig. 5. Hypertonic saline partially restores neutrophil apoptosis after TBI.

Spontaneous apoptosis was assessed as the total percentage (mean ± SEM) of Annexin-V-positive (AnV+) neutrophils (PMNs) in unstimulated whole-blood samples from healthy controls (n=20) and trauma patients resuscitated with NS (n=39), HSD (n=22), or HS (n=22) at the time of emergency department (ED) admission (≤3 h post-resuscitation) and 12 and 24 h after resuscitation. Samples were incubated at 37°C for 20 h prior to staining and flow cytometric analysis. Statistical analyses: ap < 0.05 vs. age-matched healthy controls; bp < 0.05 vs. time-matched NS-treated patients; cp < 0.05 vs. time-matched HS-treated patients, by ANOVA.
Discussion
Constitutive apoptosis regulates PMNs and their function in immune defense [24]. Inhibition of PMN apoptosis after severe injury can promote inflammation and secondary organ damage in trauma patients [24-26]. Our findings indicate that TBI significantly inhibits PMN apoptosis, which may contribute to secondary organ damage in TBI patients. Resuscitation of patients with HS - but not with HSD - partially normalized PMN apoptosis, suggesting that HS may reduce secondary organ damage. HS resuscitation blocks the production of inflammatory cytokines that have been shown to inhibit PMN apoptosis [27-30]. Therefore, inhibition of inflammatory cytokine production by HS may be one explanation for our current finding that HS partially restores PMN apoptosis in TBI patients. However, HS may also have direct effects on PMN apoptosis, possibly by modulating cAMP/PKA signaling [31-34]. HS triggers cAMP/PKA signaling via autocrine purinergic signaling processes that involve the release of cellular ATP, hydrolysis of ATP to adenosine, and the activation of adenosine receptors that modulate PMN responses [35-38]. These autocrine feedback mechanisms involve several different subtypes of the nucleotide and adenosine receptor families, collectively termed purinergic receptors, that may influence apoptosis and other PMN responses [39-44].
Our current study has shown that TBI activates PMNs in the circulation of trauma patients. Numerous previous in vitro and in vivo studies have shown that hypertonic conditions block PMN activation [35-38]. Our previous work has shown that HS resuscitation of trauma patients with hemorrhagic shock reduced PMN activation [23]. In agreement with these studies, our current data indicate that HS suppress PMN activation in TBI patients. Like in our previous study, we found that HSD significantly less effective than HS in inhibiting PMN activation. This may be due to immunostimulatory effects that have been ascribed to dextran and that could be amplified by HS [23].
While the effects of HS on PMN activation and apoptosis would be expected to decrease post-traumatic morbidity and mortality, our work has not revealed compelling evidence that clinical outcome in patients treated with HS is different from that of patients treated with HS or HSD. MODS scores, GOS-E, DRS, and overall mortality rates were similar in all three treatment groups. The parent ROC study, which included a much larger patient cohort than the present sub-study also failed to establish significant differences in clinical outcome in patients treated with HS [20]. These findings seem surprising, given the importance of PMNs in the pathological consequences of TBI. Ample evidence indicates that PMN recruitment is a characteristic event of the early inflammatory response to TBI [45]. Animal models of TBI have shown that depletion of PMNs reduces edema formation and the loss of brain tissue after TBI, suggesting that PMN recruitment to the brain is an important aspect of pathophysiological processes involved in TBI [46]. In light of these findings, we propose that the inhibitory effect on PMNs that can be achieved by pre-hospital treatment with HS resuscitation is not sufficient in extent or duration to halt the pathological processes leading to brain injury. Alternative interpretations of our results are that PMN-independent mechanisms play important roles in damaging brain tissue or that altered responses of circulating PMNs may not directly reflect the responses of PMNs in the cerebral space after TBI.
Hypertonic fluid therapy is widely used to treat TBI patients because of clinical evidence that it reduces intracranial pressure, edema formation, and morbidity after head injuries [47,48]. In previous studies, HS therapy was employed for prolonged periods after the admission of patients to the ICU. In these studies, HS was administered based on intracranial pressure measurements that were continuously monitored and maintained ≤15 mmHg [47]. In the current study, hypertonic fluids were administered as a bolus infusion in the pre-hospital setting. Under the latter circumstances, hypertonic therapy may not be able to reduce intracranial pressure and edema formation beyond the initial phase after admission of patients to the ICU, resulting in subsequent swelling and brain tissue damage. This would explain the failure of hypertonic resuscitation to improve long-term outcome in our current study, the corresponding parent clinical trial, and in previous clinical trials where pre-hospital hypertonic treatment was administered to patients with head injury [20,49]. A limitation of our current study was the lack of consistent ICP monitoring data particularly in the early phase. Many patients with ICP monitoring received their ICP monitors on average 8 h after admission to the ICU. Therefore, it was impossible to determine how hypertonic fluid resuscitation affected ICP and whether negative clinical outcome in our patients was related to ICP.
There are several other limitations to our current study. As a sub-study of the larger ROC trial, the patients for our sub-study were enrolled in only two study sites that were staffed to collect and process serial blood samples for immune studies. Thus, we could not study the full range of patients that was accessible in the larger ROC trial. For example, we could not enroll patients with severe TBI or shock that resulted in early mortality. Similarly, we were not able to enroll patients with relatively minor injuries because no serial blood samples were available due to their early discharge. As a result of these limitations, the patients represented in the current sub-study may not be fully representative of all patients who were given the three different pre-hospital fluids in the parent trial.
In a rodent model of TBI, an initial single dose of hypertonic saline treatment has been shown to improve long-term outcome and cognitive function [50]. These results seem to contradict the clinical findings with pre-hospital hypertonic fluid resuscitation mentioned above. However, in contrast to clinical studies with TBI patients, injury mechanisms and pathological processes in animal models are typically kept uniform by tightly controlling experimental conditions. Trauma patients can sustain a wide range of different injuries and treating TBI in these patients is significantly more complex compared to animal models. Therefore, treatment strategies must be adjusted to accommodate the specific needs of individual TBI patients. Our current results suggest that pre-hospital hypertonic immunomodulation with HS can reduce excessive PMN activation, but that this does not translate into better clinical outcome in TBI patients. However, it is possible that HS resuscitation combined with subsequent hyperosmotic fluid therapy to attenuate intracranial hypertension in TBI patients during their stay in the ICU could be a feasible strategy to improve longer-term neurological outcome after TBI [51,52]. We conclude that it would be worthwhile to test this concept in future studies. Since our present and previous findings [23] suggest that HSD has no protective effects and that it may exacerbate post-traumatic PMN activation, we propose that future studies should focus on the use of HS without dextran.
Acknowledgments
The authors thank M. Shiu and S. Petrongolo for their excellent laboratory work. The Resuscitation Outcome Consortium is supported by a series of cooperative agreements with 10 regional clinical centers and 1 data coordinating center (5U01 HL077863, HL077881, HL077871 HL077872, HL077866, HL077908, HL077867, HL077885, HL077887, HL077873, HL077865) from the National Heart, Lung and Blood Institute in partnership with the National Institute of Neurological Disorders and Stroke, US Army Medical Research & Material Command; The Canadian Institutes of Health Research—Institute of Circulatory and Respiratory Health; Defence Research and Development Canada, and the Heart and Stroke Foundation of Canada. In addition, this work was funded in part by grants from the National Institutes of Health, GM-51477, GM-60475, AI-072287, AI-080582, and the Congressionally Directed Medical Research Program PR043034.
References
- 1.Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–41. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- 2.Stein SC, Georgoff P, Meghan S, Mizra K, Sonnad SS. 150 years of treating severe traumatic brain injury: a systematic review of progress in mortality. J Neurotrauma. 2010;27:1343–53. doi: 10.1089/neu.2009.1206. [DOI] [PubMed] [Google Scholar]
- 3.Smania N, Avesani R, Roncari L, Ianes P, Girardi P, Varalta V, Gambini MG, Fiaschi A, Gandolfi M. Factors predicting functional and cognitive recovery following severe traumatic, anoxic, and cerebrovascular brain damage. J Head Trauma Rehabil. 2013;28:131–40. doi: 10.1097/HTR.0b013e31823c0127. [DOI] [PubMed] [Google Scholar]
- 4.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- 5.Park E, Bell JD, Baker AJ. Traumatic brain injury: can the consequences be stopped? CMAJ. 2008;178:1163–1170. doi: 10.1503/cmaj.080282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mt Sinai J Med. 2009;76:97–104. doi: 10.1002/msj.20104. [DOI] [PubMed] [Google Scholar]
- 7.Mangat HS. Severe traumatic brain injury. Continuum (Minneap Minn) 2012;18:532–546. doi: 10.1212/01.CON.0000415426.76524.e1. [DOI] [PubMed] [Google Scholar]
- 8.Zink BJ, Szmydynger-Chodobska J, Chodobski A. Emerging concepts in the pathophysiology of traumatic brain injury. Psychiatr Clin North Am. 2010;33:741–756. doi: 10.1016/j.psc.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Weed LH, McKibben PS. Experimental alteration of brain bulk. Am J Physiol. 1919;48:531–558. [Google Scholar]
- 10.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–1171. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulger EM, Hoyt DB. Hypertonic resuscitation after severe injury: is it of benefit? Adv Surg. 2012;46:73–85. doi: 10.1016/j.yasu.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Ropper AH. Hyperosmolar therapy for raised intracranial pressure. N Engl J Med. 2012;367:746–752. doi: 10.1056/NEJMct1206321. [DOI] [PubMed] [Google Scholar]
- 15.Angle N, Hoyt DB, Cabello-Passini R, Herdon-Remelius C, Loomis W, et al. Hypertonic saline resuscitation reduces neutrophil margination by suppressing neutrophil L selectin expression. J Trauma. 1998;45:7–12. doi: 10.1097/00005373-199807000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Rizoli SB, Kapus A, Fan J, Li YH, Marshall JC, et al. Immunomodulatory effects of hypertonic resuscitation on the development of lung inflammation following hemorrhagic shock. J Immunol. 1998;161:6288–6296. [PubMed] [Google Scholar]
- 17.Rizoli SB, Rhind SG, Shek PN, Inaba K, Filips D, et al. The immunomodulatory effects of hypertonic saline resuscitation in patients sustaining traumatic hemorrhagic shock: a randomized, controlled, double-blinded trial. Ann Surg. 2006;243:47–57. doi: 10.1097/01.sla.0000193608.93127.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bulger EM, Cuschieri J, Warner K, Maier RV. Hypertonic resuscitation modulates the inflammatory response in patients with traumatic hemorrhagic shock. Ann Surg. 2007;245:635–641. doi: 10.1097/01.sla.0000251367.44890.ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bulger EM, May S, Kerby JD, Emerson S, Stiell IG, et al. Out-of-hospital hypertonic resuscitation after traumatic hypovolemic shock: a randomized, placebo controlled trial. Ann Surg. 2011;253:431–441. doi: 10.1097/SLA.0b013e3181fcdb22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bulger EM, May S, Brasel KJ, Schreiber M, Kerby JD, et al. Out-of-hospital hypertonic resuscitation following severe traumatic brain injury: a randomized controlled trial. JAMA. 2010;304:1455–1464. doi: 10.1001/jama.2010.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulger EM, May S, Kerby JD, Emerson S, Stiell IG, et al. Out-of-hospital hypertonic resuscitation after traumatic hypovolemic shock: a randomized, placebo controlled trial. Ann Surg. 2011;253:431–441. doi: 10.1097/SLA.0b013e3181fcdb22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 23.Junger WG, Rhind SG, Rizoli SB, Cuschieri J, Shiu MY, Baker AJ, Li L, Shek PN, Hoyt DB, Bulger EM. Resuscitation of traumatic hemorrhagic shock patients with hypertonic saline-without dextran-inhibits neutrophil and endothelial cell activation. Shock. 2012;38:341–350. doi: 10.1097/SHK.0b013e3182635aca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Z, Sayeed MM. Activation of PI3-kinase/PKB contributes to delay in neutrophil apoptosis after thermal injury. Am J Physiol Cell Physiol. 2003;288:C1171–C1178. doi: 10.1152/ajpcell.00312.2004. [DOI] [PubMed] [Google Scholar]
- 25.Hu Z, Sayeed MM. Suppression of mitochondria-dependent neutrophil apoptosis with thermal injury. Am J Physiol Cell Physiol. 2004;286:C170–C178. doi: 10.1152/ajpcell.00187.2003. [DOI] [PubMed] [Google Scholar]
- 26.Nolan B, Collette H, Baker S, Duffy A, De M, Miller C, Bankey P. Inhibition of neutrophil apoptosis after severe trauma is NFkappabeta dependent. J Trauma. 2000;48:599–604. doi: 10.1097/00005373-200004000-00004. [DOI] [PubMed] [Google Scholar]
- 27.Cuschieri J, Gourlay D, Garcia I, Jelacic S, Maier RV. Hypertonic preconditioning inhibits macrophage responsiveness to endotoxin. J Immunol. 2002;168:1389–1396. doi: 10.4049/jimmunol.168.3.1389. [DOI] [PubMed] [Google Scholar]
- 28.Bahrami S, Zimmermann K, Szelényi Z, Hamar J, Scheiflinger F, Redl H, Junger WG. Small-volume fluid resuscitation with hypertonic saline prevents inflammation but not mortality in a rat model of hemorrhagic shock. Shock. 2006;25:283–289. doi: 10.1097/01.shk.0000208808.03148.ea. [DOI] [PubMed] [Google Scholar]
- 29.Colotta F, Re F, Polantarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80:2012–2020. [PubMed] [Google Scholar]
- 30.Simon HU. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol Rev. 2003;193:101–110. doi: 10.1034/j.1600-065x.2003.00038.x. [DOI] [PubMed] [Google Scholar]
- 31.Biffl WL, Carnaggio R, Moore EE, Ciesla DJ, Johnson JL, Silliman CC. Clinically relevant hypertonicity prevents stored blood- and lipid-mediated delayed neutrophil apoptosis independent of p38 MAPK or caspase-3 activation. Surgery. 2003;134:86–91. doi: 10.1067/msy.2003.178. [DOI] [PubMed] [Google Scholar]
- 32.Kim JY, Hong YS, Choi SH, Yoon YH, Moon SW, Lee SW. Effect of hypertonic saline on apoptosis of polymorphonuclear cells. J Surg Res. 2012;178:401–408. doi: 10.1016/j.jss.2012.01.055. [DOI] [PubMed] [Google Scholar]
- 33.Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, Caldicott A, Martinez-Losa M, Walker TR, Duffin R, Gray M, Crescenzi E, Martin MC, Brady HJ, Savill JS, Dransfield I, Haslett C. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–1064. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- 34.Xu Y, Loison F, Luo HR. Neutrophil spontaneous death is mediated by down-regulation of autocrine signaling through GPCR, PI3K, ROS, and actin. Proc Natl Acad Sci USA. 2010;107:2950–2955. doi: 10.1073/pnas.0912717107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Junger WG, Hoyt DB, Davis RE, Herdon-Remelius C, Namiki S, et al. Hypertonicity regulates the function of human neutrophils by modulating chemoattractant receptor signaling and activating mitogen-activated protein kinase p38. J Clin Invest. 1998;101:2768–2779. doi: 10.1172/JCI1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Shukla A, Namiki S, Insel PA, Junger WG. A putative osmoreceptor system that controls neutrophil function through the release of ATP, its conversion to adenosine, and activation of A2 adenosine and P2 receptors. J Leukoc Biol. 2004;76:245–253. doi: 10.1189/jlb.0204066. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Hashiguchi N, Yip L, Junger WG. Hypertonic saline enhances neutrophil elastase release through activation of P2 and A3 receptors. Am J Physiol Cell Physiol. 2006;290:C1051–C1059. doi: 10.1152/ajpcell.00216.2005. [DOI] [PubMed] [Google Scholar]
- 38.Shukla A, Hashiguchi N, Chen Y, Coimbra R, Hoyt DB, Junger WG. Osmotic regulation of cell function and possible clinical applications. Shock. 2004;21:391–400. doi: 10.1097/00024382-200405000-00001. [DOI] [PubMed] [Google Scholar]
- 39.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11:201–212. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaughan KR, Stokes L, Prince LR, Marriott HM, Meis S, Kassack MU, Bingle CD, Sabroe I, Surprenant A, Whyte MK. Inhibition of neutrophil apoptosis by ATP is mediated by the P2Y11 receptor. J Immunol. 2007;179:8544–8553. doi: 10.4049/jimmunol.179.12.8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orlic T, Loomis WH, Shreve A, Namiki S, Junger WG. Hypertonicity increases cAMP in PMN and blocks oxidative burst by PKA-dependent and -independent mechanisms. Am J Physiol Cell Physiol. 2002;282:C1261–C1269. doi: 10.1152/ajpcell.00479.2001. [DOI] [PubMed] [Google Scholar]
- 42.Inoue Y, Chen Y, Pauzenberger R, Hirsh MI, Junger WG. Hypertonic saline up-regulates A3 adenosine receptor expression of activated neutrophils and increases acute lung injury after sepsis. Crit Care Med. 2008;36:2569–2575. doi: 10.1097/CCM.0b013e3181841a91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inoue Y, Tanaka H, Sumi Y, Woehrle T, Chen Y, et al. A3 adenosine receptor inhibition improves the efficacy of hypertonic saline resuscitation. Shock. 2011;35:178–183. doi: 10.1097/SHK.0b013e3181f221fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bulger EM, Tower CM, Warner KJ, Garland T, Cuschieri J, et al. Increased neutrophil adenosine a3 receptor expression is associated with hemorrhagic shock and injury severity in trauma patients. Shock. 2011;36:435–439. doi: 10.1097/SHK.0b013e318231ee2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holmin S, Soderlund J, Biberfeld P, Mathiesen T. Intracerebral inflammation after human brain contusion. Neurosurgery. 1998;42:291–298. doi: 10.1097/00006123-199802000-00047. [DOI] [PubMed] [Google Scholar]
- 46.Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflammation. 2012;9:17. doi: 10.1186/1742-2094-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simma B, Burger R, Falk M, Sacher P, Fanconi S. A prospective, randomized, and controlled study of fluid management in children with severe head injury: lactated Ringer's solution versus hypertonic saline. Crit Care Med. 1998;26:1265–1270. doi: 10.1097/00003246-199807000-00032. [DOI] [PubMed] [Google Scholar]
- 48.Khanna S, Davis D, Peterson B, Fisher B, Tung H, O'Quigley J, Deutsch R. Use of hypertonic saline in the treatment of severe refractory posttraumatic intracranial hypertension in pediatric traumatic brain injury. Crit Care Med. 2000;28:1144–1151. doi: 10.1097/00003246-200004000-00038. [DOI] [PubMed] [Google Scholar]
- 49.Cooper DJ, Myles PS, McDermott FT, Murray LJ, Laidlaw J, et al. Prehospital hypertonic saline resuscitation of patients with hypotension and severe traumatic brain injury: a randomized controlled trial. JAMA. 2004;291:1350–1357. doi: 10.1001/jama.291.11.1350. [DOI] [PubMed] [Google Scholar]
- 50.Sell SL, Avila MA, Yu G, Vergara L, Prough DS, Grady JJ, DeWitt DS. Hypertonic resuscitation improves neuronal and behavioral outcomes after traumatic brain injury plus hemorrhage. Anesthesiology. 2008;108:873–881. doi: 10.1097/ALN.0b013e31816c8a15. [DOI] [PubMed] [Google Scholar]
- 51.Nakagawa K, Chang CW, Koenig MA, Yu M, Tokumaru S. Treatment of refractory intracranial hypertension with 23.4% saline in children with severe traumatic brain injury. J Clin Anesth. 2012;24:318–323. doi: 10.1016/j.jclinane.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 52.Eskandari R, Filtz MR, Davis GE, Hoesch RE. Effective treatment of refractory intracranial hypertension after traumatic brain injury with repeated boluses of 14.6% hypertonic saline. J Neurosurg. 2013 May 24; doi: 10.3171/2013.4.JNS121541. Epub ahead of print. [DOI] [PubMed] [Google Scholar]