

Abstract
A series of hydroxamate based HDAC inhibitors containing a phenylisoxazole as the CAP group has been synthesized using nitrile oxide cycloaddition chemistry. An HDAC6 selective inhibitor having a potency of ∼2 picomolar was identified. Some of the compounds were examined for their ability to block pancreatic cancer cell growth and found to be about 10-fold more potent than SAHA. This research provides valuable, new molecular probes for use in exploring HDAC biology.
To date, considerable research activity has focused on understanding the “histone code” and, in particular, on the design of HDACa inhibitors as novel therapeutics for the treatment of a wide range of disorders, including cancer, as well as neurodegenerative diseases, and even malaria.1 These compounds owe their action to their ability to reactivate silenced genes by modulating the condensation status of DNA. The post-translational acetylation status of chromatin is determined by the competing activities of two classes of enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs), which control the acetylation of lysine residues making up the histones. In general, HATs function to acetylate lysine groups in nuclear histones, resulting in neutralization of the charges on the histones and a more open, transcriptionally active chromatin structure, while the HDACs function to deacetylate and suppress transcription (the positively charged lysine amine group interacts with the negatively charge DNA organophosphate groups to cause compaction of the chromatin structure). A shift in the balance of acetylation on chromatin may result in changes in the regulation of patterns of gene expression.2
Because many cancers are associated with aberrant transcriptional activity and HDACs and HATs associate with both transcription activators and repressors, these enzymes have been identified as attractive targets for cancer therapy. Indeed, inhibitors of the HDACs have been shown to block tumor cell growth and induce differentiation and cell death.3 Several such inhibitory agents, including suberoylanilide hydroxamic acid (SAHA) and (E)-(1S,4S,10S,21R)-7-[(Z)-ethylidene]-4,21-di-isopropyl-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone (FR901228)1b have reached clinical trials, and SAHA has been approved by the FDA for use in cutaneous T-cell lymphoma (CTCL).4 HDACIs work by allowing the transcription and expression of genes including tumor suppressor genes. HDAC inhibitors have also been shown to enhance the therapeutic efficacy of typical cytotoxic agents when tested in combination trials.5
While a fairly extensive array of HDACIs has now been described, the majority of these fail to show any selectivity for the individual enzymes, at least as determined by using isolated enzyme assays. In most cases, such data are generally missing, as the assays were performed using a mixture of nuclear extracts. In our present efforts, and based upon other studies reported by us,6 we elected to investigate the HDAC selectivity of compounds readily assembled by use of nitrile oxide cycloaddition (NOC) chemistry (Figure 1), together with further studies of their action against pancreatic cancer cell lines. The present studies were thus designed to investigate issues of selectivity to arrive at molecular probes rather than to create candidate drugs per se. In this connection, it is now well recognized that the hydroxamate group represents a metabolic liability and may have some toxicity and that improved zinc binding groups will be needed to arrive at more druggable molecules. We have already reported some progress in this direction by employing a mercaptoacetamide group to bind to the catalytic zinc atom and shown these compounds to be superior in their neuroprotective ability in neuronal models of oxidative stress.6b
Figure 1.
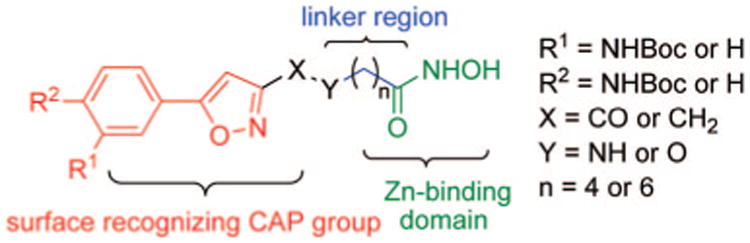
General structural features of compounds 1–11.
The synthesis of compounds 1–11 was accomplished by making use of three related building blocks, namely the 5-arylisoxazole-3-carboxylic acid ethyl esters (15–17),7 which were synthesized according to the reported procedure (Scheme 1). Saponification of the ethyl ester followed by a standard peptide coupling reaction between the resulting acid and the variably sized long chain amino acid methyl ester using HOBT-EDCI provided the corresponding methyl ester. The ether-bearing analogue 19 was synthesized from alcohol 18 by reaction with methyl 5-bromovalerate in the presence of NaH. The alcohol 18 was prepared by NaBH4 reduction of 5-phenylisoxazole-3-carboxylic ethyl ester 15. Last, these methyl esters were converted to the corresponding hydroxamic acids by heating with hydroxylamine hydrochloride in the presence of base in accord with a literature procedure.8 For those compounds bearing an amino group, Boc deprotection was carried out using TFA prior to the formation of the hydroxamic acids.
Scheme 1. Synthesis of Compounds 15–17a.
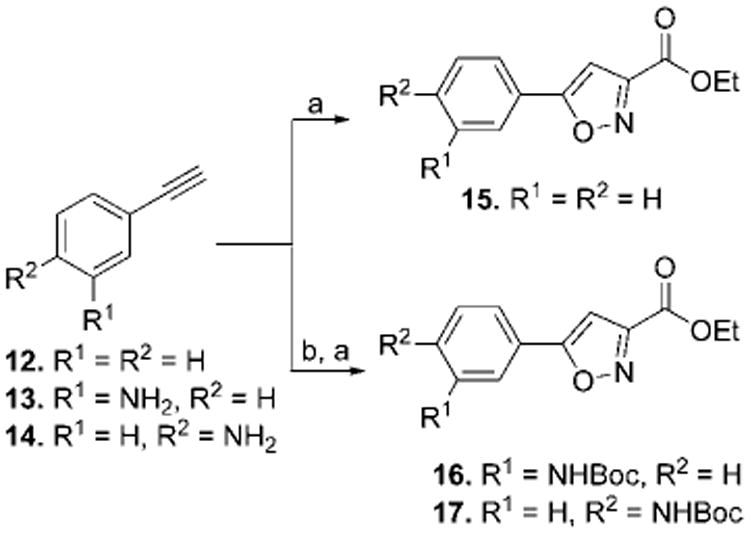
a Reactions and conditions: (a) ethyl chlorooximidoacetate, triethylamine, THF, rt, 16 h; (b) (Boc)2O, THF, reflux, 16 h.
All compounds were tested using the isolated enzymes under conditions described previously against both the class I (1, 2, 3, and 8) and class II (6 and 10) HDACs (Table 1 and Chart 1). Noteworthy is the observation that this set of structures show some selectivity for HDAC3 and 6, as they are less potent in the inhibition of HDAC1, -2, and -10, while showing only micromolar activity against HDAC8. Both the length of the linker chain and the amine-protecting group play a role in potency and selectivity. Thus compounds with a linker comprised of 6-methylene units and in their Boc-protected form (3 and 7) were highly potent and selective for HDAC3 and 6.
Table 1. In Vitro HDAC Enzyme Inhibitory Activities of Compounds 1–11 and SAHA.
| IC50 (nM) | |||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| class I | class II | ||||||
|
|
|
||||||
| compd | ClogPd | HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC6 | HDAC10 |
| 1 | 1.12 | 124 ± 3 | 133 ± 4 | 12.9 ± 0.3 | 3320 ± 282 | 49.5 ± 2.9 | 155 ± 3 |
| 2 | 1.19 | 644 ± 19 | 1690 ± 49 | 158 ± 9 | 3580 ± 181 | 61.9 ± 4.7 | 852 ± 30 |
| 3 | 3.29 | 100 ± 16 | 238 ± 3 | <0.002 | 8104 ± 681 | <0.002 | 28.2 ± 5.4 |
| 4 | 2.31 | 826 ± 33 | 2980 ± 97 | 83.6 ± 6.4 | 5810 ± 337 | 3.98 ± 0.48 | 891 ± 34 |
| 5 | 1.25 | 20.7 ± 3.2 | 56.7 ± 3.4 | 4.04 ± 0.18 | 938 ± 43.6 | 2.54 ± 0.22 | 17.5 ± 1.2 |
| 6 | 0.27 | 716 ± 32 | 1940 ± 71 | 144 ± 10 | 3430 ± 151 | 72.2 ± 5.6 | 840 ± 37 |
| 7 | 3.29 | 271 ± 23 | 252 ± 10 | 0.42 ± 0.08 | 6851 ± 707 | 0.002 | 90.7 ± 12 |
| 8 | 2.31 | 628 ± 23 | 1740 ± 77 | 115 ± 6 | 3650 ± 196 | 31.6 ± 1.6 | 690 ± 27 |
| 9 | 1.25 | 56.3 ± 4.9 | 147 ± 4 | 10.2 ± 0.4 | 3458 ± 259 | 5.86 ± 0.16 | 44.9 ± 9 |
| 10 | 0.27 | 360 ± 12 | 1050 ± 32 | 65.8 ± 2.1 | 2830 ± 128 | 35.1 ± 2.0 | 401 ± 10 |
| 11 | 1.28 | 64.5 ± 3.6 | 131 ± 6 | 18 ± 1 | 4900 ± 253 | 6.81 ± 0.28 | 73 ± 3 |
| SAHAa | 1.44 | 68 ± 14 | 164 ± 45 | 48 ± 17 | 1524 ± 463 | 90 ± 26 | b |
| SAHAc | 1.44 | 96 | 282 | 17 | 1140 | 14 | 72 |
| TSA | 2.23 | 4 | 14 | 2 | 1.38 | 1 | 5 |
SAHA data from ref 9.
Not available.
SAHA and TSA data provided by Amphora Inc.
The partition coefficients (ClogP) were calculated according to the fragment-based program KOWWIN 1.63 http://www.syrres.com/esc/est_kowdemo.htm.
Chart 1. Comparison of IC50 (nM) Values of Compounds 1–11 and SAHA against Class I and Class II HDACs.
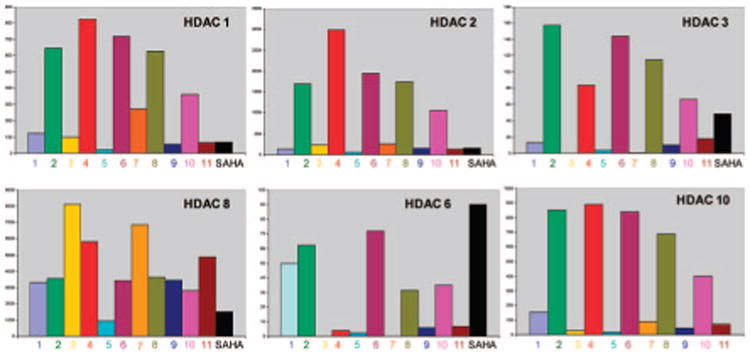
In fact, compound 7 shows an impressive potency of 2 picomoles against HDAC6 coupled with a superb level of enzyme selectivity. Given the importance of HDAC6 in the deacetylation of tubulin, the design of inhibitors against this target is of considerable interest.10 Replacement of the Boc group of 7 by acetyl as in 11 led to a considerable drop in potency, suggesting the impact of this large lipophilic group on surface recognition. Compounds bearing an unsubstituted phenyl ring as in 1 and 2 show a similar trend in selectivity against HDAC3 and -6, although they are less potent, thus demonstrating that the phenylisoxazoles are readily amenable to structural modifications to refine their potency and selectivity.
Figure 2 shows inhibitor 7 docked in the binding site of our current homology model of HDAC6. The HDAC6 homology model was built using the X-ray crystal structure of HDAC7 (2PQP) as a template.11 The ZBG (zinc-binding group) hydroxamic acid group and the six methylene groups bind deep inside the narrow binding pocket. The CAP region of the bound ligand appears to interact with only one side of the protein. The carbonyl group of the Boc group might interact with His499, which may be important in positioning the CAP residue on the surface of the HDAC protein. Such interactions are absent with the other inhibitors included in this study and may thus explain the higher potency observed for this compound. Preliminary investigations of other class I and class II HDAC homology models indicate that the loop areas surrounding the CAP residues show significant differences among the various HDACs and may eventually account for the enzyme selectivity that has been observed.
Figure 2.
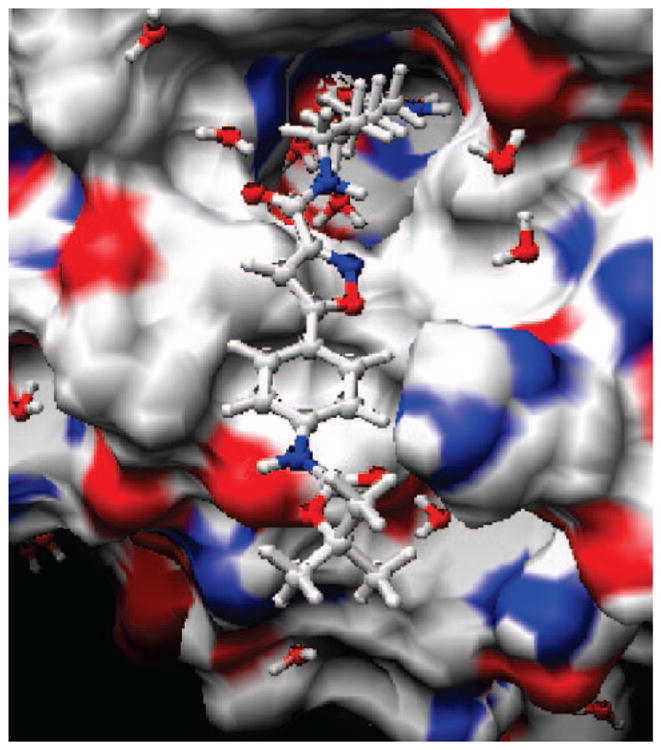
Compound 7 docked into the binding site of the current HDAC6 homology model.
To further validate the utility of these HDACIs at the cellular level, we investigated their ability to block cancer cell growth. Because the ClogP values of these compounds are actually better than that of SAHA (Table 1), we believed that these ligands would be able to permeate the cell membrane. As pancreatic cancer is the fifth leading cause of cancer death in the U.S. and essentially remains an incurable disease with the incidence nearly equaling mortality, we chose these cell lines for study.12 These cancers fail to respond to widely used chemotherapeutic drugs like 5-fluorouracil and gemcitabine,13 and thus there is a clear need for more effective drugs. The present HDACIs were tested against five transformed cell lines as well as against two normal cell lines. These data are provided in Table 2 along with comparison data using the now marketed drug SAHA. As is apparent, some of these isoxazoles show very good IC50 values in the nanomolar range, and are more active than SAHA. Compounds 7 and 9, for example, are active against both the Mia Paca-2 and Panc04.03 cell lines at the 100 nM level and at the 200–300 nM level against HupT3, whereas compound 5 is active against HupT3, Mia Paca-2 and SU86.86 at the 100–200 nM level. Of particular interest to the present study is the high activity shown by the relatively HDAC6 selective compound 7, especially in view of its weaker HDAC1 activity, which is believed essential to the induction of apoptosis.14 Compounds 8, 10, and 11 which show poorer HDAC inhibition, show much weaker activity against these cell lines. Also, it should be noted that the activity of these isoxazoles against the transformed and nontransformed cell lines (HMEC and HPDE6c7) is no better than that shown by SAHA. Given the different levels of expression of the HDACs in the pancreatic cancer cells, the formation of multiprotein repressor complexes, as well as issues of distribution and metabolism, it is unlikely that one should expect a high correlation between the in vitro enzyme inhibitory activity and cellular cytotoxicity.
Table 2. In Vitro Antiproliferative Activity of Compounds 1 – 11 against Pancreatic Cancer Cell Lines.
| IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| compd | BxPC-3 | HupT3 | Mia Paca-2 | Panc 04.03 | SU.86.86 | HMECa | HPDE6c7a |
| 1 | 7 | 0.4 | 1 | 1.5 | 1 | b | b |
| 2 | 28 | 5 | 4 | 5 | 5 | b | b |
| 3 | 1 | 0.8 | 0.7 | 1 | 1 | 7 | 1.5 |
| 4 | 36 | 14 | 8 | 14 | 22 | b | b |
| 5 | 0.7 | 0.1 | 0.1 | 0.9 | 0.2 | <1 | 0.2 |
| 6 | 38 | 37 | 25 | >50 | 25 | b | b |
| 7 | 1 | 0.3 | 0.1 | 0.1 | 0.6 | <1 | 0.5 |
| 8 | 25 | 10 | 12 | 16 | 24 | b | b |
| 9 | 0.8 | 0.2 | 0.1 | 0.1 | 0.8 | <1 | 0.4 |
| 10 | 33 | 7 | 10 | 17 | 11 | b | b |
| 11 | 6 | 1 | 0.9 | 4 | 1 | b | b |
| SAHA | 5 | 0.8 | 1 | 1 | 1 | 1.3 | 1.3 |
Normal cell lines.
Not detected.
In summary, the present work demonstrates the ability of NOC chemistry to quickly generate a novel series of HDAC inhibitors, showing features of high potency and selectivity, as well as ability to inhibit pancreatic cancer cell growth. While human xenograft studies using these compounds are now underway, we anticipate that some of these compounds will become valuable molecular probes for further elucidating HDAC6 biology.
Supplementary Material
Scheme 2. Synthesis of Compounds 1 and 2a.
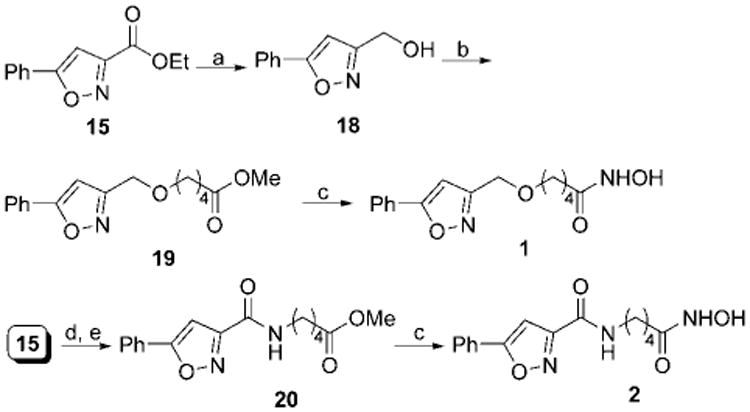
a Reagents and conditions: (a) NaBH4, MeOH, rt, 30 min; (b) NaH (60%), DMF, 0 °C to rt, 1 h then methyl 5-bromovalerate, 0 to 70 °C, 12 h; (c) NH2OH·HCl, KOH, MeOH, rt, 15 min; (d) LiOH, THF, MeOH, H2O, 0 °C to rt, 1 h; (e) HCl·NH2(CH2)4COOMe, EDCI, HOBT, DIEA, DMF, 0 °C to rt, 12 h.
Scheme 3. Synthesis of Compounds 3–11a.
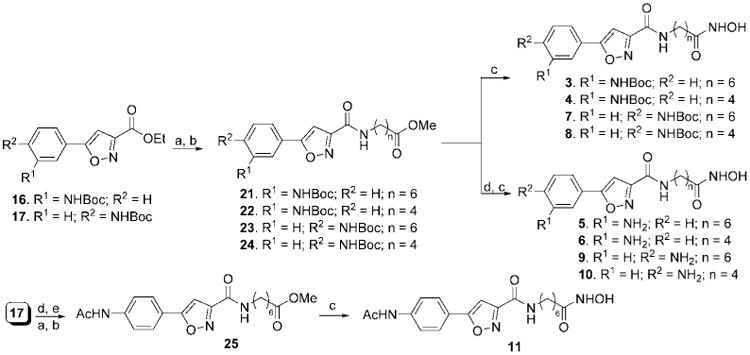
a Reagents and conditions: (a) LiOH, THF, MeOH, H2O, 0 °C to rt, 1 h; (b) HCl·NH2(CH2)nCOOMe, EDCI, HOBT, DIEA, DMF, 0 °C to rt, 12 h; (c) NH2OH·HCl, KOH, MeOH, rt, 15 min; (d) TFA, CH2Cl2, 0 °C to rt, 2 h; (e) Ac2O, pyridine, CH2Cl2, 0 °C to rt, 5 h.
Acknowledgments
This manuscript is dedicated to Professor Elias J. Corey on the occasion of his 80th birthday. We are indebted to Dr. Rong He for assisting in the preparation of the manuscript. This work was supported in part by gift funds from an anonymous donor, by a grant (no. 271210) from the ADDF/Elan, and by the Mayo Foundation and the Pancreatic Cancer SPORE P50 CA102701.
Footnotes
Abbreviations: Boc, tert-butyloxycarbonyl; CTCL, cutaneous T-cell lymphoma; DNA, deoxyribonucleic acid; EDCI, 1-ethyl-3-(3′-dimethyl-aminopropyl)carbodiimide; FDA, Food & Drug Administration; HATs, histone acetyltransferases; HDAC, histone deacetylase; HDACs, histone deacetylases; HDACIs, histone deacetylase inhibitors; HOBT, 1-hydroxy-benzotriazole; NOC, nitrile oxide cycloaddition; SAHA, suberoylanilide hydroxamic acid; TFA, trifluoroacetic acid; ZBG, zinc-binding group.
Supporting Information Available: Experimental details and HPLC data of compounds 1–11. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Butler KV, Kozikowski AP. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr Pharm Des. 2008;14:505–528. doi: 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]; (b) Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discovery. 2006;5:769–784. doi: 10.1038/nrd2133. and references cited therein. [DOI] [PubMed] [Google Scholar]; (c) Paris M, Porcelloni M, Binaschi M, Fattori D. Histone Deacetylase Inhibitors: From Bench to Clinic. J Med Chem. 2008;51:1505–1528. doi: 10.1021/jm7011408. [DOI] [PubMed] [Google Scholar]
- 2.(a) Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]; (b) Wolffe AP, Guschin D. Review: Chromatin structural features and targets that regulate transcription. J Struct Biol. 2000;129:102–122. doi: 10.1006/jsbi.2000.4217. [DOI] [PubMed] [Google Scholar]; (c) Kurdistani SK, Grunstein M, Grunstein M. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol. 2003;4:276–284. doi: 10.1038/nrm1075. [DOI] [PubMed] [Google Scholar]; (d) Struhl K, Moqtaderi Z. The TAFs in the HAT. Cell. 1998;94:1–4. doi: 10.1016/s0092-8674(00)81213-1. [DOI] [PubMed] [Google Scholar]
- 3.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kelly WK, Zhu AX, Scher H, Curley T, Fallon M, Slovin S, Schwartz L, Larson S, Tong W, Hartley-Asp B, Pellizzoni C, Rocchetti M. Dose escalation study of intravenous estramustine phosphate in combination with Paclitaxel and Carboplatin in patients with advanced prostate cancer. Clin Cancer Res. 2003;9:2098. [PubMed] [Google Scholar]; (b) Carducci MA, Gilbert J, Bowling MK, Noe D, Eisenberger MA, Sinibaldi V, Zabelina Y, Chen TL, Grochow LB, Donehower RC. Phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120 h infusion schedule. Clin Cancer Res. 2001;7:3047–3055. [PubMed] [Google Scholar]; (c) Sasakawa Y, Naoe Y, Inoue T, Sasakawa T, Matsuo M, Manda T, Mutoh S. Effects of FK228, a novel histone deacetylase inhibitor, on human lymphoma U-937 cells in vitro and in vivo. Biochem Pharmacol. 2002;64:1079–1090. doi: 10.1016/s0006-2952(02)01261-3. [DOI] [PubMed] [Google Scholar]
- 5.(a) Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv Cancer Res. 2004;91:137–168. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]; (b) Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kozikowski AP, Chen YF, Gaysin AM, Savoy DN, Billadeau DD, Kim KH. Chemistry, Biology, and QSAR Studies of Substituted Biaryl Hydroxamates and Mercaptoacetamides as HDAC Inhibitors-Nanomolar-Potency Inhibitors of Pancreatic Cancer Cell Growth. ChemMedChem. 2008;3:487–501. doi: 10.1002/cmdc.200700314. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kozikowski AP, Chen YF, Gaysin A, Chen B, D'Annibale MA, Suto CM, Langley BC. Functional Differences in Epigenetic Modulators: Superiority of Mercaptoacetamide-Based Histone Deacetylase Inhibitors Relative to Hydroxamates in Cortical Neuron Neuroprotection Studies. J Med Chem. 2007;50:3054–3061. doi: 10.1021/jm070178x. [DOI] [PubMed] [Google Scholar]; (c) Chen YF, Lopex-Sanchez M, Savoy DN, Billadieu DD, Dow GS, Kozikowski AP. A series of potent and selective, triazolylphenyl-based histone deacetylase inhibitors [HDACIs] with activity against pancreatic cancer cells and plasmodium falciparum. J Med Chem. 2008 doi: 10.1021/jm701606b. in press. [DOI] [PubMed] [Google Scholar]
- 7.(a) Xin Z, Liu G, Pei Z, Szczepankiewicz BG, Serby MD, Zhao H. U S Pat Appl Publ. Abbott Laboratories; USA: 2004. Preparation of arylisoxazolecarboxylates as protein-tyrosine phosphatase (PTP1B) inhibitors. [Google Scholar]; (b) Zhao H, Liu G, Xin Z, Serby MD, Pei Z, Szczepankiewicz BG, Hajduk PJ, Abad-Zapatero C, Hutchins CW, Lubben TH, Ballaron SJ, Haasch DL, Kaszubska W, Rondinone CM, Trevillyan JM, Jirousek MR. Isoxazole carboxylic acids as protein tyrosine phosphatase 1B (PTP1B) inhibitors. Bioorg Med Chem Lett. 2004;14:5543–5546. doi: 10.1016/j.bmcl.2004.08.063. [DOI] [PubMed] [Google Scholar]
- 8.Gediya LK, Chopra P, Purushottamachar P, Maheshwari N, Njar VC. A new simple and high-yield synthesis of suberoylanilide hydroxamic acid and its inhibitory effect alone or in combination with retinoids on proliferation of human prostate cancer cells. J Med Chem. 2005;48:5047–5051. doi: 10.1021/jm058214k. [DOI] [PubMed] [Google Scholar]
- 9.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. Determination of the class and isoform selectivity of small molecule HDAC inhibotors. Biochem J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 10.(a) Boyault C, Sadoul K, Pabion M, Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene. 2007;26:5468–5476. doi: 10.1038/sj.onc.1210614. [DOI] [PubMed] [Google Scholar]; (b) Dompierre JP, Godin JD, Charrin BC, Cordeliáeres FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuetz A, Min J, Allali-Hassani A, Schapira M, Shuen M, Loppnau P, Mazitschek R, Kwiatkowski NP, Lewis TA, Maglathin RL, McLean TH, Bochkarev A, Plotnikov AN, Vedadi M, Arrowsmith CH. Human HDAC7 harbors a class IIa HDAC-specific zinc binding motif and cryptic deacetylase activity. J Biol Chem. 2008 Feb 19; doi: 10.1074/jbc.M707362200. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1053. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 13.Haller DG. New perspectives in the management of pancreas cancer. Semin Oncol. 2003;30:3–10. doi: 10.1016/s0093-7754(03)00296-3. [DOI] [PubMed] [Google Scholar]
- 14.Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, et al. Neuroprotection by histone deacetylase-related protein. Mol Cell Biol. 2006;26:3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.