

Abstract
The signal transduction and enzyme activity were investigated in biosensors based on the glucose oxidase (GOx) and carbon nanotubes (CNT) embedded in a bio-adhesive film of chitosan (CHIT). The voltammetric studies showed that, regardless of CHIT matrix, the GOx adsorbed on CNT yielding a pair of surface-confined current peaks at -0.48 V. The anodic peak did not increase in the presence of glucose in an O2-free solution indicating the lack of direct electron transfer (DET) between the enzymatically active GOx and CNT. The voltammetric peaks were due to the redox of enzyme cofactor flavin adenine dinucleotide (FAD), which was not the part of active enzyme. The presented data suggest that DET may not be happening for any type of GOx/CNT-based sensor. The biosensor was sensitive to glucose in air-equilibrated solutions indicating the O2-mediated enzymatic oxidation of glucose. The signal transduction relied on the net drop in a biosensor current that was caused by a decrease in a 4-e- O2 reduction current and an increase in a 2-e- H2O2 reduction current. The enzyme assays showed that CNT nearly doubled the retention of GOx in a biosensor while decreasing the average enzymatic activity of retained enzyme by a factor of 4-5. Such inhibition should be considered when using a protein-assisted solubilization of CNT in water for biomedical applications. The proposed analytical protocols can be also applied to study the effects of nanoparticles on proteins in assessing the health risks associated with the use of nanomaterials.
Keywords: Biosensors, Glucose Oxidase, Carbon Nanotubes, Chitosan, Enzyme Assays
Introduction
The direct electron transfer (DET) between the active redox enzymes and conductive nanomaterials is important for the development of electrochemical devices for sensing and energy conversion. In particular, the DET-based detection is central to the advancement of mediatorless biosensors, i.e. devices that do not require any extra reagents in a sample to detect enzyme’s substrate. However, the DET is not a common phenomenon. This is in part because the redox active centers of enzymes are often shielded from the electrodes by a glycoprotein coat preventing the efficient electron tunneling.
In the past decade, the DET has been reported in the case of electrodes based on the enzyme glucose oxidase (GOx) and carbon nanotubes (CNT).1-28 It has been explained by hypothesizing that either CNT plug into a GOx molecule or GOx partially unfolds facilitating the electrical contact between the enzyme’s cofactor flavin adenine dinucleotide (FAD) and CNT.1-5, 21, 22 The DET-based electrochemical glucose sensing at GOx/CNT-based electrodes has also been debated with arguments on both sides of the issue depending on the specifics of experiments.18-32
The present study focuses on the role of DET in glucose sensing at a GOx/CNT hybrid that was embedded in a bio-adhesive film of polysaccharide chitosan (CHIT) on the electrode surface. The CHIT provided a biocompatible, inert, and protective matrix for GOx and served as an effective dispersant for CNT.33-35 The interactions between the GOx and CNT in the CHIT matrix and aqueous solutions were also probed. The three aspects of the GOx/CNT system were studied including (i) the role of reactions relevant to the electrochemical glucose sensing in the generation of current flowing through a GOx/CNT-based biosensor, (2) the effect of CNT on the retention and enzymatic activity of GOx in CHIT films, and (3) the enzymatic activity of GOx in aqueous suspensions of CNT. The enzyme retention and activity were analyzed using the newly developed internally calibrated electrochemical continuous enzyme assay (ICECEA).36 The assay relied on the quantification of enzyme activity via enzyme-free calibration in one short continuous experiment.
EXPERIMENTAL SECTION
Reagents
The glucose oxidase (GOx, from Aspergillus niger, E.C. 1.1.3.4, 210 units mg-1) and chitosan (CHIT, MW ~1 ×106 Da, 80% deacetylation) were purchased from Sigma-Aldrich. The flavin adenine dinucleotide (FAD) was from Alfa Aesar (A14495). The different batches of multiwalled carbon nanotubes (CNT, synthesized via chemical vapor deposition, ~95% nominal purity) were purchased from Nanolab (Brighton, MA) and used as received. The CNT had a diameter of 15 ± 5 nm and length of 1-5 μm. Other chemicals including the NaH2PO4·H2O, Na2HPO4·H2O, HCl, and NaOH were purchased from Fisher. The 0.10 wt. % chitosan (CHIT) solutions were prepared by dissolving chitosan flakes in hot (80-95 °C) aqueous solutions of 0.050 M HCl, cooling to room temperature, adjusting the pH to ~4.5 with NaOH solution, and filtering with a 0.45-μm Millex-HA syringe filter. A glucose stock solution (1.0 M) was prepared in pH 7.40 phosphate buffer (0.050 M) and was allowed to mutarotate for 24 h at room temperature before use. The suspensions of CNT (1.0 mg mL-1) were prepared in 0.10 wt. % CHIT solutions by 20-min sonication. All solutions were prepared using 18-MΩ-cm deionized water that was purified with a Barnstead NANOpure cartridge system. They were stored in a refrigerator (~4°C) when not in use.
Electrochemical Measurements
The cyclic voltammograms and amperograms were recorded by using a CHI 832B workstation (CH Instruments, Inc.). The measurements were performed in the three-electrode system. The 3.0-mm diameter glassy carbon (GC) disc or 1.6-mm dia. Pt disc electrode served as a working electrode, a platinum wire was used as an auxiliary electrode, and Ag/AgCl/3MNaCl (BAS) served as a reference electrode. The working electrodes were wet polished on an Alpha A polishing cloth (Mark V Lab) with successively smaller particles (0.3 and 0.05 μm diameter) of alumina. The alumina slurry was removed from the electrode surface by 30-s ultrasonications in methanol and water.
All experiments were performed at room temperature (21 ± 1°C) using a pH 7.40 phosphate buffer solution (0.050 M) as a background electrolyte. The O2-free solutions were prepared by purging them with high purity argon for 30 min and maintaining argon blanket above a solution during a measurement. This reduced the oxygen concentration in a solution to <5 μM as determined by an YSI dissolved oxygen meter,37, 38 Such low oxygen concentration did not support a sustainable current due to the O2-mediated oxidation of glucose by GOx. The experiments were repeated at least three times, and the results are presented with relative standard deviation (RSD).
Preparation of Film Electrodes
The film electrodes were prepared by casting the following solutions on the electrode surface: (A) 0.10 wt. % CHIT solution containing GOx, and (B) 0.10 wt. % CHIT solution containing CNT (1.0 mg mL-1). The well-adhering surface films were formed after evaporating water for 2h at room temperature. All films were cast on the surface of a GC electrode with an exception of a film that was used to record amperograms shown in Figure 4. It was cast on a Pt electrode.
Figure 4.
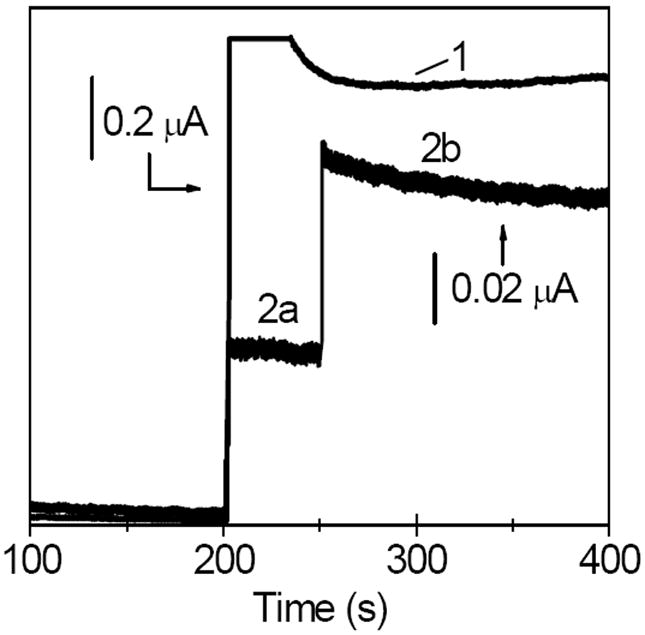
Amperometric traces (E=0.40 V) recorded at a Pt/GOx/CNT film electrode during the addition of aliquots (1) 0.50 mM H2O2 at 200 s, (2a) 2.0 mM glucose at 200 s, and (2b) 10.0 mM glucose at 250 s. All films contained chitosan as an immobilization matrix. Background electrolyte, air-equilibrated pH 7.40 phosphate buffer (0.050 M).
The CHIT-GOx/CNT film electrodes were prepared by mixing together the 10.0-μL aliquots of solutions A and B (vide supra). The initial enzyme load of the films was equal to 10 U of GOx for the voltammetric and amperometric experiments and 0.10, 0.30, 0.60, and 1.0 U for enzyme assays and potentiometric measurements. The CHIT-GOx and CHIT/CNT film electrodes were prepared by casting a 10.0-μL aliquot of solution A and B, respectively, on the electrode surface. The CHIT-FAD/CNT film electrodes were prepared by casting 10.0 μL of solution B, evaporating water, adding 10.0 μL of 10 mM FAD solution, and again evaporating water.
For the sake of brevity, the CHIT acronym will be omitted from films’ abbreviated names. For example, instead of “CHIT-GOx/CNT film” a shorter name “GOx/CNT film” will be used, instead of “CHIT-GOx film” we will use “GOx film” etc. The film electrodes were soaked in a stirred pH 7.40 phosphate buffer solution for 30 min before their first use. They were rinsed with water, air dried, and stored at 4°C when not in use.
RESULTS AND DISCUSSION
The electrochemical sensing of glucose at a GOx/CNT-based biosensor may involve the following reactions:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
where the GOx(FAD)CNT and GOx(FAD) denote the enzyme molecules that are within the electron tunneling distance from CNT and those that are remote from the CNT, respectively. The role of each of these reactions in the glucose detection at a GOx/CNT-based biosensor was investigated.
Electroactivity of the GOx/CNT System in O2-free Glucose Solutions (reactions 1 and 2)
The sequence of reactions 1 and 2 represents the much sought-after bioelectrocatalysis where GOx oxidizes glucose and is directly recycled at the electrode surface with no need for any redox mediator. The bioelectrocatalysis in the GOx/CNT system was probed by using cyclic voltammetry. Figure 1A (line 1) shows a typical cyclic voltammogram that was recorded at a GOx/CNT film electrode in an O2-free phosphate buffer solution at pH 7.40. Such voltammograms displayed background/capacitive currents that were dependent on a particular batch of CNT, but they all had a common feature which was a pair of current peaks at a formal (midpoint) potential of -0.48 ± 0.01 V. The symmetrical shape of the peaks and their proportionality to the potential scan rate indicated that the electrode process involved the redox species that were adsorbed on CNT. Apparently, the presence of CHIT matrix did not prevent the GOx from adsorbing on CNT. The control experiments with the film electrodes containing either CNT or GOx yielded featureless voltammograms (Figure 1, lines 2 and 3), which showed that the presence of both GOx and CNT on the electrode surface was necessary to observe the current peaks. The lack of electroactivity in GOx films (line 3) argued against a notion39 that chitosan chains act as the molecular wires enabling the electron transfer between the GOx and the surface of GC electrode.
Figure 1.
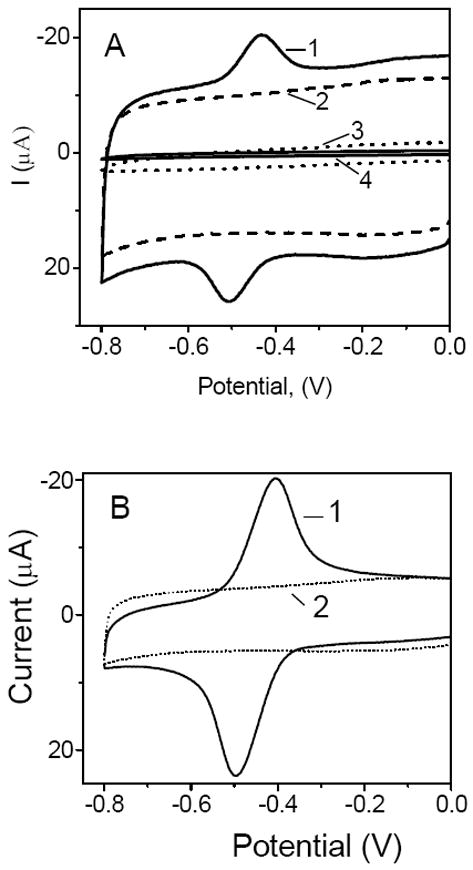
Cyclic voltammograms recorded at (A) (1) GOx/CNT, (2) CNT, and (3) GOx film electrodes, and (4) bare glassy carbon electrode; (B) (1) FAD/CNT, and (2) CNT film electrodes. All films contained chitosan as an immobilization matrix. The voltammograms were recorded in the O2-free pH 7.40 phosphate buffer solutions. Scan rate, 50 mV s-1.
The control experiments with the FAD/CNT film electrodes yielded a pair of current peaks (Figure 1B, line 1), which were due to the surface-confined electrode process
| (8) |
These peaks were located at the same formal potential (-0.48 V) as that recorded at the GOx/CNT film electrodes, which indicated that the electroactivity of GOx/CNT films was due to the redox of enzyme cofactor FAD.
Figure 2 shows that the addition of glucose to an O2-free solution had no effect on the electroactivity of GOx/CNT films, even at a very high glucose concentration (100 mM). The lack of sensitivity to glucose clearly indicated that the current peaks at -0.48 V were due to the electroactive FAD that was not the part of enzymatically active GOx, i.e. reactions 1 and 2 were not taking place in the present GOx/CNT-based biosensor. Similar lack of bioelectrocatalysis at electrodes containing CNT and GOx has been recently reported.30-32 The voltammetric data in Figure 2 are also in line with the recent studies, which reported the loss of activity of GOx upon its adsorption on the nanostructured surfaces including carbon nanofibers.40,41 The loss of protein function has been correlated with a high degree of GOx unfolding, increase of β-sheet and a reduction of α-helical secondary structure, and unfavorable orientation of the enzyme upon surface contact.40
Figure 2.
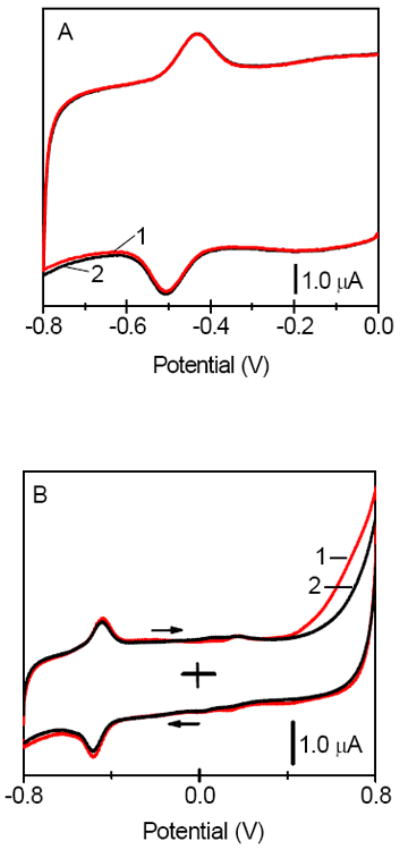
Cyclic voltammograms recorded at the GOx/CNT film electrodes in (A) (1) absence and (2) presence 0.60 mM glucose; (B) (1) absence and (2) presence of 100 mM glucose in a solution. All films contained chitosan as an immobilization matrix. The voltammograms were recorded in the O2-free pH 7.40 phosphate buffer solutions. Scan rate, 5.0 mV s-1.
Electroactivity of the GOx/CNT System in O2-containing Solutions (reactions 3 and 4)
In the air-equilibrated solutions, the current due to the reduction of O2 at CNT (Figure 3A, line 1, reaction 3) overlapped with the current peaks at -0.48 V (Figure 3A, line 2) complicating the analysis. The control experiment showed that a CNT electrode displayed a featureless voltammogram in the absence of both O2 and GOx in a solution (Figure 3A, line 3). In the air-equilibrated solutions, a GOx/CNT film electrode yielded a voltammogram 3 (Figure 3B), which was the superposition of currents due to the reduction of O2 (Figure 3A, line 1) and the redox of FAD (Figure 3A, line 2). The alternative interpretation of the voltammogram 3 based on the reversed reaction 2 and forward reaction 4 lacked an experimental support. According to these reactions, the cathodic and anodic peaks at -0.48 V should increase and decrease, respectively, in the presence of oxygen in a solution. However this was not observed based to the integration of these peaks after a digital subtraction of voltammograms that were recorded at GOx/CNT and CNT film electrodes, respectively (IGOx/CNT − ICNT). The oxygen had no effect on the background-corrected cathodic and anodic peaks at -0.48 V.
Figure 3.
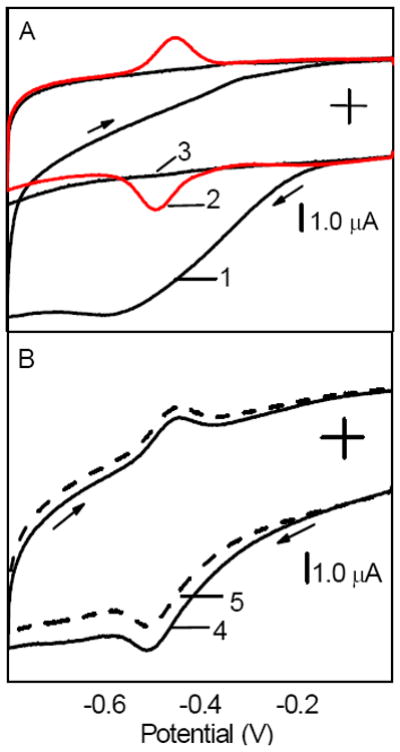
(A) Cyclic voltammograms recorded at (1) CNT film electrode in air-equilibrated solution, (2) GOx/CNT film electrode in O2-free solution, and (3) CNT film electrode in O2-free solution. (B) Cyclic voltammograms recorded at GOx/CNT film electrode in air-equilibrated solution containing (4) 0 and (5) 0.60 mM glucose. All films contained chitosan as an immobilization matrix. Background electrolyte, pH 7.40 phosphate buffer solution. Scan rate, 50 mV s-1.
The addition of glucose to an air-equilibrated solution resulted in the shift of the entire voltammogram 3 toward lower cathodic currents (Figure 3B, line 4). Again, the shift did not affect the background-corrected current peaks at -0.48 V. Therefore, the shift was ascribed to the decrease in O2 reduction current due to the depletion of O2 in a biosensor by the mediated enzymatic process (reactions 5 and 6).
Mediated Detection of Glucose at a GOx/CNT-based Biosensor (reactions 5-6)
The sequence of reactions 5 and 6 represents the mediated detection scheme where GOx oxidizes glucose and is recycled by its natural redox mediator oxygen. The existence of enzymatically active GOx molecules in the GOx/CNT system was confirmed by the detection of H2O2 that was produced by the sequence of reactions 5 and 6. To this end, the GOx/CNT film was cast on the surface of Pt electrode, which has a good activity toward the oxidation of H2O2. Figure 4 shows a current step (line 1), which is a response of Pt electrode to the addition of H2O2 to a stirred air-equilibrated solution. The analogous current steps (line 2a-2b) were recorded when the experiment was repeated by adding glucose instead of H2O2 to a solution.
The control experiment showed that glucose was not directly oxidized at the Pt electrode under such conditions. Therefore, the current steps 2a and 2b were due to the oxidation of H2O2 that was produced in the enzymatic process inside the surface film. Apparently, the glucose added to a solution reacted with a subset of enzymatically active GOx(FAD) molecules (reaction 5) yielding the reduced form of enzyme (GOx(FADH2)), which was subsequently oxidized by O2 (reaction 6) producing the H2O2. The model of multilayer adsorption suggests that the GOx molecules that retained their enzymatic activity toward glucose were not directly adsorbed on CNT.
Figure 3 (line 1) and Figure 5 show that O2 and H2O2 were reduced at CNT in the same potential window (< -0.1 V). Consequently, the O2-mediated glucose detection in such potential window involved the reduction of both O2 and H2O2 at a GOx/CNT-based biosensor. Therefore, the signal transduction relied on the net current decrease (Figure 3B) that was caused by the decrease in a current due to the 4-e- O2 reduction (reaction 3) and an increase in a current due to the 2-e- H2O2 reduction (reaction 7).
Figure 5.
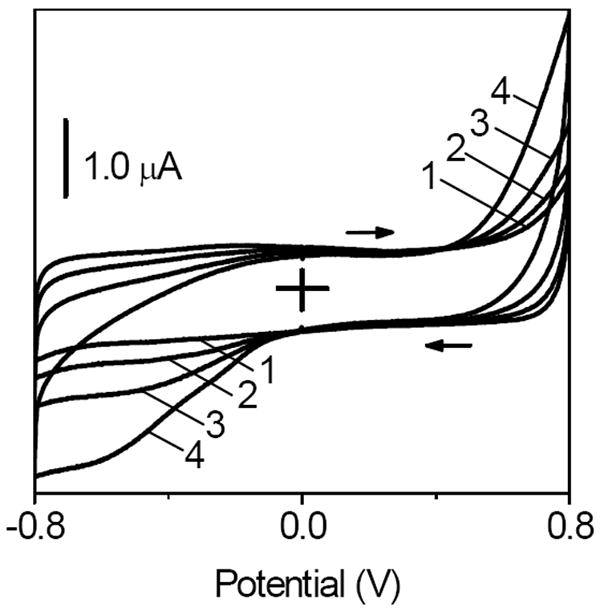
Cyclic voltammograms recorded at a CNT film electrode in O2-free solution that contained (1) 0, (2) 0.2, (3) 1.0, and (4) 4.0 mM H2O2. All films contained chitosan as an immobilization matrix. Background electrolyte, pH 7.40 phosphate buffer. Scan rate, 50 mV s-1.
CNT Effect on the Retention and Activity of GOx in CHIT Films
The retention and activity of GOx in CHIT films were determined by using the internally calibrated electrochemical continuous enzyme assay (ICECEA). The assay consisted of two phases, a calibration and actual assay phase, which were performed consecutively in the same solution and at the same working electrode. The activity unit (U) of GOx was determined by measuring the initial rate of the overall enzymatic reaction
| (9) |
Under the condition of enzyme being a limiting reagent, the initial rate is the measure of enzyme activity, which is defined as the amount of enzyme’s substrate that is converted to a product per unit time (1 U = μmole min-1). The initial rate was determined by measuring the increase in the concentration of H2O2 that was formed in the reaction 9 over time. The assay required three solutions: (A) β-D-glucose solution in a pH 7.40 phosphate buffer, (B) aqueous solution of H2O2, and (C) aqueous solution of GOx. The ICECEA was performed at a bare Pt electrode (Figure 6). After recording a baseline current in a stirred solution A, the three aliquots of a solution B were added (arrows “a”) in order to record the three current steps that were used to calculate a calibration slope (CS). This was followed by the addition of an aliquot of solution C, or by dipping a GOx or GOx/CNT film (arrow “b”), in order to trigger the enzymatic reaction 9 and record a rising current-time segment. The assay slope (AS) of this segment represented the initial reaction rate. Dividing the assay slope AS (μA s-1) by the calibration slope CS (μA μM-1) and multiplying by 60 (s min-1) yielded the enzyme activity in U L-1.
Figure 6.
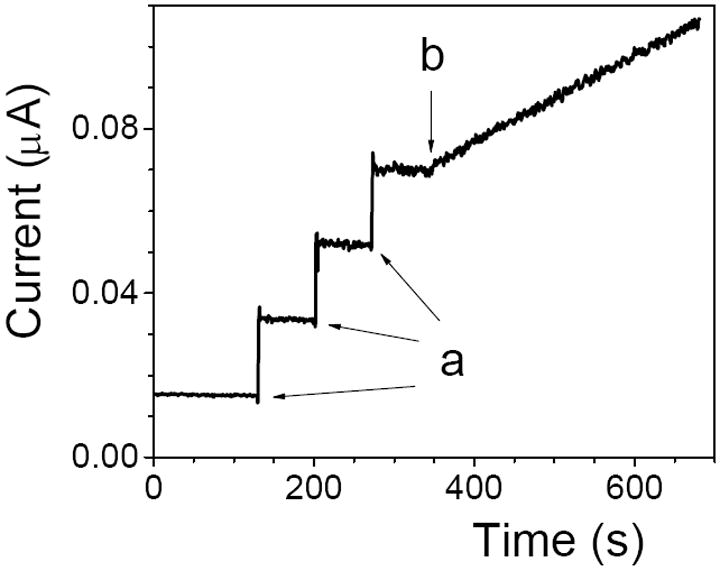
ICECEA current-time trace (E=0.60 V) recorded at a bare Pt electrode in a stirred solution of 10 mM β-D-glucose in pH 7.40 phosphate buffer (solution A). The arrows “a” indicate the moment of addition of 20.0-μL aliquots of 2.0 mM H2O2 (solution B). The arrow “b” shows the moment when a GOx film was immersed in the solution. The angled I-t segment is due to the oxidation of H2O2 that was produced in the enzymatic reaction 9.
The ICECEA provided a good accuracy (relative error <3%) and precision (RSD <3%, N =3) for the determination of enzyme activity. The low relative error and RSD resulted from the unique integration of enzyme-free calibration with actual enzyme assay in one continuous experiment using the same electrode and solution. This eliminated the need for transferring the electrodes between the calibration and assay solutions providing a high degree of consistency between the calibration and assay phases of individual runs.
The amount of GOx that was retained in a surface film was expressed in terms of the enzyme activity units “U retained”. The “U retained” were determined by subtracting the amount of enzyme that leached from a film into a solution (“U leached”) from the amount of enzyme that was initially loaded into a surface film (“U loaded”). The “U loaded” were calculated based on the known aliquot of GOx that was cast on the electrode surface. The selected range of initial enzyme loading was lower (0.10-1.0 U) than that used in the earlier voltammetric experiments (10 U) in order to better discern the effects of CNT on the activity of GOx. The amount of CNT in each film was always the same (10.0 μg).
In order to determine the “U leached”, a GOx or GOx/CNT film was soaked in a stirred buffer solution (1.00 mL) for 30 min. The 0.200-mL aliquot of this solution was then used as the solution C in the ICECEA analysis. The recorded assay slope AS was multiplied by five to get a total number of “U leached”. The activity of GOx in a film (“U active”) was determined by dipping a GOx or GOx/CNT film in a stirred assay mixture (solutions A+B) and recording the assay slope AS (Figure 6). The control experiments showed that the assay slope was not dependent on the increase in the rate of solution stirring, which suggested that the concentration of glucose and oxygen were sufficient to avoid diffusional limitations to the enzymatic reaction in a surface film.
The assay data showed several trends. The “U retained” increased proportionally with the increase in the initial enzyme loading. The slopes of the linear plots of “U retained” vs. “U loaded” revealed that the retention of GOx was equal to 30% (R2 = 0.865) and 55% (R2 = 0.971) in the GOx and GOx/CNT films, respectively. The increase of enzyme retention in the films containing CNT can be ascribed to the adsorption of GOx on CNT. Such adsorption was also seen in the earlier voltammetric studies (Figures 1 and 2), which showed the symmetrical surface-confined redox peaks in cyclic voltammograms that were recorded at GOx/CNT film electrodes. The CNT-enhanced enzyme retention in surface films further strengthened the argument that CHIT matrix did not preclude the strong interactions between the GOx and CNT.
The similar level of enzyme retention in CHIT films has been observed before in the case of lactate oxidase (22%)38 and glutamate oxidase (57%).42 It has been also observed that the retention can be as high as >99% when the enzyme is crosslinked with CHIT by using the glutaric dialdehyde.43
The ICECEA analysis also revealed that the “U active” increased proportionally with the increase in both the “U retained” and “U loaded”. Figure 7 shows the dependence of ratio “U active”/“U retained” vs. “U loaded”. The main trend in the data is that the average enzymatic activity of GOx dropped in the presence of CNT in a film. The drop in the U ratio was from 11% to 2% and from 22% to 5% in the case of the films that were initially loaded with 0.10 and 1.0 U GOx, respectively. This negative impact of CNT on the activity of GOx correlates with the voltammetric studies, which showed that the current peaks at -0.48 V (Figure 1) were not due to the enzymatically active GOx.
Figure 7.
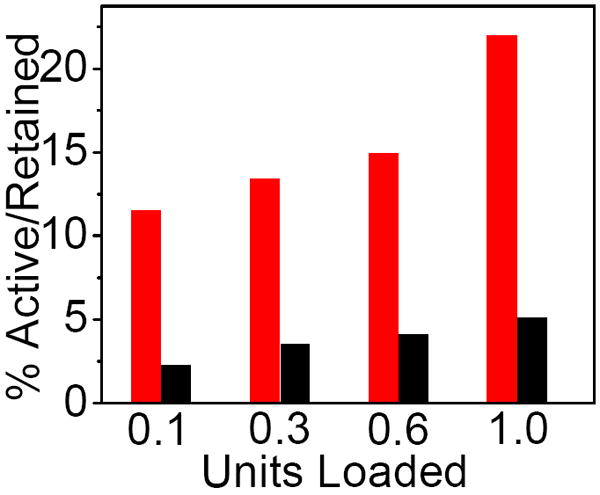
The enzymatic activity of GOx in CHIT films (red bars) and CHIT/CNT films (black bars) that were loaded with different enzyme units.
Activity of GOx in Aqueous Suspensions of CNT
The impact of CNT suspensions on the activity of GOx in aqueous solutions was investigated by monitoring the pH change during the enzymatic reaction 9. According to this reaction, the pH of a reaction mixture should decrease in time because of the generation of gluconic acid. Based on the results of enzyme assays, the expectation was that CNT would slow down the rate of generation of gluconic acid in a solution.
Indeed, Figure 8 shows that the decrease in pH was slower in the presence of CNT suspension in a solution (trace a1) than that in the absence of CNT (trace a). This was ascribed to the slower production of gluconic acid due to the lower enzymatic activity of GOx molecules interacting with CNT. The pH decrease was not affected by the increase in the rate of solution stirring indicating the lack of transport limitations during the measurements. The difference in pH between the solutions with and without CNT (ΔpH) was larger when the smaller amount of GOx was dissolved in a solution. For example, Figure 8 shows that the ΔpH at the end of the experiment was ~40% larger for 0.10 U (traces a1-a) than for 1.0 U of GOx (traces b1-b). Apparently, a larger relative fraction of GOx molecules was negatively impacted by their interactions with CNT in a solution containing less enzyme. In summary, the voltammetric analysis, amperometric enzyme assays, and amperometric measurements all showed that the GOx displayed lower enzymatic activity in the presence of CNT.
Figure 8.
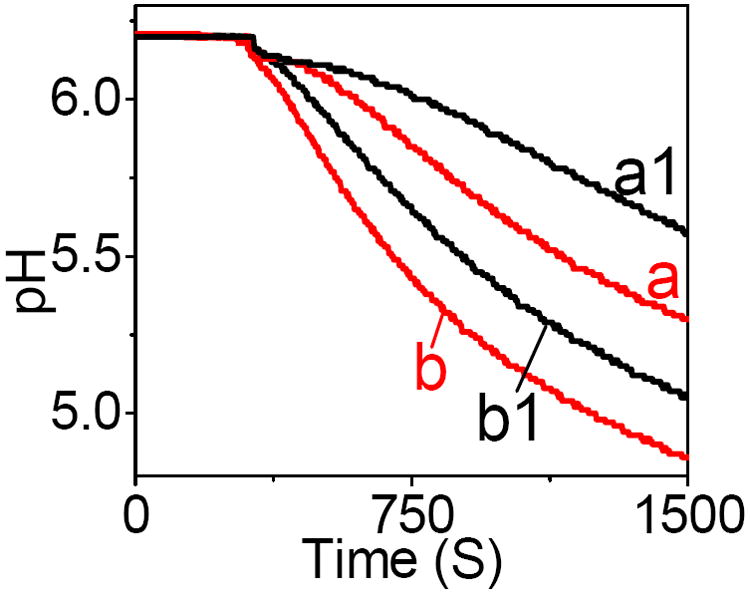
The pH change in a stirred solution containing (a) 0.10 U, and (b) 1.0 U of GOx after the addition of glucose (0.10 M) at 370 s. The traces a1 and b1 were recorded in the presence of CNT (5.0 mg) in a solution a and b, respectively. Background electrolyte, 0.10 M KNO3 solution.
CONCLUSIONS
The DET was not the mechanistic basis for glucose sensing at a GOx/CNT-based biosensor because no increase in the anodic current was observed when glucose was added to a deoxygenated solution. This indicated that GOx molecules that were within the electron tunneling distance from CNT were not enzymatically active toward glucose. The presented data are consistent with the hypothesis that adsorption of GOx on CNT affects the active conformation of the enzyme.40, 41 Indeed, such adsorption was seen in the enzymatic assays, which showed that CNT nearly doubled the retention of GOx in a biosensor. The enzyme assays and potentiometric measurements also indicated that CNT significantly decreased the average enzymatic activity of GOx. The GOx molecules that remained active in the GOx/CNT system required O2 to mediate the enzymatic oxidation of glucose. Despite considerable efforts, no DET-based commercial biosensors for glucose have been developed thus far.
Acknowledgments
This work was supported by the NIH/MBRS/SCORE Grant GM 08194 and The Welch Foundation Departmental Research Grant AX-0026.
References
- 1.Guiseppi-Elie A, Lei C, Baughman RH. Nanotchenology. 2002;13:559–564. [Google Scholar]
- 2.Patolsky F, Weizmann Y, Willner I. Angew Chem Int Ed. 2004;43:2113–2117. doi: 10.1002/anie.200353275. [DOI] [PubMed] [Google Scholar]
- 3.Liu J, Chou A, Rahmat W, Paddon-Row, Gooding JJ. Electroanalysis. 2005;17:38–46. [Google Scholar]
- 4.Liu Y, Wang M, Zhao F, Xu Z, Dong S. Biosens Bioelectron. 2005;21:984–988. doi: 10.1016/j.bios.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Zhao H-Z, Sun J-J, Song J, Yang Q-Z. Carbon. 2010;48:1508–1514. [Google Scholar]
- 6.Zhao YD, Zhang WD, Chen H, Luo QM. Anal Sci. 2002;18:939–941. doi: 10.2116/analsci.18.939. [DOI] [PubMed] [Google Scholar]
- 7.Liang W, Zhuobin Y. Sensors. 2003;3:544–554. [Google Scholar]
- 8.Luong JHT, Hrapovic S, Wang D, Bensebaa F, Simard B. Electroanalysis. 2004;16:132–139. [Google Scholar]
- 9.Cai C, Chen J. Anal Biochem. 2004;332:75–83. doi: 10.1016/j.ab.2004.05.057. [DOI] [PubMed] [Google Scholar]
- 10.Gong K, Yan Y, Zhang M, Su L, Xiong S, Mao L. Anal Sci. 2005;21:1383–1393. doi: 10.2116/analsci.21.1383. [DOI] [PubMed] [Google Scholar]
- 11.Jia N, Liu L, Zhou Q, Wang L, Yan M, Jiang Z. Electrochim Acta. 2005;51:611–618. [Google Scholar]
- 12.Ivnitski D, Artyushkova K, Rincon RA, Atanassov P, Luckarift HR, Johnson GR. Small. 2008;4:357–364. doi: 10.1002/smll.200700725. [DOI] [PubMed] [Google Scholar]
- 13.Jia F, Shan C, Li F, Niu L. Biosens Bioelectron. 2008;24:945–950. doi: 10.1016/j.bios.2008.07.057. [DOI] [PubMed] [Google Scholar]
- 14.Yao Y-L, Shiu K-K. Electroanalysis. 2008;20:1542–1548. [Google Scholar]
- 15.Li J, Zhao F, Wang G, Gui Z, Xiao F, Zeng B. Electroanalysis. 2009;21:150–156. [Google Scholar]
- 16.Gao R, Zheng J. Electrochem Comm. 2009;11:608–611. [Google Scholar]
- 17.Ghica ME, Pauliukaite R, Fatibello-Filho O, Brett CMA. Sens Actuators B. 2009;142:308–315. [Google Scholar]
- 18.Deng C, Chen J, Nie Z, Si S. Biosens Bioelectron. 2010;26:213–219. doi: 10.1016/j.bios.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 19.Deng S, Jian G, Lei J, Hu Z, Ju H. Biosens Bioelectron. 2009;25:373–377. doi: 10.1016/j.bios.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 20.Li F, Song J, Li F, Wang X, Zhang Q, Han D, Ivaska A, Niu L. Biosens Bioelectron. 2009;25:883–888. doi: 10.1016/j.bios.2009.08.044. [DOI] [PubMed] [Google Scholar]
- 21.Deng C, Chen J, Chen X, Xiao C, Nie L, Yao S. Biosens Bioelectron. 2008;23:1272–1277. doi: 10.1016/j.bios.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Liu Q, Lu X, Li J, Yao X, Li J. Biosens Bioelectron. 2007;22:3203–3209. doi: 10.1016/j.bios.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Feng M, Tachikawa H. Biosens Bioelectron. 2007;22:3036–3041. doi: 10.1016/j.bios.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 24.Nejadnik MR, Deepak FL, Garcia CD. Electroanalysis. 2011;23:1462–1469. doi: 10.1002/elan.201000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo X, Killiard AJ, Smyth MR. Electroanalysis. 2006;18:1131–1134. [Google Scholar]
- 26.Jose MV, Marx S, Murata H, Koepsel RR, Russell AJ. Carbon. 2012;50:4010–4020. [Google Scholar]
- 27.Maghsoodi S, Gholami Z, Chourchian H, Mortazavi Y, Khodadadi AA. Sens Actuators B. 2009;141:526–531. [Google Scholar]
- 28.Ryu J, Kim H, Lee S, Hahn HT, Lashmore D. J Nanosci Nanotechnol. 2010;10:941–947. doi: 10.1166/jnn.2010.1892. [DOI] [PubMed] [Google Scholar]
- 29.Milton RD, Baur J, Varcoe JR, Thumser AE, Slade RCT. Phys Chem Chem Phys. 2012;14:9582–9585. doi: 10.1039/c2cp41651d. [DOI] [PubMed] [Google Scholar]
- 30.Wooten M. Electrochemical Sensors Based on Enzymes, Biopolymers and Nanoparticles. ProQuest UMI; Ann Arbor: 2010. [Google Scholar]
- 31.Wang Y, Yao Y. Microchim Acta. 2012;176:271–277. [Google Scholar]
- 32.Goran JM, Mantilla SM, Stevenson KJ. Anal Chem. 2013;85:1571–1581. doi: 10.1021/ac3028036. [DOI] [PubMed] [Google Scholar]
- 33.Wooten M, Gorski W. Anal Chem. 2010;82:1299–1304. doi: 10.1021/ac902301b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang M, Mullens C, Gorski W. Anal Chem. 2007;79:2446–2450. doi: 10.1021/ac061698n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang M, Smith A, Gorski W. Anal Chem. 2004;76:5045–5050. doi: 10.1021/ac049519u. [DOI] [PubMed] [Google Scholar]
- 36.Zhang M, Karra S, Gorski W. Anal Chem. 2013;85:6026–6032. doi: 10.1021/ac4008557. [DOI] [PubMed] [Google Scholar]
- 37.Wei X, Cruz J, Gorski W. Anal Chem. 2002;74:5039–46. doi: 10.1021/ac020216e. [DOI] [PubMed] [Google Scholar]
- 38.Wei X, Zhang M, Gorski W. Anal Chem. 2003;75:2060–64. doi: 10.1021/ac020765k. [DOI] [PubMed] [Google Scholar]
- 39.Zhao C, Meng Y, Shao C, Wan L, Jiao K. Electroanalysis. 2008;20:520–526. [Google Scholar]
- 40.Seehuber A, Dahint R. J Phys Chem B. 2013;117:6980–6989. doi: 10.1021/jp401906h. [DOI] [PubMed] [Google Scholar]
- 41.Olenic L, Lupu D, Garabagiu S, Biris AS. J Mater Sci : Mater Med. 2009;20:177–183. doi: 10.1007/s10856-008-3550-y. [DOI] [PubMed] [Google Scholar]
- 42.Zhang M, Mullens C, Gorski W. Electrochim Acta. 2006;51:4528–4532. [Google Scholar]
- 43.Zhang M, Mullens C, Gorski W. Electroanalysis. 2005;17:2114–2120. [Google Scholar]