

Abstract
Cardiomyocyte apoptosis contributes toward the loss of muscle mass in myocardial pathologies. Previous reports have implicated type I cAMP-dependent protein kinase (PKA) and p90 ribosomal S6 kinase (RSK) in cardiomyocyte apoptosis. However, the precise mechanisms and the isoform of RSK involved in this process remain undefined. Using adult rat ventricular myocytes and mouse-derived cardiac HL-1 cardiomyocytes, we demonstrate that hypoxia/reoxygenation (H/R)-induced apoptosis is accompanied by a decrease in the type I PKA regulatory subunit (PKARIα) and activation of RSK1. As previously described by us for other cell types, in cardiomyocytes, inactive RSK1 also interacts with PKARIα, whereas the active RSK1 interacts with the catalytic subunit of PKA. Additionally, small interfering (siRNA)-mediated silencing of PKARIα or disrupting the RSK1/PKARIα interactions with a small, cell-permeable peptide activates RSK1 and recapitulates the H/R-induced apoptosis. Inhibition of RSK1 or siRNA-mediated silencing of RSK1 attenuates H/R-induced apoptosis, demonstrating the role of RSK1 in cardiomyocyte apoptosis. Furthermore, silencing of RSK1 decreases the H/R-induced phosphorylation of sodium–hydrogen exchanger 1 (NHE1), and inhibition of NHE1 with 5′-N-ethyl-N-isopropyl-amiloride blocks H/R induced apoptosis, indicating the involvement of NHE1 in apoptosis. Overall, our findings demonstrate that H/R-mediated decrease in PKARIα protein levels leads to activation of RSK1, which via phosphorylation of NHE1 induces cardiomyocyte apoptosis.
Introduction
It is now well established that cardiomyocytes undergo apoptosis and that this process is enhanced upon an insult such as cardiac ischemia followed by reperfusion of the ischemic zone. This hypoxia/reperfusion (H/R)-induced cardiomyocyte apoptosis impedes repair of the heart muscle and contributes toward deterioration of heart function and progression toward end-stage heart failure. Clearly, therefore, it is important to understand the precise molecular mechanisms that lead to cardiomyocyte apoptosis.
Previous studies have suggested that cAMP-dependent protein kinase (PKA) contributes to cardiac hypertrophy induced by prostaglandin E2 (PGE2) and angiotensin II (Enns et al., 2010; He et al., 2010). PKA is a heterotetramer composed of a dimer of regulatory subunits (PKAR) and two catalytic subunits (PKAc). There are two forms of PKAR (PKARI and PKARII), and each of these has two isoforms: PKARIα, PKARIβ, PKARIIα, and PKARIIβ (Doskeland et al., 1993; Skalhegg and Tasken, 1997). PKA is classified as type I or type II, depending on the PKAR subtype (PKARI or PKARII) to which the PKAc is bound (Doskeland et al., 1993; Skalhegg and Tasken, 1997). cAMP binds the PKAR subunits and dissociates them from PKAc, resulting in alleviation of inhibition and activation of PKAc. Previously, it was assumed that in cardiomyocytes, type II PKA is more organized by A kinase–anchoring proteins (AKAPs), whereas type I PKA is mainly cytoplasmic. However, it has been shown that the distribution of both type I and type II PKA in cardiomyocytes is highly organized and that the two types of PKA respond differently to agonists and also phosphorylate different intracellular proteins (Wong and Scott, 2004). For instance, β-adrenergic receptors activate type II PKA, whereas PGE2 activates type I PKA (Wong and Scott, 2004). Because PGE2 can induce cardiac hypertrophy (He et al., 2010), it is possible that type I PKA plays a role in this pathology. PKA has also been suggested to play a role in oxidative stress- or phenylephrine-induced cardiomyocyte apoptosis (Valks et al., 2002; Cieslak and Lazou, 2007).
The four forms of p90 ribosomal S6 kinases (RSK1–RSK4) belong to a family of proteins with two kinase domains (Frodin and Gammeltoft, 1999; Anjum and Blenis, 2008; Cargnello and Roux, 2011). RSK1, RSK2, and RSK3 share sequence similarities, and these isoforms are immediately downstream of, and activated by, ERK1/2 (Frodin and Gammeltoft, 1999; Anjum and Blenis, 2008). RSK4 is a longer protein with different functions (Frodin and Gammeltoft, 1999; Dummler et al., 2005; Anjum and Blenis, 2008). Although RSK1 shares similarity in sequence with its other isoforms, RSK2 and RSK3, as shown by studies in RSK2 null mice, the three RSKs are not redundant (Zeniou et al., 2002). Differentiation of PC12 cells is induced by RSK1, but not RSK2, showing a lack of redundancy (Silverman et al., 2004). By phosphorylating its substrates, RSK1 has been implicated in multiple cellular processes, including cell proliferation, growth, and survival. In some noncardiac cells, such as HEK293 cells, B82L cells, and hematopoietic cell line 32D, it has been shown that RSK1 phosphorylates and inactivates proapoptotic proteins, such as Bcl-xL/Bcl-2–associated death promoter and death-associated protein kinase, and thereby plays an antiapoptotic role (Shimamura et al., 2000; Chaturvedi et al., 2006, 2009). However, RSK1 has also been reported to phosphorylate Nur77 in T cells and promote apoptosis (Wang et al., 2009). In cardiomyocytes, indirect evidence using a dominant negative form of RSK1 has implicated RSK1 in cardiomyocyte apoptosis (Maekawa et al., 2006).
Previous studies from our laboratory have shown that the inactive and active forms of RSK1, but not RSK2 or RSK3, interact with subunits of type I PKA and that these interactions regulate the activities of both PKA as well as RSK1 (Chaturvedi et al., 2006, 2009; Gao and Patel, 2009; Gao et al., 2010). Because RSK1 and PKA have been implicated in cardiomyocyte apoptosis (Valks et al., 2002; Maekawa et al., 2006; Cieslak and Lazou, 2007) and because inactive and active forms of RSK1 interact with PKARIα and PKAc, respectively (Chaturvedi et al., 2006, 2009; Gao and Patel, 2009; Gao et al., 2010), we investigated the physiologic relevance of the RSK1/PKA subunit interactions in mediating H/R-induced apoptosis in adult rat ventricular myocytes (ARVMs) and in the mouse cardiac cell line HL-1 cells. We demonstrate that H/R decreases PKARIα protein levels and leads to activation of RSK1. Silencing of PKARIα recapitulates the hypoxia/reoxygenation-mediated activation of RSK1 and apoptosis. Additionally, active RSK1-mediated phosphorylation of the sodium–hydrogen exchanger 1 (NHE1) mediates apoptosis resulting from H/R.
Materials and Methods
Reagents.
Antibodies against phospho-PKA substrate, cleaved caspase 7, cleaved PARP, and phospho-Ser-14-3-3 binding motif were purchased from Cell Signaling (Beverly, MA). Anti–phospho-RSK1/2 (Ser221) antibody from R&D Systems (Minneapolis, MN), anti-actin antibody (MP Biomedicals, Aurora, OH), and anti-RSK1 and rabbit anti-PKAc antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were also used. The anti-PKARIα and monoclonal anti-PKAc antibodies were from BD Biosciences (Palo Alto, CA). Anti–phosphoS732-RSK1 was raised against a peptide (sequence: AQRRVRKLP(pS)TTL) by Rockland (Gilbertsville, PA). Anti-NHE1 antibody was from Millipore (Temecula, CA). N-terminally palmitoylated peptides (PS and Mut PS), corresponding to PKARIα pseudosubstrate region (amino acids 91–99), was synthesized by New England Peptide (Gardner, MA) (Gao et al., 2010). The RSK inhibitor, SL0101 [kaempferol-3-O-(3″,4″-di-O-acetyl-α-L-rhamnopyranoside)] was purchased from R&D Systems (Minneapolis, MN). EIPA (5′-N-ethyl-N-isopropyl-amiloride) was from Sigma-Aldrich (Saint Louis, MO).
Isolation of Adult Rat Ventricular Myocytes.
The ARVMs were isolated by our core facility according to the protocols reviewed and approved by the Animal Care and Use Committee of Loyola University Chicago, Stritch School of Medicine. The method used was that described by Guggilam et al. (2013). Briefly, the rats were anesthetized, and their hearts were excised and mounted on a Langendorff apparatus. The hearts were retrograde perfused through the aorta with perfusion buffer and an enzymatic digestion buffer for 15–20 minutes [perfusion buffer plus 12.5 μM CaCl2, 0.85 mg/ml collagenase II (Worthington Biochemical Corp., Lakewood, NJ)]. The ventricles were minced and myocytes mechanically dispersed in digestion buffer and filtered. Cells were then resuspended in increasing concentrations of CaCl2 over 50 minutes to achieve a final concentration of 1 mM of Ca2+. The isolated myocytes were plated on laminin-coated dishes in Dulbecco’s modified Eagle’s medium supplemented with 5 mM taurine, 5 mM creatine, 2 mM l-carnitine, 25 mM HEPES, 20 U per liter of insulin, and 100 U/ml penicillin-streptomycin. After 1 hour of plating, the cells were replaced with fresh medium and used for the experiments 5 hours later.
HL-1 Cell Culture.
The mouse heart cell line (HL-1) was the gift from Dr. Claycomb (Louisiana State University Health Sciences Center, New Orleans, LA). HL-1 cells were plated on fibronectin/gelatin-coated plates and cultured with Claycomb media (Sigma-Aldrich) supplemented with 10% fetal bovine serum, 10 mM norepinephrine, 2 mM l-glutamine, and penicillin/streptomycin in 5% CO2 incubator at 37°C. Before hypoxia, the cells were incubated overnight in Claycomb medium minus fetal bovine serum and norepinephrine.
Hypoxia/Reoxygenation.
ARVM and HL-1 cells were exposed to 94% N2, 5% CO2, and 1% O2 for 24 hours followed by reoxygenation in 5% CO2 and 95% air for 1 hour.
RSK1 and PKARIα Silencing with Small Interfering RNAs.
HL-1 cells were plated at 3 × 105 cells/3.5 cm dish in complete Claycomb medium. The next day, cells were transfected with 40 nM of mutant or wild types of small interfering (siRNAs) targeted against PKARIα or RSK1 using Transit TKO (Mirus). Cells were incubated 48 hours before experimentation. ARVMs (0.5–1.0 × 105 cells/3.5 cm dish) were transfected with 30 nM of mutant or wild types of siRNAs against RSK1 and incubated 16 hours before experimentation. The RSK1-specific siRNA sequence was as follows: sense, GGA CCA AGA UGG AGA GAG ACA UCC T; antisense, AGG AUG UCU CUC UCC AUC UUG GUC CGA. PKARIα-specific siRNA sequence was sense, GGA GGA GGC AAG ACA GAU UCA GUG UCU AC; antisense, AGA CAC UGA AUC UGU CUU GCC UCC UCC UU.
Treatment of Cells.
To study the role of RSK1 in apoptosis, ARVMs or HL-1 cells were incubated with or without RSK inhibitor SL0101 (50 μM) for the entire duration of the control or H/R period before lysing cells. The palmitoylated PKARIα peptide (peptide PS, sequence: KGRRRRGAI) or its mutant form (Mut-PS, sequence: KGAARRGAI) (Gao et al., 2010) was transduced in serum-starved HL-1 cells by incubating cells with 2 μM concentration of the peptides for the experimental periods. Similarly, HL-1 cells were incubated with the PKA inhibitor H89 (N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide) (10 μM) for the entire duration of the control or H/R protocol. To inhibit NHE1, EIPA (5 μM) was added to ARVMs or HL-1 cells during the last 5 hours of exposure to normoxia or H/R.
Cellular Apoptosis.
Apoptosis was analyzed by Western blotting and terminal dUTP nick end labeling (TUNEL) assays after HR or other treatments. For Western blotting, cell lysates were probed with anticleaved PARP and anticleaved caspase 7 antibodies. The densities of protein bands were quantified and normalized with loading control actin. For TUNEL assay, HL-1 cells were fixed with formalin and then analyzed for apoptosis with the DeadEnd Fluorometric TUNEL System (Promega, Madison, WI) following the manufacturer’s instructions. The apoptotic nuclei were stained with fluorescein-labeled dUTP. In every experiment, 10 fields were randomly recorded for every condition. Both the TUNEL-positive nuclei and total nuclei were counted, and the results are shown as a percent of nuclei that stained positive for TUNEL.
Immunoprecipitations and Western Analysis.
To examine the interactions between RSK1 and PKA subunits, HL-1 cells were washed twice with ice-cold PBS and scraped into lysis buffer [20 mM Hepes, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 2.5 mM MgCl2, 0.1 mM ATP, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, and protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN)]. Cell lysates were cleared by centrifugation at 20,000g for 15 minutes. The supernatants (500 μg of protein) were incubated for 2 hours at 4°C with 2 μg of anti-PKAc antibody together with 15 μl of protein G–conjugated agarose beads. After three thorough washes with lysis buffer, proteins in the immunoprecipitates were eluted with Laemmli sample buffer and subjected to SDS-PAGE for Western analyses.
To detect RSK1 phosphorylation, HL-1 cells that had been exposed to H/R were lysed in a buffer containing 50 mM Hepes, pH 7.4, 1% Triton X-100, 150 mM NaCl, 1 mM dithiothreitol, 1 mM sodium ortho-vanadate, 10 mM NaF, 5 mM sodium pyrophosphate, 10 mM β-glycerophosphate, 0.1 mM phenylmethylsulfonyl fluoride, and 1 μg/ml each of pepstatin A, aprotinin, and leupeptin. RSK1 was immunoprecipitated with anti-RSK1 antibody as described, and the phosphorylation status of RSK1 was monitored using anti–phospho-Ser-221 RSK1 antibody.
To monitor phosphorylation of NHE1 by RSK1, we took advantage of the findings that the RSK phosphorylation site Ser703 on NHE1 forms a 14-3-3 protein binding motif and can be detected in immunoprecipitated NHE1 using the anti-14-3-3 motif binding antibody (Garciarena et al., 2009; Lucien et al., 2011). Essentially, cells were lysed in buffer (pH 7.4) containing 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium ortho-vanadate, 50 mM NaF, 1% Triton X-100, 0.1% SDS, and protease inhibitors. The cell lysates were immunoprecipitated with anti-NHE1 antibody, and then the phospho-NHE1 in the immunoprecipitate was detected by Western blotting with anti–14-3-3 binding motif antibody.
Results
H/R Decreases PKARIα Levels and Changes the Interactions of RSK1 with PKA Subunits.
To mimic the ischemia and reperfusion-mediated cardiomyocyte apoptosis observed in human hearts after ischemia reperfusion (Kang and Izumo, 2000), we exposed ARVMs and HL-1 cells (Fig. 1) to either normoxia (controls) or hypoxia (1% O2) for 24 hours, followed by reoxygenation for 1 hour. In ARVM and HL-1 cells, H/R increased the levels of cleaved caspase 7 by 19.2- and 5-fold, respectively, and also elevated cleaved PARP levels by 18.2- and 7.5-fold, respectively (Fig. 1, A and B). Thus, HL-1 cells and ARVMs underwent significant apoptosis after H/R. Consistently, as monitored by the TUNEL assay, H/R increased apoptosis by 3-fold in HL-1 cells (Fig. 1C). Additionally, in both ARVM and HL-1 cells, H/R resulted in a significant (57%) decrease in the levels of PKARIα protein without altering the levels of PKAc or RSK1 (Fig. 1A; Supplemental Fig. 1). Because we previously observed that decreases in PKARIα levels are accompanied by activation of RSK1 (Chaturvedi et al., 2009), we immunoprecipitated RSK1 and monitored the phosphorylation status on Ser221, which is the final phosphorylation on RSK1 that fully activates the enzyme (Jensen et al., 1999). As shown in Fig. 1D, lower panel, H/R enhanced the phosphorylation of RSK1 on Ser221. Likewise, in ARVM H/R increased the phosphorylation of Ser732 on RSK1 by nearly 9-fold (Fig. 1D, top panel). Ser732 on RSK1 is autophosphorylated by the fully active RSK1 (Smith et al., 1999; Roux et al., 2003) and, therefore, is indicative of RSK1 activity. These findings demonstrate that in ARVM and HL-1 cells, H/R decreases PKARIα levels and also activates RSK1. Using B82L and HeLa cells, we previously showed that RSK1, via its interactions with PKA subunits, is indirectly associated with AKAPs, such as dual-specificity AKAP1, which also bind the catalytic subunit of protein phosphatase 2A (PP2A) (Chaturvedi et al., 2009). Disruption of RSK1 interactions with PKA subunits, as, for example, by decreasing PKARIα levels, removes RSK1 from proximity to PP2A, which dephosphorylates RSK1 and leads to activation of RSK1 (Chaturvedi et al., 2009; Gao et al., 2010). Since H/R decreases PKARIα levels and because RSK1 has been implicated in cardiac apoptosis (Maekawa et al., 2006), we determined whether the interactions of RSK1 with PKA subunits are altered by H/R. In control HL-1 cells grown in normoxia, immunoprecipitates of PKAc contained RSK1 together with PKARIα (Fig. 1E). However, after H/R, less PKARIα and inactive RSK1 were present in complex with PKAc and more of the phosphorylated, active RSK1 was present (Fig. 1E). These results are consistent with our previous finding in other cell types in that inactive RSK1 binds to PKARIα and the active RSK1 binds PKAc (Chaturvedi et al., 2006, 2009; Gao and Patel, 2009; Gao et al., 2010). Thus, when PKARIα is decreased and RSK1 is activated after H/R, less of the PKARIα is present in the PKAc immunocomplex and more of the active RSK1 is bound to PKAc. Thus, consistent with our findings by silencing of PKARIα in B82L (Chaturvedi et al., 2009), a decrease in PKARIα levels in HL-1 cells by H/R activates RSK1 and alters its interactions with the PKA subunits.
Fig. 1.

H/R-mediated cardiomyocyte apoptosis is accompanied by a decrease in PKARIα and an increase in RSK1 activity. ARVMs (A) or mouse cardiac myocyte HL-1 cells (B) were exposed to normoxia or hypoxia (1% O2) for 24 hours, followed by reoxygenation for 1 hour (H/R). Cell lysates were subjected to Western analysis for protein expression. (C) HL-1 cells were exposed to normoxia or H/R as in B and were analyzed with TUNEL assay for apoptosis. Apoptotic nuclei are shown as green. The number of TUNEL-positive nuclei were counted and presented as percent of total nuclei. Scale bar: 20 μm. (D). H/R results in RSK1 phosphorylation. Upper panel is Western analysis of the whole-cell lysates from ARVMs; lower panel is Western blotting of immunoprecipitated RSK1 from HL-1 cell lysates probed with anti–phospho-S221 RSK antibody. (E) RSK1 interacts with PKA subunits, and exposure to H/R alters the interaction pattern in HL-1 cells. Cells were exposed to normoxia or H/R as in B and then harvested for immunoprecipitation with anti-PKAc antibody. N, normoxia; IP, immunoprecipitate; WCL, whole-cell lysate. The panels with bar graphs in A–D are quantification of data. Data shown are means ± S.E.M. from at least three independent experiments. *P < 0.05. Protein expression levels are normalized for loading control actin. (E) Representative of three similar experiments.
Decrease in PKARIα Leads to RSK1 Activation and Apoptosis.
To determine whether the decrease in PKARIα observed after H/R contributes to RSK1 activation and induction of apoptosis, we silenced PKARIα in normoxic HL-1 cells. As shown in Fig. 2, siRNA against PKARIα effectively decreased PKARIα expression by 74% without altering the expression of PKAc or RSK1 (see quantifications of controls in Supplemental Fig. 2). Additionally, as determined by phosphorylation of Ser732, the site that active RSK1 autophosphorylates (Smith et al., 1999; Roux et al., 2003) silencing of PKARIα increased the amounts of active, phospho-S732 RSK1 by 22-fold (Fig. 2, A and B). Moreover, similar to the observations after H/R, PKARIα silencing increased the amounts of cleaved PARP and cleaved caspase 7 by 10- and 3-fold, respectively (Fig. 2, A and B). Thus, the decrease in PKARIα levels is sufficient to activate RSK1 and induce apoptosis in HL-1 cells.
Fig. 2.
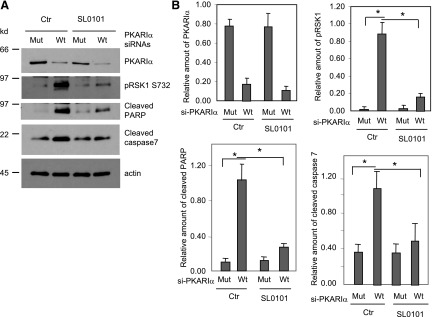
Silencing of PKARIα in HL-1 cells induces RSK1-mediated apoptosis. (A) 48 hours after transfection with mutant (Mut) or Wt siRNA against PKARIα, HL-1 cells were serum starved and incubated with or without SL0101 (50 μM) for 24 hours. Cell lysates were subjected to Western analysis. (B) Quantitative data from Western blots of four different experiments normalized for actin (loading control). Data shown are means ± S.E.M. *P < 0.05. Ctr, control.
Previously, we showed that inactive RSK1 binds to the pseudosubstrate region of PKARIα and that a cell-permeable peptide corresponding to this region can disrupt the interactions between endogenous RSK1 and PKARIα in intact cells (Gao et al., 2010). Therefore, as an alternate strategy to decrease PKARIα/RSK1 interactions, we treated normoxic HL-1 cells with the palmitoylated, cell-permeable peptide corresponding to the pseudosubstrate region of PKARIα (peptide Wt-PS, sequence: KGRRRRGAI) (Gao et al., 2010). A mutant palmitoylated, cell-permeable, control peptide (Mut-PS, sequence: KGAARRGAI) does not interfere with the interactions between endogenous RSK1 and PKARIα (Gao et al., 2010). Compared with Mut-PS, transduction of Wt-PS into HL-1 cells increased the amounts of active, phospho-S732 RSK1 by 14-fold (Fig. 3, A and B) without altering the total levels of RSK1, PKARIα or PKAc (Fig. 3A). Moreover, peptide Wt-PS increased cell apoptosis as monitored by 8- and 3-fold increases in amounts of cleaved PARP and cleaved caspase 7, respectively (Fig. 3, A and B). Notably, Wt-PS and Mut-PS did not alter the levels of phospho-PKA substrates, indicating that at the concentrations used, these peptides do not alter interactions between PKARIα and PKAc (Fig. 3, A and B). These data demonstrate that the disruption of PKARIα/RSK1 interactions in HL-1 cells is sufficient to increase the activity of RSK1 and induce apoptosis.
Fig. 3.
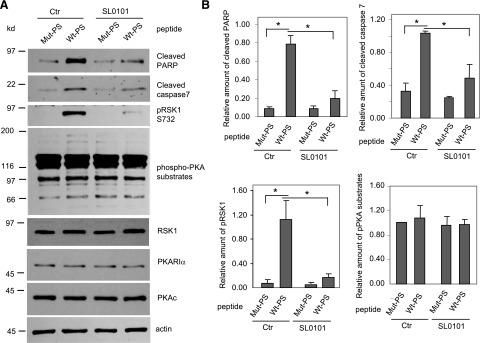
A cell-permeable peptide (Wt-PS) that disrupts interactions between PKARIα and RSK1 activates RSK1 and induces apoptosis in HL-1 cardiomyocytes. (A) Serum-starved HL-1 cells were incubated with 2 μM of the peptide corresponding to the pseudosubstrate region of PKARIα (Wt-PS) or the same peptide-harboring mutations (Mut-PS) either in the presence or absence of SL0101 (50 μM) for 24 hours. Cell lysates were subjected to Western analysis for the various indicated proteins as well as phospho-S732 RSK1 and phospho-PKA substrates. (B) Quantification of data from Western blots of three different experiments normalized for actin (loading control). For phospho-PKA substrate, the 66-kDa band was quantified. Data shown are means ± S.E.M. *P < 0.05. Ctr, control.
RSK1 Activation Induces Cardiomyocyte Apoptosis.
We next examined the role of active RSK1 in apoptosis that is induced by silencing of PKARIα, by disruption of PKARIα/RSK1 interactions, or by H/R. First, we used the RSK inhibitor SL0101 (Smith et al., 2005) to determine the role of the activated RSK in apoptosis induced by silencing of PKARIα or by the Wt-PS peptide, which disrupts PKARIα/RSK1 interactions. As evident by the decreased phospho-Ser732 RSK1 levels (Figs. 2, A and B, and 3, A and B), SL-0101 attenuated PKARIα siRNA- or Wt-PS–mediated activation of RSK1 by 88%. Additionally, in PKARIα siRNA- or Wt-PS–treated cells, SL0101 significantly decreased the levels of cleaved PARP by 70% (Fig. 2B) and 75% (Fig. 3B), respectively. Likewise, SL0101 also decreased cleaved caspase 7 by 55% (Fig. 2B) and 52% (Fig. 3B) in PKARIα siRNA– or Wt-PS–treated cells, respectively. Additionally, the RSK inhibitor SL0101 reduced H/R-induced activation of RSK1 (phospho-S732 RSK1) in ARVMs and HL-1 cells by 75 and 69%, respectively (Fig. 4, A and B). The inhibition of RSK1 in AVRM and HL-1 cells was accompanied by decreased levels of cleaved caspase 7 (70 and 50.4%, respectively) and cleaved PARP (71 and 69.3%, respectively) (Fig. 4, A and B). Notably, the RSK inhibitor SL0101, by itself, did not alter the levels of RSK1, PKARIα PKAc in ARVMs (Supplemental Fig. 3).
Fig. 4.
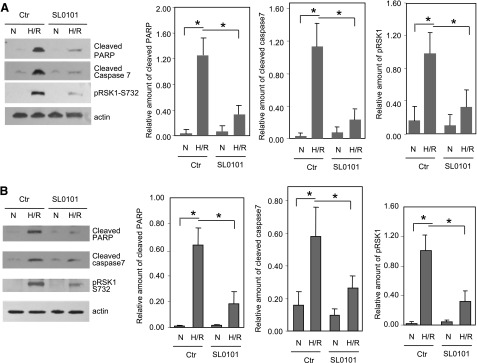
H/R-induced apoptosis is attenuated by RSK1 inhibitor SL0101. ARVMs (A) or HL-1 cells (B) were treated with or without SL0101 (50 μM) and exposed to normoxia or hypoxia (1% O2) for 24 hours, followed by reoxygenation for 1 hour. Cell lysates were subjected to Western analysis. Panels with bar graphs are quantitative data from Western blots of five (for ARVMs) or four (for HL-1) different experiments normalized for actin (loading control). Data shown are means ± S.E.M. *P < 0.05. Ctr, control; N, normoxia.
Since SL-0101 inhibits all isoforms of RSK, to determine whether RSK1 plays a role in cardiomyocyte apoptosis, we transfected ARVMs and HL-1 cells with mutant or wild-type siRNA against RSK1. Silencing of RSK1 with siRNA decreased the total amounts of RSK1 in ARVMs by 86% and in HL-1 cells by 77% (Fig. 5, A and B), without altering PKAc or PKARIα levels (Supplemental Fig. 4, A and B). H/R decreased PKARIα levels in ARVM and HL-1 cells in the presence of control or RSK1-specific siRNA (Supplemental Fig. 4). However, RSK1-specific siRNA decreased cleaved caspase 7 levels by 60% in both cell types, decreased cleaved PARP levels by 51% in ARVM, and by 60% in HL-1 cells; it also attenuated the number of TUNEL-positive cells by 46% (Fig. 5, A–C). These data (Fig. 5) show that in ARVM and HL-1 cells, RSK1 mediates H/R-induced apoptosis.
Fig. 5.

H/R-induced apoptosis is attenuated by RSK1 specific siRNA. ARVMs (A) or HL-1 cells (B and C) were transfected with RSK1-specific siRNA or its mutant (Mut). Cells were subjected to normoxia or hypoxia (1% O2) for 24 hours, followed by reoxygenation for 1 hour. Apoptosis was analyzed by Western blotting (A and B) or TUNEL assay (C). (A and B) Western analysis for the indicated proteins. Panels with bar graphs are quantitative data on Western blots from four different experiments normalized for loading control actin. (C) TUNEL assay. Apoptotic nuclei were stained green. Scale bar: 20 μm. The number of apoptotic nuclei were counted and presented as a percent of total nuclei. Results from three independent experiments were analyzed. Data shown are means ± S.E.M. *P < 0.05. DAPI, 4′,6-diamidino-2-phenylindole; N, normoxia.
Elevated PKA Activity Does Not Contribute toward H/R-Induced Cardiomyocyte Apoptosis.
Since H/R decreases PKARIα levels, it would be expected that PKAc would be dissociated from PKARIα and is, therefore, activated. Presently, the role of PKA in cardiomyocyte apoptosis remains unclear. Studies have reported that PKA plays both an apoptotic and an antiapoptotic role (Valks et al., 2002; Cieslak and Lazou, 2007). Therefore, we examined whether PKA activity is altered after H/R and whether PKA contributes to cardiomyocyte apoptosis. We treated HL-1 cells with PKA inhibitor H89 during the course of H/R. Figure 6 shows that, indeed, H/R increased PKA activity in HL-1 cells as indicated by 2.5-fold increase in phospho-PKA substrates (Fig. 6, A and B). As shown by a 72% decrease in phospho-PKA substrates (Fig. 6, A and B), H89 treatment significantly decreased elevation in PKA activity after H/R. However, the levels of the apoptosis markers, cleaved PARP and cleaved caspase 7, remained unaltered in the presence or absence of H89 (Fig. 6, A and B). These data suggest that although PKA is activated by H/R, PKA activation is not involved in the H/R-induced cardiomyocyte apoptosis. H89 is reported to have nonspecific effects on kinases other than PKA. Therefore, we determined whether H89 altered H/R-mediated activation of RSK1. As monitored by RSK1 phosphorylation on S732, the site that is autophosphorylated by the fully active RSK1, H89 did not alter H/R-induced RSK1 activation (Fig. 6, A and B). Thus, H/R-induced apoptosis in HL-1 cells that results from decreased PKARIα levels (Figs. 1 and 2) is not caused by elevations in PKA activity.
Fig. 6.
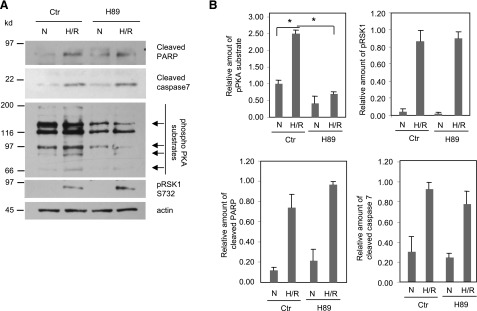
Elevated PKA activity does not contribute toward the H/R-induced apoptosis in cardiomyocytes. (A) After serum starvation, HL-1 cells were treated with or without H89 (10 μM) and exposed to normoxia or hypoxia (1% O2) for 24 hours, followed by reoxygenation for 1 hour. Cell lysates were subjected to Western analysis for the indicated proteins. Arrows depict the changes in intensity of the PKA substrate bands. (B) Quantitative data from Western blots of three different experiments normalized for actin (loading control). To quantify the amount of phospho-PKA substrates, the 66-kDa PKA substrate bands were scanned and normalized for actin. Data shown are means ± S.E.M. *P < 0.05. Ctr, control; N, normoxia.
RSK1 Induces Apoptosis via Phosphorylation of NHE1.
Previous studies have shown that RSK (isoform unknown) can phosphorylate NHE1 on Ser703 and increase its activity (Phan et al., 1997; Moor and Fliegel, 1999; Takahashi et al., 1999). Additionally, studies have shown that phosphorylated NHE1 can induce cardiomyocyte apoptosis (Garciarena et al., 2009). Therefore, we investigated whether NHE1 is phosphorylated by RSK1 and is involved in RSK1-mediated apoptosis by H/R. First, we determined whether NHE1 is phosphorylated by RSK1 after H/R. As shown in Fig. 7A, NHE1 was phosphorylated in H/R, and when RSK1 expression was silenced by RSK1-specific siRNA, the phosphorylation of NHE1 was significantly decreased by 61%, demonstrating that RSK1 phosphorylates NHE1 after H/R. Next, to determine whether NHE1 mediated H/R-induced apoptosis in ARVM and HL-1 cells, we incubated the cells with the NHE1 inhibitor EIPA. As shown in Fig. 7, B and C, in ARVM and HL-1 cells, EIPA decreased amounts of cleaved PARP by 70 and 58%, respectively, and also attenuated levels of cleaved caspase 7 by 78 and 59%, respectively. Thus, inhibition of NHE1 attenuates HR-induced apoptosis in ARVM and HL-1 cells. Moreover, the results in Fig. 7 suggest that RSK1-mediated phosphorylation and activation of NHE1 result in the apoptosis of cardiomyocytes observed after H/R.
Fig. 7.

NHE1 mediates cardiomyocyte apoptosis in H/R. (A) NHE1 is phosphorylated in HL-1 cells in H/R and the silenting of RSK1 blocks NHE1 phosphorylation. HL-1 cells were transfected with RSK1-specific siRNA or its mutant (Mut). After serum starvation, cells were subjected to normoxia or hypoxia (1% O2) for 24 hours, followed by reoxygenation for 1 hour. Cell lysates were immunoprecipitated with anti-NHE1 antibody. The NHE1 phosphorylation was then examined with anti–phospho-14-3-3 binding motif antibody. (B and C) NHE1 inhibitor EIPA attenuates apoptosis in HL-1 cells (B) or ARVMs (C) in H/R. EIPA (5 μM) or vehicle was added to the cells during the last 5 hours of normoxia or H/R periods. Cell lysates were subjected to Western analysis. Panels on the right show quantification of Western blots from three (A) or four (B and C) different experiments were quantified and normalized for actin (loading control). Data shown are means ± S.E.M. *P < 0.05. Ctr, control; N, normoxia.
Discussion
For a long time, it was thought that cardiomyocytes do not undergo apoptosis or proliferation. However, pioneering work in these areas from the Anversa laboratory and others showed that apoptosis and proliferation are essential features of cardiomyocytes both under normal and pathologic conditions (Anversa et al., 2006; Kajstura et al., 2010). Hence, it is estimated that between the ages of 20 and 100 in men and women, cardiomyocytes turn over 11–15 times (Kajstura et al., 2010). The rate of cardiomyocyte apoptosis is enhanced after myocardial infarcts or ischemia-reperfusion of the myocardium, and because the proliferation rate of cardiomyocytes does not keep pace with apoptosis under these conditions, fibroblasts replace the necrotic myocardium, resulting in fibrosis and decreased cardiac function (Kang and Izumo, 2000; Kajstura et al., 2010). Thus, an understanding of the precise mechanisms that contribute toward cardiomyocyte apoptosis is essential to curtail this event in cardiac pathology.
Using ARVMs and HL-1 cells, in this report, we provide evidence for a new paradigm that contributes to cardiomyocyte apoptosis after (H/R). This paradigm involves an H/R-mediated decrease in PKARIα protein level that leads to activation of RSK1 and, ultimately, RSK1-mediated phosphorylation of NHE1, which induces apoptosis of cardiomyocytes. The following experimental evidence supports this novel mode of regulation. First, a reduction of endogenous PKARIα by siRNA mimics the actions of H/R in the activation of RSK1 as well as induction of apoptosis (i.e., increased cleaved PARP and cleaved caspase 7 levels) (Fig. 2). Second, disruption of the PKARIα/RSK1 interactions using the cell-permeable peptide (Wt-PS) also results in activation of RSK1 and induction of apoptosis as observed after H/R (Fig. 3). Thus, our data demonstrate that either a decrease in levels of PKARIα or dissociation of PKARIα from RSK1 is sufficient to activate RSK1 and induce apoptosis in cardiomyoctes. Although H/R, by reducing the amounts of PKARIα, elevates PKAc activity, our data show that H/R-induced cardiomyocyte apoptosis is not the result of increase in PKAc activity.
Upon reduction of PKARIα levels or disruption of PKARIα/RSK1 interactions, RSK1 is activated because it is no longer tethered (via PKARIα) with AKAPs, such as dual-specificity AKAP1, which also binds the catalytic subunit of PP2A, the phosphatase that dephosphorylates and inactivates RSK1 (Chaturvedi et al., 2009). The necessity for elevations in RSK1 activity to induce cardiomyocyte apoptosis is supported by our findings that the RSK inhibitor and siRNA targeted specifically against RSK1 attenuate NHE1 phosphorylation, as well as apoptosis after H/R. Notably, we have previously shown that the siRNA used in these experiments does not alter the levels of RSK2 or RSK3 and that neither RSK2 nor RSK3 interact with PKARIα (Chaturvedi et al., 2006; Gao and Patel, 2009).
Our findings that activation of RSK1 is necessary to induce apoptosis in cardiomyocytes cells differs from findings in previous reports from our laboratory, as well as from those of others for noncontractile cells in which RSK1, by phosphorylating the apoptotic protein Bcl-xL/Bcl-2–associated death promoter, dissociates it from Bcl-2 and inhibits apoptosis (Shimamura et al., 2000; Chaturvedi et al., 2009). Thus, RSK1 may prevent or enhance apoptosis in a cell type–specific manner, depending on the apoptotic stimuli and major apoptotic or antiapoptotic substrates of RSK1 expressed in a given cell type. In this study, we found that in HL-1 cells, RSK1 phosphorylates NHE1 and NHE1 mediates the proapoptotic effects of RSK1 in HL-1 cardiomyocytes after H/R. Thus, inhibition of NHE1 might help alleviate the cardiac muscle damage from ischemia and reperfusion.
RSK1 regulates the transcription of a number of genes and, by phosphorylating tuberous sclerosis complex 2, activates mTOR and protein translational machinery. Therefore, it has been suggested that RSK1 plays an important role in the onset of cardiac hypertrophy (Proud, 2004; Rolfe et al., 2005). However, the role of RSK1 in cardiac hypertrophy has been challenged (Fonseca et al., 2011), and a recent study has shown that RSK3 plays an important role in cardiac hypertrophy (Li et al., 2013). Regions of the hypertrophied heart are also exposed to hypoxic conditions resulting from overgrowth of cardiac tissue and insufficient blood supply (Kang and Izumo, 2000; Fortuno et al., 2001), and the chronic hypoxic condition can induce cardiomyocyte apoptosis and the loss of cells. Our findings presented here suggest a direct role of RSK1 in augmenting apoptosis. Thus, it is possible that a combination of RSK isoforms contributes to the pathology of the hypertrophied heart and muscle damage. In this context, blockade of RSK1 and RSK3 would be very beneficial in attenuating cardiomyocyte apoptosis and cardiac hypertrophy.
In conclusion, the studies presented here demonstrate that H/R of cardiac myocytes results in decreased PKARIα levels with a resultant activation of RSK1. The active RSK1 via phosphorylation of NHE1 contributes to onset of apoptosis. The evidence presented here also demonstrates the importance of the RSK1 interactions with PKA subunits in regulating apoptosis in the heart. In this context, therapeutic strategies to preserve PKARIα protein levels or retain RSK1/PKARIα interactions after H/R would be beneficial in preventing cardiac muscle damage.
Supplementary Material
Acknowledgments
The authors Drs. Xiang Ji (Loyola University Chicago) for help with the isolation of ARVMs and Dr. William Claycomb (Louisiana State University Health Science Center) for the HL-1 cell line.
Abbreviations
- AKAP
PKA–anchoring proteins
- ARVMs
adult rat ventricular myocytes
- EIPA
5′-N-ethyl-N-isopropyl-amiloride
- H/R
hypoxia/reoxygenation
- H89
N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide
- NHE1
sodium–hydrogen exchanger 1
- PGE2
prostaglandin E2
- PKA
protein kinase A
- PKAc
catalytic subunit of PKA
- PKAR
PKA regulatory subunit
- PP2A
protein phosphatase 2A
- RSK
p90 ribosomal S6 kinase
- siRNA
small interfering RNA
- SLO101
kaempferol-3-O-(3″,4″-di-O-acetyl-α-L-rhamnopyranoside)
- TUNEL
terminal dUTP nick end labeling
Authorship Contributions
Participated in research design: Patel, Gao.
Conducted experiments: Gao, Lin, Sadayappan.
Performed data analysis: Patel, Gao.
Wrote or contributed to the writing of the manuscript: Patel, Gao.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM079226]; and the National Institutes of Health National Heart, Lung, and Blood Institute [Grants R01-HL105826 and K02-HL114749].
 This article has supplemental material available at mol.pharm.aspetjournals.org.
This article has supplemental material available at mol.pharm.aspetjournals.org.
References
- Anjum R, Blenis J. (2008) The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol 9:747–758 [DOI] [PubMed] [Google Scholar]
- Anversa P, Kajstura J, Leri A, Bolli R. (2006) Life and death of cardiac stem cells: a paradigm shift in cardiac biology. Circulation 113:1451–1463 [DOI] [PubMed] [Google Scholar]
- Cargnello M, Roux PP. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75:50–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi D, Cohen MS, Taunton J, Patel TB. (2009) The PKARIalpha subunit of protein kinase A modulates the activation of p90RSK1 and its function. J Biol Chem 284:23670–23681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi D, Poppleton HM, Stringfield T, Barbier A, Patel TB. (2006) Subcellular localization and biological actions of activated RSK1 are determined by its interactions with subunits of cyclic AMP-dependent protein kinase. Mol Cell Biol 26:4586–4600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslak D, Lazou A. (2007) Regulation of BAD protein by PKA, PKCdelta and phosphatases in adult rat cardiac myocytes subjected to oxidative stress. Mol Cells 24:224–231 [PubMed] [Google Scholar]
- Døskeland SO, Maronde E, Gjertsen BT. (1993) The genetic subtypes of cAMP-dependent protein kinase: functionally different or redundant? Biochim Biophys Acta 1178:249–258 [DOI] [PubMed] [Google Scholar]
- Dümmler BA, Hauge C, Silber J, Yntema HG, Kruse LS, Kofoed B, Hemmings BA, Alessi DR, Frödin M. (2005) Functional characterization of human RSK4, a new 90-kDa ribosomal S6 kinase, reveals constitutive activation in most cell types. J Biol Chem 280:13304–13314 [DOI] [PubMed] [Google Scholar]
- Enns LC, Bible KL, Emond MJ, Ladiges WC. (2010) Mice lacking the Cβ subunit of PKA are resistant to angiotensin II-induced cardiac hypertrophy and dysfunction. BMC Res Notes 3:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca BD, Alain T, Finestone LK, Huang BP, Rolfe M, Jiang T, Yao Z, Hernandez G, Bennett CF, Proud CG. (2011) Pharmacological and genetic evaluation of proposed roles of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK), extracellular signal-regulated kinase (ERK), and p90(RSK) in the control of mTORC1 protein signaling by phorbol esters. J Biol Chem 286:27111–27122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortuño MA, Ravassa S, Fortuño A, Zalba G, Díez J. (2001) Cardiomyocyte apoptotic cell death in arterial hypertension: mechanisms and potential management. Hypertension 38:1406–1412 [DOI] [PubMed] [Google Scholar]
- Frödin M, Gammeltoft S. (1999) Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol 151:65–77 [DOI] [PubMed] [Google Scholar]
- Gao X, Chaturvedi D, Patel TB. (2010) p90 ribosomal S6 kinase 1 (RSK1) and the catalytic subunit of protein kinase A (PKA) compete for binding the pseudosubstrate region of PKAR1alpha: role in the regulation of PKA and RSK1 activities. J Biol Chem 285:6970–6979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Patel TB. (2009) Regulation of protein kinase A activity by p90 ribosomal S6 kinase 1. J Biol Chem 284:33070–33078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garciarena CD, Caldiz CI, Portiansky EL, Chiappe de Cingolani GE, Ennis IL. (2009) Chronic NHE-1 blockade induces an antiapoptotic effect in the hypertrophied heart. J Appl Physiol (1985) 106:1325–1331 [DOI] [PubMed] [Google Scholar]
- Guggilam A, Hutchinson KR, West TA, Kelly AP, Galantowicz ML, Davidoff AJ, Sadayappan S, Lucchesi PA. (2013) In vivo and in vitro cardiac responses to beta-adrenergic stimulation in volume-overload heart failure. J Mol Cell Cardiol 57:47–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Harding P, LaPointe MC. (2010) PKA, Rap1, ERK1/2, and p90RSK mediate PGE2 and EP4 signaling in neonatal ventricular myocytes. Am J Physiol Heart Circ Physiol 298:H136–H143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen CJ, Buch MB, Krag TO, Hemmings BA, Gammeltoft S, Frödin M. (1999) 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J Biol Chem 274:27168–27176 [DOI] [PubMed] [Google Scholar]
- Kajstura J, Gurusamy N, Ogórek B, Goichberg P, Clavo-Rondon C, Hosoda T, D’Amario D, Bardelli S, Beltrami AP, Cesselli D, et al. (2010) Myocyte turnover in the aging human heart. Circ Res 107:1374–1386 [DOI] [PubMed] [Google Scholar]
- Kang PM, Izumo S. (2000) Apoptosis and heart failure: a critical review of the literature. Circ Res 86:1107–1113 [DOI] [PubMed] [Google Scholar]
- Li J, Kritzer MD, Michel JJ, Le A, Thakur H, Gayanilo M, Passariello CL, Negro A, Danial JB, Oskouei B, et al. (2013) Anchored p90 ribosomal S6 kinase 3 is required for cardiac myocyte hypertrophy. Circ Res 112:128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucien F, Brochu-Gaudreau K, Arsenault D, Harper K, Dubois CM. (2011) Hypoxia-induced invadopodia formation involves activation of NHE-1 by the p90 ribosomal S6 kinase (p90RSK). PLoS ONE 6:e28851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa N, Abe J, Shishido T, Itoh S, Ding B, Sharma VK, Sheu SS, Blaxall BC, Berk BC. (2006) Inhibiting p90 ribosomal S6 kinase prevents (Na+)-H+ exchanger-mediated cardiac ischemia-reperfusion injury. Circulation 113:2516–2523 [DOI] [PubMed] [Google Scholar]
- Moor AN, Fliegel L. (1999) Protein kinase-mediated regulation of the Na(+)/H(+) exchanger in the rat myocardium by mitogen-activated protein kinase-dependent pathways. J Biol Chem 274:22985–22992 [DOI] [PubMed] [Google Scholar]
- Phan VN, Kusuhara M, Lucchesi PA, Berk BC. (1997) A 90-kD Na(+)-H+ exchanger kinase has increased activity in spontaneously hypertensive rat vascular smooth muscle cells. Hypertension 29:1265–1272 [DOI] [PubMed] [Google Scholar]
- Proud CG. (2004) Ras, PI3-kinase and mTOR signaling in cardiac hypertrophy. Cardiovasc Res 63:403–413 [DOI] [PubMed] [Google Scholar]
- Rolfe M, McLeod LE, Pratt PF, Proud CG. (2005) Activation of protein synthesis in cardiomyocytes by the hypertrophic agent phenylephrine requires the activation of ERK and involves phosphorylation of tuberous sclerosis complex 2 (TSC2). Biochem J 388:973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Richards SA, Blenis J. (2003) Phosphorylation of p90 ribosomal S6 kinase (RSK) regulates extracellular signal-regulated kinase docking and RSK activity. Mol Cell Biol 23:4796–4804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura A, Ballif BA, Richards SA, Blenis J. (2000) Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr Biol 10:127–135 [DOI] [PubMed] [Google Scholar]
- Silverman E, Frödin M, Gammeltoft S, Maller JL. (2004) Activation of p90 Rsk1 is sufficient for differentiation of PC12 cells. Mol Cell Biol 24:10573–10583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skålhegg BS, Taskén K. (1997) Specificity in the cAMP/PKA signaling pathway. differential expression, regulation, and subcellular localization of subunits of PKA. Front Biosci 2:d331–d342 [DOI] [PubMed] [Google Scholar]
- Smith JA, Poteet-Smith CE, Malarkey K, Sturgill TW. (1999) Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J Biol Chem 274:2893–2898 [DOI] [PubMed] [Google Scholar]
- Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM, Lannigan DA. (2005) Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res 65:1027–1034 [PubMed] [Google Scholar]
- Takahashi E, Abe J, Gallis B, Aebersold R, Spring DJ, Krebs EG, Berk BC. (1999) p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J Biol Chem 274:20206–20214 [DOI] [PubMed] [Google Scholar]
- Valks DM, Cook SA, Pham FH, Morrison PR, Clerk A, Sugden PH. (2002) Phenylephrine promotes phosphorylation of Bad in cardiac myocytes through the extracellular signal-regulated kinases 1/2 and protein kinase A. J Mol Cell Cardiol 34:749–763 [DOI] [PubMed] [Google Scholar]
- Wang A, Rud J, Olson CM, Jr, Anguita J, Osborne BA. (2009) Phosphorylation of Nur77 by the MEK-ERK-RSK cascade induces mitochondrial translocation and apoptosis in T cells. J Immunol 183:3268–3277 [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD. (2004) AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol 5:959–970 [DOI] [PubMed] [Google Scholar]
- Zeniou M, Ding T, Trivier E, Hanauer A. (2002) Expression analysis of RSK gene family members: the RSK2 gene, mutated in Coffin-Lowry syndrome, is prominently expressed in brain structures essential for cognitive function and learning. Hum Mol Genet 11:2929–2940 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.