

Abstract
Species C human adenovirus serotype 5 (HAdV-C5) is widely used as a vector for cancer gene therapy, because it efficiently transduces target cells. A variety of HAdV-C5 vectors have been developed and tested in vitro and in vivo for cancer gene therapy. While clinical trials with HAdV-C5 vectors resulted in effective responses in many cancer patients, administration of HAdV-C5 vectors to solid tumors showed responses in a limited area. A biological barrier in tumor mass is considered to hinder viral spread of HAdV-C5 vectors from infected cells. Therefore, efficient virus-spread from an infected tumor cell to surrounding tumor cells is required for successful cancer gene therapy. In this study, we compared HAdV-C5 to sixteen other HAdV serotypes selected from species A to G for virus-spread ability in vitro. HAdV-D9 showed better virus-spread ability than other serotypes, and its viral progeny were efficiently released from infected cells during viral replication. Although the HAdV-D9 fiber protein contains a binding site for coxsackie B virus and adenovirus receptor (CAR), HAdV-D9 showed expanded tropism for infection due to human CAR (hCAR)-independent attachment to target cells. HAdV-D9 infection effectively killed hCAR-negative cancer cells as well as hCAR-positive cancer cells. These results suggest that HADV-D9, with its better virus-spread ability, could have improved therapeutic efficacy in solid tumors compared to HAdV-C5.
Introduction
Cancer is a major public health problem in the United States. Approximately 1.64 million new cancer cases and approximately 580,000 deaths were predicted to occur in the United States in 2012 [1]. Given this, some local therapies (surgery and radiation therapy) and systemic therapies (chemotherapy, hormone therapy, and immunotherapy) have greatly reduced the mortality of cancer disease [1]. However, primary tumors will often relapse and subsequently metastasize to other organs [2]. These considerations warrant the development of new treatment strategies for cancer.
Cancer gene therapy is one feasible treatment that involves introducing genetic material into human cells to eradicate cancer [3]. Virotherapy in particular represents a promising new direction for cancer gene therapy [4], [5]. This approach utilizes viruses designed to specifically kill tumor cells but not normal cells [6], [7]. Because the biology of HAdV-C5 is relatively well characterized [8], it has emerged as one of the most attractive candidate virotherapy agents, which are called oncolytic HAdVs [4]. A number of strategies to specifically kill tumor cells are based on the interacting molecular mechanisms between adenoviral biology and cellular signaling pathways associated with human cancers [5]. A variety of oncolytic HAdVs have been tested in vitro [9]–[14] and caused effective responses in clinical trials [15]–[23]. On the other hand, oncolytic HAdVs utilizing HAdV-C5 showed responses in limited area [24]–[26]. As a biological barrier in tumor mass hinders virus spread from infected cells [12], [27], [28], we need to develop more efficacious oncolytic HAdVs.
A number of transcriptional targeting strategies using tumor-specific promoters have been tried to improve replication specificity to limit toxicity [29]–[31]. Numerous infectivity enhancement strategies were tested for targeting cancer cells with no human coxsackie B virus and adenovirus receptor (hCAR) as a primary receptor for HAdV-C5 [32], [33]. In addition, several HAdV serotypes were tested to examine antitumor efficacy to solid tumors by in vivo [34]. Thus, many types of advanced oncolytic HAdVs have been reported for cancer gene therapy. We need HAdVs with better virus-spread ability to overcome a biological barrier in tumor mass. However, strategies to enhance virus-spread ability of HAdV-C5 are limited [35], [36].
A total of over 50 serotypes of HAdVs, divided into species A to G, have been identified [37]. These wild type HAdVs produce different sizes of plaques on cells [38], thus, we hypothesized that HAdVs with better virus-spread ability can be identified from alternative serotypes by comparing the size of the plaques. We examined viral spread ability in vitro of sixteen HAdV serotypes by plaque assay as compared with that of HAdV-C5. In this study, we report the biological and physical properties of HAdVs in vitro, highlighting HAdV-D9 as a candidate for cancer gene therapy in solid tumor.
Materials and Methods
Cell Lines
Human embryonic kidney 293 (HEK293), human lung adenocarcinoma cell line A549, human ovarian adenocarcinoma cell lines SKOV-3 and OVCAR-3, human pancreatic carcinoma cell lines BxPC-3 and MIA-PaCa-2, human breast cancer cell lines AU-565, MCF-7 and ZR-75-1, human malignant mesothelioma cell lines H2052, H2452 and MSTO-211H, human prostate cancer cell line PC-3, were obtained from the American Type Culture Collection (ATCC; Manassas, VA). Human embryonic kidney 293A was obtained from Life Technologies (Carlsbad, CA, USA). Chinese hamster ovary (CHO) cells and CHO-hCAR cells stably expressing human CAR were provided by Jeffrey M. Bergelson [39]. All cells described above except AU-565, ZR-75-1, three kinds of malignant mesothelioma cell lines, CHO, and CHO-hCAR were cultured in Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 Ham, (DMEM/F12; Sigma-Aldrich, St. Louis, MO) containing 10% fetal bovine serum (FBS; Hyclone; Logan, UT), 2 mM L-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin (Mediatech, Inc., Herndon, VA). The other cell lines were maintained in culture medium recommended by each supplier. All cells were incubated at 37°C in an atmosphere of 5% CO2 in air. Also, infected cells were maintained with medium containing 2% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin.
Adenoviruses, Propagation, and Purification
Human adenoviruses (HAdVs), HAdV-C2, B3, E4, C5, D9, D10, B14, B16, D20, B21, A31, B34, B35, D37, F40, F41, and D51 were obtained from ATCC and were propagated in A549 or HEK293 cells. We purified HAdVs in ten T175-cm2 flasks of infected A549 or infected HEK293 cells by two rounds of cesium chloride (CsCl) gradient ultracentrifugation [40]. CsCl was removed by dialysis against phosphate-buffered saline (PBS [pH 7.4]) containing 10% glycerol. Purified HAdVs were stored at −80°C prior to experiments.
Titration
Plaque assay was performed to determine the infectious titer (plaque forming units; PFU/ml) of purified HAdVs on 293A cells or A549 cells [41]. The particle titer (viral particles [VP]/ml) was determined by A 260 absorbance of purified virus particles and by assuming that 1.1×1012 VP/ml has an absorbance of 1.0 at 260 nm [42]. In brief, the particle titer is calculated based on the protein amount of the purified adenoviruses observed at absorbance at 260 nm under denaturing conditions using SDS (an OD260 of 1.0 corresponds to 0.28 mg/ml protein in 0.1% [w/v] SDS buffer). A value of 1.77×108 daltons (Da) for the molecular mass of the VP is obtained by dividing 2.3×107 Da (the molecular mass of adenovirus DNA) by 0.13 because 13% of an adenoviral particle is DNA. Next, 1.54×108 Da, the molecular mass of the adenoviral proteins that compose the VP, is calculated by subtracting 2.3×107 Da (the molecular mass of the adenovirus DNA) from 1.77×108 Da (the molecular mass of the VP). Finally, 1.1×1012 particles/ml/OD260 unit is obtained by dividing 2.8×10−4 g/ml protein by 1.54×108 Da (the molecular mass of the adenoviral proteins) and multiplying it with Avogadro’s number (NA) (6.02×1023). In order to calculate the HAdV genome titer, we extracted the adenovirus genome from purified HAdV. In brief, 100 µl of purified adenovirus were mixed with sodium dodecyl sulfate (SDS, Sigma-Aldrich) and proteinase K (Sigma-Aldrich) at final concentrations of 0.1% (w/v) and 0.1 mg/ml, respectively. The mixture was incubated at 56°C for 1 hour and subsequently cooled at room temperature. Purified adenovirus genome was extracted by a standard method with phenol and chloroform [43] and dissolved with 100 µl of sterilized H2O. The titer of the adenovirus genome was calculated on the assumption that a solution of 50 µg/ml of purified DNA has an absorbance of 1.0 at 260 nm [44]. We then calculated the concentration of the adenoviral genome using the following formula: adenoviral genome titer at an absorbance of 1.0 at 260 nm = (5.0×10−5 g/ml)/molecular mass of the double stranded adenoviral genome×Avogadro’s number (NA) = 6.02×1023 mol−1 [44]. We summarized accession numbers of the HAdV genomes which were obtained from GenBank and each genome titer per 1OD at 260 nm in Table S1.
Measurement of Plaque Sizes
We performed plaque assay as described elsewhere [41] and measured the sizes of individual single plaques by Traceable Certificate of Calibration for Digital Caliper (Control Company, Friendswood, TX, USA). In brief, a solution of purified HAdVs was subjected to 10-fold serial dilution and A549 cells or 293A cells were infected with the diluted HAdVs in a six-well plate. After 1 hour post-infection, each solution of HAdVs was removed and 2 ml of fresh DMEM/F12 that contained 2% FBS and 0.75% BD Difco agar (BD Diagnostic Systems, Sparks, MD, USA) was laid over infected cells. Subsequently we incubated infected cells and laid over 2 ml of fresh DMEM/F12 that contained 2% FBS and 0.75% agar at 4 and 8 days post-infection. We culture infected cells another six days and laid over 2 ml of fresh DMEM/F12 that contained 2% FBS, 0.75% agar, and 0.033% Neutral red (Life Technologies, Carlsbad, CA, USA). We measured sizes of 10 individual single plaques which were visualized by neutral red staining and calculated the mean of plaque sizes. In cases of HAdV-B16, F40 and F41, we were not able to measure the sizes of plaques by a caliper due to their very small sizes. Therefore, we counted the number of plaques produced with these HAdVs on cells using an inverted IX-70 microscope (Olympus Corporation, Melville, NY) equipped with a DP71 digital camera (Olympus Corporation) and software of DP Controller and DP Manager (Olympus Corporation).
One-step Growth Curve Analysis of HAdVs
A549 cells were grown to 80% confluence in 6-well plates and the number of cells was counted to determine a multiplicity of infection (MOI). Cells were infected with HAdV at an MOI of 10 PFU/cell. Infected cells were maintained in 3 ml of medium containing 2% FBS and both infected cells and culture medium were harvested by using a cell scraper at various hours post-infection. We disrupted infected cells along with culture medium by three cycles of freeze and thaw and removed cell debris by centrifugation at 1,000×g for 5 minutes (min) at 4°C. The supernatant was used as the sample of whole HAdV contained in the fraction of both infected cells and culture medium. Additionally, we separated culture medium and infected cells by centrifugation at 1,000×g for 5 min at 4°C. The culture medium was used as a solution of HAdV which was released from infected cells. The infected cell pellet was resuspended in 2 ml of fresh medium and disrupted by three cycles of freeze and thaw. The lysates were centrifuged at 3,500×g for 10 min at 4°C and the supernatant was used as a sample of HAdV which was held in infected cells. Solutions of HAdVs prepared from each fraction were stored at −80°C prior to titration.
Cellular Uptake of HAdV
Cells were grown to 80% confluency in 24-well plate and the number of cells was counted before viral infection. We infected cells with HAdV at an MOI of 100 genomes/cell and maintained infected cells in 1 ml of medium for 30 or 60 min. After 30 or 60 min post-infection, infected cells were harvested and centrifuged 1,000×g for 5 min at 4°C. For a blocking experiment, CHO-hCAR cells were grown to 80% confluency in 24-well plate and removed medium. We incubated CHO-hCAR cells with 1 ml of FBS-free medium containing recombinant HAdV-C5 fiber knob protein [45] at a final concentration of 0.5, 5.0 or 50 µg/ml at 4°C for 1 hour. No blocking agent was added to the control wells. After 1 hour post-incubation, we removed medium and infected cells with HAdV-C5 or HAdV-D9 at an MOI of 100 genomes/cell for 1 hour. Subsequently, we harvested infected cells and medium, centrifuged 1,000×g for 5 min at 4°C, and removed supernatant. Total cell DNA containing adenoviral genome was extracted from infected cells by a method described previously [46] and stored at −80°C prior to quantitative PCR (qPCR) analysis.
Quantitative PCR (qPCR) Analysis
Position 1 refers to the left end of wild-type HAdV-C5 genome (GenBank accession number AC_000008.1), wild-type HAdV-D9 genome (accession number AJ854486.1). Oligonucleotides corresponding to the sense strand of the HAdV-C5 L4 region (5′-GAGCGCTCAGGAATCTTGC-3′: 25,576–25,594 nucleotide position [nt]), the antisense strand of the L4 region (5′-CGCGGTACTTAATGGGCAC-3′; 25,645–25,627 nt), the sense strand of the HAdV-D9 E3 region (5′-GTCCCATGGTGACTCTGCT-3′: 26,186–26,204 nt), the antisense strand of the E3 region (5′-CAGTGGTCCAGATGCCTCA-3′; 26,236–26,218 nt). The qPCR condition was as follows: 40 cycles of denaturation at 94°C for 15 seconds (sec) and annealing and extension at 60°C for 60 sec. Each virus genome extracted from purified HAdVs was used to generate a standard curve to calculate the adenoviral L4 or E3 DNA copy number. We calculated a value to convert one copy number of the HAdV-C5 genome to the copy number of the HAdV-D9 genome by using the standard curves of HAdV-genomes (Figure S1). As a result, one copy number of the HAdV-C5 genome is equivalent to 1.04 of the HAdV-D9. We normalized cellular uptakes of HAdVs in CHO and CHO-hCAR as a fold of genome transfer detected in HAdV-C5-infected CHO cells at 1 hour post-infection. Triplicates of analysis were performed with 50 ng of total DNA extracted from infected cells by using SYBR Select Master Mix (Life Technologies).
Flow Cytometry
Cells were cultured in a 75-cm2 tissue culture flask, harvested with PBS [pH 7.4] containing 0.53 mM EDTA, and washed with wash buffer (PBS [pH 7.4] containing 0.1% bovine serum albumin [BSA]). We incubated 5.0×104 cells with anti-human CAR monoclonal antibody RmcB [47] which was kindly provided from Dr. Douglas [48] for 1 hour. Following incubation, the cells were washed three times with wash buffer and incubated with Alexa Fluor 488-conjugated goat anti-mouse immunoglobulin G (Invitrogen). After two additional washing steps, the cells were analyzed using MACSQuant Analyzer (Milteryl Biotec Inc. Auburn, CA) or Becton Dickinson Fluorescence Activated Cell Sorter (FACS aria) (BD Biosciences, Franklin Lakes, NJ).
Two-dimensional Cell Viability Assay
Cells were seeded at a density of 5.0×103 cells per well in 96-well culture plates and infected with adenoviruses at MOIs of serial PFU/cell. We assessed cytopathic effect induced with HAdV infection at 6 days post-infection by using CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay Kit (Promega, Madison, WI) in accordance with the manufacture’s instruction. We measured the absorbance of the formazan product at 490 nm and the absorbance at 630 nm as a reference by a microplate reader (PowerWave HT 340, BioTek, Winooski, VT) and eliminated the value obtained at 630 nm as a background from that obtained at 490 nm. Cell killing activity induced with the HAdV infection was represented as relative value to uninfected cells by using GraphPad Prism 6 (GraphPad Software, Inc, La Jolla, CA).
Three-dimensional Cell Viability Assay
We measured cell killing activity of HAdVs in spheroids formed by Cultrex 3-D Spheroid Colorimetric Proliferation/Viability Assay (Trevigen, Inc. Gaithersburg, MD). In brief, cells were seeded at a density of 5.0×103 cells per well in a 3-D culture qualified 96-well spheroid formation plate, centrifuged at 200×g for 3 minutes at room temperature in a swinging bucket rotor. We incubated cells at 37°C in an atmosphere of 5% CO2 in air for 72 hours for spheroid formation. We counted cell numbers by trypsinizing spheroids and infected spheroids with adenovirus at various MOIs. We assessed cytopathic effect induced with HAdV infection at 12 days post-infection in accordance with the manufacture’s instruction. We measured the absorbance of the formazan product at 560 nm and the absorbance at 630 nm as a reference by PowerWave HT 340 microplate reader (BioTek) and eliminated the value obtained at 630 nm as a background from that obtained at 560 nm. Cell killing activity induced with the HAdV infection was represented as relative value to uninfected cells by using GraphPad Prism 6 (GraphPad Software).
Statistical Analysis
The data were expressed as mean+standard deviation (SD) or mean + standard error of the mean (SEM). Unpaired student t-test was used for the statistical analysis.
Results
Biological and Physical Properties of HAdVs
We used 16 serotypes (HAdV-C2, B3, E4, D9, D10, B14, B16, B21, D20, A31, B34, B35, D37, F40, F41, and D51) selected from species A to G in order to examine virus-spread ability as compared to HAdV-C5. We summarized cellular receptors for HAdVs which were suggested due to being experimentally examined by using some HAdVs in Table S2. We first propagated HAdVs in a human lung carcinoma cell line, A549 cells, which is a permissive cell line to propagate HAdVs. Of the tested HAdVs, 10 serotypes (HAdV-C2, B3, E4, D9, D10, A31, B34, B35, D37, and D51) as well as HAdV-C5 were propagated well in A549 cells, but 6 kinds of HAdVs (HAdV-B14, B16, B21, D20, F40, and F41) were not. In contrast, these HAdVs (HAdV-B14, B16, B21, D20, F40, and F41) were propagated well in HEK293 cells (data not shown). Both A549 and HEK293 cell lines express the tested HAdVs’ receptors [49], hCAR, human CD46 (hCD46), and αv-integrins [49]. Nevertheless, the propagation of 6 kinds of HAdVs (HAdV-B14, B16, B21, D20, F40, and F41) was restricted in A549 cells. Thus, these experimental observations suggested that 6 kinds of HAdVs propagated in HEK293 cells had host range diversity for productive infection. We purified HAdVs which were propagated in ten T175-cm2 flasks of A549 or HEK293 cells and measured biological and physical titers of purified HAdVs. Because 6 serotypes (HAdV-B14, B16, B21, D20, F40, and F41) were not propagated well in A549 cells, we used 293A cells to measure those infectious titers by plaque assay. We were able to count the number of plaques on A549 or 293A cells infected with most HAdVs by a caliper, but not able to count the plaques of HAdV-B16, F40, and F41 due to being very small sizes (data not shown). Therefore, we counted the numbers of plaques of these HAdVs observed by microscopy (Figures S2 and S3) and then calculated their infectious titers (Table 1). The infectious titers of most HAdVs were similar to that of HAdV-C5, but those of HAdV-B3, HAdV-B16, A31, F40, and F41 were 20- to 890-fold lower as compared to HAdV-C5 (Table 1). The genome titers of most HAdVs showed similar values to that of HAdV-C5, and the ratios of genomes to PFU in HAdVs except HAdV-B3, F40, and F41 were the range of 10 to 520 (Table 1). Moreover, the values of particle titers of most HAdVs were similar to that of HAdV-C5, but the particle titers of HAdV-B16 and F40 showed 10- to 40-fold lower values than that of HAdV-C5 (Table 1). Thus, the ratios of particles to PFU in HAdVs except HAdV-A31, B3, F40, and F41 were in the range of 10 to 360 (Table 1). The analyses of the biological and physical properties of HAdVs purified in this study were reproducible. Dr. Green et al. have reported that the ratios of particles to PFU of HAdV-C1 to D30 which were purified from infected KB cells were the ranges from 11∶1 to 2300∶1 [38]. Thus, we obtained similar ratios of particles to PFU in HAdVs except HAdV-B3 and D21 as compared with data reported by Dr. Green et al. [38]. Together, our results indicated that some HAdV serotypes have host range diversity and most HAdVs were effectively purified from A549 or 293 cells (Table 1).
Table 1. Summary of biological and physical titers of wild type HAdVs used in this study.
| Host cell line | Species | Serotypesa | PFU/ml | Total PFUb | Genomes/ml | Genomes/PFU | VP/ml | VP/PFU |
| A549 | A | 31 | 1.17×109±7.64×108 | 3.97×109 | 4.48×1011 | 384 | 2.26×1011 | 2,513 |
| B1 | 3 | 2.83×107±2.02×107 | 9.63×107 | 1.14×1012 | 40,305 | 3.79×1011 | 75,800 | |
| B2 | 34 | 7.17×109±1.26×109 | 3.50×1010 | 3.92×1011 | 55 | 4.09×1011 | 57 | |
| 35 | 6.00×109±5.00×108 | 3.00×1010 | 6.90×1011 | 115 | 1.90×1011 | 32 | ||
| C | 2 | 4.17×109±5.77×108 | 1.08×1010 | 2.27×1011 | 55 | 1.78×1011 | 43 | |
| 5 | 2.53×1010±1.53×109 | 1.01×1011 | 5.12×1011 | 20 | 1.46×1012 | 58 | ||
| D | 9 | 2.33×1010±1.76×1010 | 1.87×1011 | 4.64×1012 | 199 | 3.52×1012 | 151 | |
| 10 | 4.83×109±2.89×108 | 1.11×1010 | 2.49×1012 | 514 | 1.73×1012 | 357 | ||
| 37 | 2.03×1010±4.16×109 | 7.12×1010 | 1.37×1012 | 67 | 1.31×1012 | 65 | ||
| 51 | 9.00×109±1.00×109 | 6.75×1010 | 2.49×1012 | 277 | 2.38×1012 | 264 | ||
| E | 4 | 2.60×1010±2.60×109 | 1.56×1011 | 1.65×1012 | 64 | 1.49×1012 | 57 | |
| 293 | B1 | 16 | 5.00×108±5.00×108 | 3.75×109 | 1.46×1011 | 292 | 3.60×1010 | 72 |
| 21 | 6.00×1010±1.32×1010 | 3.24×1011 | 1.26×1012 | 21 | 7.63×1011 | 13 | ||
| B2 | 14 | 1.48×1010±6.33×109 | 1.30×1011 | 3.20×1012 | 216 | 2.28×1012 | 154 | |
| D | 20 | 9.33×1010±3.01×1010 | 9.33×1011 | 1.17×1012 | 13 | 9.74×1011 | 10 | |
| F | 40 | 8.50×107±5.63×107 | 2.30×108 | 1.28×1012 | 15,069 | 1.00×1011 | 1,278 | |
| 41 | 1.23×108±4.04×107 | 8.26×108 | 7.20×1011 | 5,837 | 5.05×1011 | 5,504 |
Numbers refer to the serotypes of HAdVs.
Total PFU was calculated with the volume of purified HAdVs.
HAdV-D9 and D51 have Better Virus-spread Ability than HAdV-C5
In order to develop new oncolytic HAdVs with better virus-spread ability than HAdV-C5, we require HAdVs which are capable of replicating well in tumors and efficiently spreading from infected cancer cells to surrounding cancer cells. Because each serotype produces different sizes of plaques on monolayer cells [38], plaque assay is suitable for assessing virus-spread ability in vitro [36], [38], [50], [51]. We performed plaque assay to identify HAdVs with better virus-spread ability and summarized the plaque sizes of HAdVs which were measured by a caliper in Table 2. HAdV-D9 (mean and standard deviation of the plaque size; 4.45±0.67 mm, n = 10) and HAdV-D51 (mean and standard deviation of the plaque size; 4.38±0.53 mm, n = 10) produced larger plaques on A549 cells than other serotypes including HAdV-C5 (Figure 1 and Table 2). Also, we observed clear plaque morphologies of HAdV-D9 and D51 with larger sizes by microscopy (Figure S2). On the other hand, the sizes of the plaques of HAdV-A31, HAdV-B3, B34, B35, D10, D37, and E4 on A549 cells were smaller than those of HAdV-C5 (Figure 1 and Table 2). As expected, we did not observe the plaques of HAdV-F41 on A549 cells (Figure 1). Although we tested plaque assay of HAdV-F41 at the range of dilution between 5.0×10−4 to 5.0×10−9 on A549 cells, we did not detect any plaques of HAdV-F41 on A549 cells by microscopy (data not shown). In contrast to A549 cells, we detected the plaques of HAdV-F41 on 293A cells (Figure S3). Because HAdV-F41 lacked virus-spread ability on A549 cells, it is not suitable for an oncolytic vector to solid tumors. Thus, our data demonstrated that HAdV-D9 and D51 have better virus-spread ability than HAdV-C5. Although HAdV-D51 as well as HAdV-D9 similarly killed in most cancer cell lines, cell killing activity by HAdV-D51 was restricted in pancreatic cell lines, BxPC-3 and MIA-PaCa-2 cells as compared to HAdV-C5 and D9 (Figure S4). While the nucleotide sequences for the HAdV-D9 genome as well as the HAdV-C5 genome were available from GenBank, the HAdV-D51 genome sequence was not determined yet. Since HAdV-D9 induced cell killing at low concentrations in broad ranges of cancer cell lines, we selected HAdV-D9 rather than HAdV-D51. We further analyzed the biological properties of HAdV-D9 with better virus-spread ability as compared to HAdV-C5.
Table 2. Host cell lines used for viral propagation and plaque sizes of HAdVs.
| Host cell line | Species | Serotypesa | Plaque sizeb (mm) |
| A549 | A | 31 | 0.50±0.58 |
| B1 | 3 | 0.78±0.38 | |
| B2 | 34 | 1.63±0.18 | |
| 35 | 1.45±0.52 | ||
| C | 2 | 2.86±1.36 | |
| 5 | 2.19±0.33 | ||
| D | 9 | 4.45±0.67 | |
| 10 | 0.73±0.59 | ||
| 37 | 1.13±0.32 | ||
| 51 | 4.38±0.53 | ||
| E | 4 | 1.58±0.30 | |
| HEK293 | B1 | 16 | unmeasurablec |
| 21 | 4.06±0.93 | ||
| B2 | 14 | 1.35±0.28 | |
| D | 20 | 4.52±0.51 | |
| F | 40 | unmeasurablec | |
| 41 | unmeasurablec |
Numbers refer to the serotypes of HAdVs.
Plaque sizes of HAdVs were represented as the average of the values obtained from ten individual single plaques.
Plaque sizes of these viruses were not able to be measured by a caliper.
Figure 1. Plaque morphology of HAdVs on A549 cells.
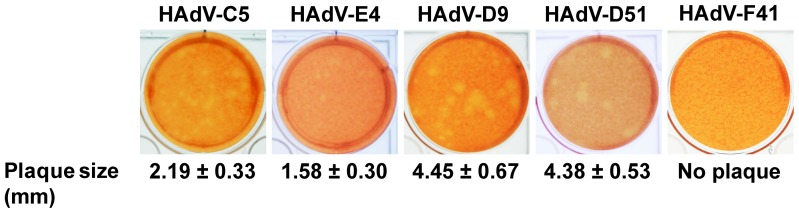
HAdVs (HAdV-C5, D9, D51, E4, and F41) were serially diluted with medium containing 2% FBS and A549 cells were infected with HAdV at the range of dilution of 5.0×10−8 and 5.0×10−9 for 1 hour. We performed plaque assay as described in the Materials and Methods section. At 14 days post-infection, we stained cells with 2 ml of medium containing 0.75% agar and 0.033% neutral red in order to visualize individual single plaques on A549 cells. Photographs of plaque morphology of HAdVs were taken with a digital camera.
HAdV-D9 is Efficiently Released from Infected Cells
The plaque sizes of HAdV-D9 on A549 cells were larger than those of HAdV-C5 (Figure 1 and Table 2). We supposed that the release of the HAdV-D9 progeny from infected cells is better than that of HAdV-C5. Therefore, we measured infectious titers of HAdVs which were released from infected cells to culture medium at various times post-infection by plaque assay. Also, we determined infectious titers of HAdVs in infected cells and the whole of infected cells and culture medium. The replication of HAdV-D9 illustrated similar kinetics to that of HAdV-C5 in the whole of both infected cells and culture medium (Figure 2A). Also, the replication ability of HAdV-D9 in infected cells was comparable to that of HAdV-C5 (Figure 2B). As expected, HAdV-D9 was efficiently released by an earlier time point from infected cells to culture medium as compared with HAdV-C5 (Figure 2C). Moreover, the amounts of infectious HAdV-D9 which were detected in culture medium continued to be significantly higher than those of infectious HAdV-C5 during viral replication observed by 12 hours post-infection (Figure 2B and 2C). Thus, these results demonstrated that the HAdV-D9 progeny were more efficiently released from infected cells to culture medium as compared to HAdV-C5.
Figure 2. Release of HAdV-D9 from infected cells to culture medium.

A549 cells were infected with HAdV-C5 or HAdV-D9 at an MOI of 10 PFU/cell and harvested at various time points. Samples to measure the infectious titer of HAdVs were prepared from infected cells harvested along with culture medium (A), a fraction extracted from infected cells without culture medium (B), and culture medium (C). Infectious titers of HAdVs contained in each fraction were measured by triplicate TCID50 assays. Data points represent mean+standard error of the mean (n = 3). Unpaired student t-test analysis was performed with respect to HAdV-C5 at each time point and significance is indicated by *P<0.05.
HAdV-D9 Infection is Independent of hCAR
Species D HAdVs are considered to use several cellular receptors: hCAR, hCD46, αv-integrins, and/or sialic acids [52], [53]. Since HAdV-D9 expresses the fiber protein which contains a binding site for hCAR [54] and is capable of binding to hCAR [55], it is considered to utilize hCAR as a receptor for binding to target cells. On the other hand, HAdV-D9 is considered to possess the ability to interact with target cells independently of hCAR [53]. In addition, it has been suggested that the RGD motif of penton-base interacts directly with an integrin without the association of the fiber and hCAR [56]. Therefore, we examined the modality of HAdV-D9 infection in CHO and CHO-hCAR cells which stably express full length hCAR by quantifying the HAdV genomes. One copy number of the HAdV-C5 genome is equivalent to 1.04 copies of the HAdV-D9 genome. HAdV-D9 as well as HAdV-C5 was incorporated into CHO-hCAR cells (Figure 3A). Of note, the cellular uptake of the HAdV-D9 genome in CHO cells was approximately 15-fold higher than that of HAdV-C5 (Figure 3A). Thus, these data suggested that HAdV-D9 was more efficiently incorporated into CHO cells as compared to HAdV-C5 (Figure 3A). Also, the cellular uptake of the HAdV-D9 genome in CHO and CHO-hCAR cells was observed in a time-dependent manner (Figure 3B and 3C). On the other hand, the HAdV-C5 genome was readily detected in CHO-hCAR cells, but not in CHO cells (Figure 3B and 3C). Therefore, these results suggested that HAdV-D9 infection to target cells is independent of hCAR. We also investigated HAdV-D9 binding to CHO-hCAR cells with or without a blocking of the HAdV-C5 recombinant knob protein. While the recombinant protein blocked attachment of HAdV-C5 to CHO-hCAR cells in a dose dependent manner, it only partially inhibited that of HAdV-D9 (Figure 3D). This result was identical with an observation previously reported in an hCAR-positive cancer cell line [56]. Thus, the blocking of hCAR mediated by the HAdV-C5 recombinant knob protein did not inhibit HAdV-D9 infection. Moreover, we assessed whether HAdV-D9 targets human cancer cells expressing little or no hCAR as well as hCAR-positive A549 cells. The infection of HAdV-D9 in A549 cells was approximately 1.3-fold higher than that of HAdV-C5. In PC-3, AU-565 and MCF-7 cell lines, which express little or no hCAR [49], [57], it was approximately 2- to 4-fold higher than that of HAdV-C5 (Figure 3E). Thus, HAdV-D9 was incorporated into not only cancer cell lines which express little or no hCAR but also hCAR-positive cancer cells (Figure 3E). This data also supported the concept that the gene transfer mediated by HAdV-D9 is achieved at an hCAR-independent manner. Taken together, HAdV-D9 infection demonstrated hCAR-independent tropism in cancer cells.
Figure 3. Cellular uptake of HAdV-D9 independently of human CAR.

Cells (CHO and CHO-hCAR) were infected with HAdV-C5 or HAdV-D9 at an MOI of 100 genomes/cell, and total cell DNA extracted from infected cells were used for qPCR analysis. (A) Cellular uptakes of HAdVs in CHO and CHO-hCAR were represented as a fold of genome transfer in HAdV-C5-infected CHO cells at 1 hour post-infection and normalized by cellular uptake of HAdV-C5 in CHO cells; HAdV-C5 (black bars) and HAdV-D9 (white bars). (B and C) Cellular uptakes of HAdVs in a time-dependent manner in CHO (B) and CHO-hCAR (C). Total cell DNA was extracted from infected cells at 0, 30, and 60 min post-infection and used for qPCR analysis; HAdV-C5 (black squares) and HAdV-D9 (white squares). Data points represent mean+standard error of the mean (n = 3). CHO; human CAR negative CHO cells and CHO-hCAR; CHO cells stably expressing human CAR. (D) CHO-hCAR cells were treated with the HAdV-C5 fiber knob protein at a final concentration of 0, 0.5, 5.0 or 50 µg/ml at 4°C for 1 hour and then infected with HAdV at an MOI of 100 genomes/cell at 37°C for 1 hour. HAdV-C5 (black bars) and HAdV-D9 (white bars). (E) Cellular uptakes of HAdV-D9 in cancer cells which express little or no hCAR. Cells were infected with HAdV-C5 or HAdV-D9 at an MOI of 100 genomes/cell for 1 hour post-infection, and total cell DNA extracted from infected cells was analyzed by qPCR. HAdV-C5 (black bars) and HAdV-D9 (white bars). Cellular uptakes of HAdVs in cell lines were represented as (A) a fold of genome transfer, (B, C and D) a percentage of genome transfer, and (E) Genome copy numbers per 50 ng of total DNA of each HAdV. Data points represent mean+standard error of the mean (n = 3).
HAdV-D9 Exhibits Anticancer Effects in Variety of Cancer Cell Lines and a Spheroid Model
HAdV-D9 exhibits increased cellular uptake in the absence of hCAR and demonstrated hCAR-independent tropism in cancer cells (Figure 3). To examine whether HAdV-D9 infection kills hCAR-negative cancer cells, we performed in vitro cell killing assay in a broad range of cancer cell lines including hCAR-positive cancer cell lines. Cell killing activity of HAdV-D9 in these cell lines was determined by measuring remaining cell viability at 6 days post-infection. We first tested hCAR expression in cancer cell lines by flow cytometry using anti-hCAR, clone RmcB [58]. A549, OVCAR-3, BxPC-3, and H2452 cells expressed hCAR at high levels (Figure 4A). While MIA-PaCa-2 and AU-565 cells expressed hCAR at middle levels, MCF-7, ZR-75-1, and H2052 cells expressed hCAR at very low levels [59] (Figure 4A). On the other hand, hCAR expression in SKOV-3, MSTO-211H, and PC-3 cells was undetectable (Figure 4A). HAdV-D9 was able to induce cell killing at smaller amounts of infectious viruses in BxPC-3, AU-565, MCF-7, ZR-75-1, H2052 and PC-3 as compared to HAdV-C5 (Figure 4B). Also, HAdV-D9 as well as HAdV-C5 similarly killed the other cancer cell lines (Figure 4B). These data demonstrated that HAdV-D9 infection effectively kills cancer cells with attenuated hCAR and as well as hCAR-positive. Moreover, we evaluated cell killing activity of HAdV-D9 in spheroids of A549 or PC-3 cells. HAdV-D9 induced cell killing at smaller amounts of infectious viruses in PC-3 spheroids as well as A549 spheroids (Figure 4C), whereas HAdV-C5 showed attenuated cell killing activity in hCAR-negative spheroids (Figure 4C). Collectively, our data demonstrated that HAdV-D9 exhibits anticancer efficacy in broad ranges of cancer cell lines and in an in vitro 3-D model mimicking solid tumors.
Figure 4. Analysis of hCAR expression and cell killing activity of HAdV-D9 in cancer cell lines.

(A) Analysis of hCAR expression in various cancer cell lines by flow cytometry. Filled, gray histograms indicate stained cells; open, white histograms indicate unstained control cells. (B) Two-dimensional cell viability assay. Cancer cell lines were infected with HAdV-C5 (black squares) or HAdV-D9 (white squares) at indicated MOIs. Cell survival in each well was measured at 6 days post-infection using an MTS assay and plotted on y-axis as the percentage of the control values obtained from uninfected cells. (C) Three-dimensional cell viability assay. Spheroids were infected with HAdV-C5 (black squares) or HAdV-D9 (white squares) at indicated MOIs. Cell survival in each well was measured at 12 days post-infection using an MTT assay and plotted on y-axis as the percentage of the control values obtained from uninfected cells. Data points represent mean+standard error of the mean (n = 3).
Discussion
A variety of oncolytic HAdVs engineered using HAdV-C5 have been characterized in vitro and in vivo [9]–[11]. In particular, 12 kinds of oncolytic HAdVs tested in patients with a variety of cancers were well tolerated [12], [15]–[23]. Most patients showed stable disease defined in Response Evaluation Criteria in Solid Tumor (RECIST) [12], [15]–[23]. On the other hand, intratumoral administration to solid tumors of an oncolytic HAdV seemed to be inefficient because tumor sizes in patients were not reduced [24]. To explain the inefficiency, a biological barrier in tumor mass is considered to hinder viral spread from infected cells [12], [27], [28]. Adenovirus mutants with improved replication ability and enhanced virus-spread ability were developed by bioselection approaches [35], [36]. Also, several HAdV serotypes were tested for identifying new HAdVs with lower seroprevalence and equal or better antitumor efficacy to solid tumors by in vitro cell killing assay [34]. Although HAdV-C6 was suggested as a means to address to solid tumor treatment, it showed anti-cancer activity comparable to HAdV-C5 [34].
In this study, we focused on virus-spread ability of HAdVs to overcome a biological barrier in tumor mass. We tested 16 serotypes along with HAdV-C5 in order to examine virus-spread ability. Ten of 16 serotypes, as well as HAdV-C5, propagated well in A549 cells (Table 1), but the others did not propagate (data not shown). Although HAdV-B3 was propagated in A549 cells, it showed higher values in the ratios of genomes to PFU and VP to PFU (Table 1). Our data suggested that larger numbers of the HAdV-B3 particles are needed to produce one plaque as compared with HAdV-C5. Previous reports demonstrated that larger amounts of incomplete HAdV-B3 particles are produced in infected cells [60]. Also, it is difficult to separate incomplete and infectious virus particles by purification [60]. Thus, purified HAdV-B3 which we prepared in this study may contain larger numbers of incomplete viral particles. Moreover, our data showed that HAdV-B3 and B16 were purified at very low infectious titers as compared to HAdV-C5 (Table 1). HAdV-B3 and B16 had attenuated virus-spread ability evidenced by the production of small plaques (Table 2). On the other hand, species F HAdVs show host range diversity in cell culture, and it is difficult to propagate and amplify species F HAdVs [61], [62]. Also, we had difficulty propagating species F HAdVs as well as B14, B16, D20, and B21 in A549 cells. Collectively, nine serotypes (HAdV-C2, E4, D9, D10, A31, B34, B35, D37, and D51), as well as HAdV-C5, were efficiently purified at high infectious titers from infected A549 cells and seemed to merit careful investigation for virus-spread ability.
We then investigated whether HAdV serotypes have better ability with regard to virus-spread by comparing plaque morphology with HAdV-C5 and found that HAdV-D9 and D51 generated larger plaques than those of other serotypes tested (Figure 1 and Figure S2). HAdV-D9 and D51 were screened as the candidates to make oncolytic HAdVs due to abilities of efficient production and better virus spread in cancer cells. Moreover, a seroprevalence study in a Belgian population demonstrated that HAdV-D9 and D51, as well as HAdV-C6, shows low seroprevalence as compared to HAdV-C5 [63]. These HAdVs may be suitable as gene delivery vectors for treating patients with pre-existing immunity [63]–[65]. On the other hand, HAdV-D9 was isolated from stool in 1957 [66], and HAdV-D51 was isolated from a stool specimen of an AIDS patient with fever, Pneumocystis jirovecii pneumonia, and diarrhea in 1989 [67]. Recently, HAdV-D9 was suggested to be associated with acute gastroenteritis in Bangladesh, as it was isolated from stools of infants and children with acute gastroenteritis at a frequency of 0.7% (6/917 patients) along with HAdV-F40 (0.8% [7/917 patients]) and HAdV-D10 (0.4% [4/917 patients]) [68]. Thus, both viruses seem to be associated with diarrhea in children and AIDS-patients. However, there are no serious diseases associated with HAdV-D9, HAdV-D51 nor HAdV-C5 [69]. Although some serotypes in species D (HAdV-D8, D19, and D37) cause epidemic keratoconjunctivitis (EKC) in humans, HAdV-D9 does not [70], [71]. Thus, HAdV-D9 is considered to be distinct from EKC-associated HAdVs [72]. The pathogenic significance of AIDS-associated HAdV infections, including HAdV-D51 in AIDS patients is unclear [73]–[75]. Since the complete nucleotide sequence of the HAdV-D9 genome, but not the HAdV-D51 genome, has been available from GenBank, the HAdV-D9 genome is suitable for characterization of the biology and vector construction. In addition, HAdV-D51 attenuated cell killing activity in pancreatic cancer cell lines as compared to HAdV-D9 (Figure S4). Based on these considerations, we characterized the biology of HAdV-D9 identified as a candidate of oncolytic HAdV vectors. HAdV-D9 as well as HAdV-C5 replicated well in A549 cells and the viral progeny was efficiently released from infected cells as compared to HAdV-C5 (Figure 2). Our results demonstrated that better ability of HAdV-D9-spread was correlated with viral release from infected cells to culture medium (Figures 1 and 2).
HAdVs involved in species D utilize several cellular receptor(s) which are hCAR, hCD46, integrins, and/or sialic acids for attachment to target cells [53]. Although several serotypes involved in species D use sialic acid as a cellular receptor [76], [77], HAdV-D9 does not use it as a receptor [76]–[78]. The interaction to hCD46 of HAdV-D9 by comparing species B HAdVs was not shown [79]. Computational analysis of the HAdV-D9 genome suggested that the fiber protein contains an hCAR-binding domain [55]. While HAdV-D9 uses hCAR at low affinity for attachment as compared to that of HAdV-C5 [55], [56], it is able to attach to αv-integrins through its penton-base at fiber-independent manner [56]. Thus, HAdV-D9 is considered to utilize both hCAR and αv-integrins for attachment [56] and preferentially use αv-integrins for attachment [54]. Although HAdV-D9 binding to hCAR-positive cells was partially blocked by the HAdV-C5 recombinant knob protein as compared to HAdV-C5 (Figure 3D) as previously reported [56], our qPCR analysis of HAdV binding to CHO cell lines and cancer cell lines with low hCAR expression demonstrated that HAdV-D9 was capable of binding to target cells independently of hCAR (Figure 3). Thus, we obtained a result that HAdV-D9 uses cellular receptor(s) other than hCAR for attachment.
One of the problems using oncolytic HAdV-C5 vectors is a low infectivity to hCAR-negative cancer cells [39], [80], [81]. The hCAR expression levels tend to be low in the majority of advanced tumors [82]–[89]. In addition, the hCAR expression in tumors exhibits marked distributional heterogeneity [89]. Thus, the varied and insufficient expression of hCAR hampers uniform HAdV-C5 transduction throughout tumors, especially in advanced tumors. Therefore, we need to target not only hCAR-positive cancer cells but also hCAR-negative cancer cells. Since HAdV-D9 efficiently killed hCAR-negative cancer cell lines as compared to HAdV-C5 (Figure 4), the expanded tropism of HAdV-D9 with better virus-spread ability will be useful for treatments of solid tumors. Another of the problems is that the majority of the HAdV-C5 vectors infect the liver by coagulation factor X, which is a vitamin K-dependent coagulation factor and the major factor in hCAR-independent uptake by hepatocytes, when they are systemically delivered [90]. However, HAdV-D9 does not bind to coagulation factor X [91], giving HAdV-D9 another advantage as an oncolytic vector over HAdV-C5, in addition to the better virus spread ability and expanded tropism of HAdV-D9.
In summary, we have selected HAdV-D9 from 16 serotypes of HAdV by comparing in vitro virus-spread ability as well as the in vitro propagation property between a cancer cell line and a cell line immortalized by the HAdV-C5 E1 gene region. Also, we revealed that a broad range of cancer cell lines were infected with HAdV-D9 independently of hCAR status. HAdV-D9 infection efficiently induced cytopathic effect in 2- and 3-D culture cells. In this study, we examined HAdV-D9 biology in vitro for a vector development of cancer gene therapy. Construction of the HAdV-D9 vectors is under way in our laboratory in order to overcome the limitation of HAdV-C5 vector. Our experimental results provided great impact to the development of HAdV-D9-based vectors in human gene therapy as well as the elucidation of HAdV-D9 biology on the molecular mechanism of its infection.
Supporting Information
Standard curves of the HAdV-C5 and D9 genomes for qPCR. The copy numbers of purified HAdV genomes was calculated as described in the Materials and Methods section. A 100-fold serial dilution of purified HAdV genomes ranging from 102 to 108 copies per reaction was used to generate both plots; HAdV-C5 (black squares) and HAdV-D9 (white squares). Each data point represents the threshold cycle (Ct) average of samples prepared in triplicate. One copy number of the HAdV-C5 genome is equivalent to 1.04 copies of the HAdV-D9 genome.
(TIF)
Comparative analysis of plaque morphology of HAdVs on A549 cells. Plaque assay was performed as described in the Materials and Methods section. Monolayers of A549 cells in a six-well plate were infected with HAdVs, overlaid with 0.75% agar in growth medium containing 2% FBS, and stained with 0.033% neutral red at 14 days post-infection. The pictures showed microscopic view of three individual plaques formed on A549 cells infected with HAdVs.
(TIF)
Comparative analysis of plaque morphology of HAdVs on 293A cells. Monolayers of 293A cells in a six-well plate were infected with HAdVs which were propagated in 293A cells. After 1 hour post-infection, infected 293A cells were overlaid with medium containing 0.75% agar and stained with 0.033% neutral red at 14 days post-infection. The pictures showed microscopic view of three individual plaques formed on 293A cells infected with HAdVs.
(TIF)
Cell killing activity of HAdV-D9 and D51 in cancer cell lines. Nine cancer cell lines were infected with HAdV-C5 (black squares), HAdV-D9 (white squares) or HAdV-D51 (black diamonds) at indicated MOIs. Cell survival in each well was measured at 6 days post-infection using MTS assay and plotted on y-axis as the percentage of the control values obtained from uninfected cells. Data points represent mean + standard error of the mean (n = 3).
(TIF)
Genome copy numbers of HAdVs at an absorbance of 1.0 at 260 nm.
(DOC)
Classification and cellular receptors of HAdVs.
(DOC)
Acknowledgments
We thank Drs. Barbara Muz and Jesse J. Parry for flow cytometry analysis.
Funding Statement
This work was supported by grants from Susan G. Komen for the Cure KG100194 (Drs. David T. Curiel and Hideyo Ugai), and the National Institutes of Health 5R01CA154697 (Drs. David T. Curiel and Michael J. Mathis). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. CA: a cancer journal for clinicians 62: 10–29. [DOI] [PubMed] [Google Scholar]
- 2. Nguyen DX, Bos PD, Massague J (2009) Metastasis: from dissemination to organ-specific colonization. Nature reviews Cancer 9: 274–284. [DOI] [PubMed] [Google Scholar]
- 3. McCormick F (2001) Cancer gene therapy: fringe or cutting edge? Nature reviews Cancer 1: 130–141. [DOI] [PubMed] [Google Scholar]
- 4. Chiocca EA (2002) Oncolytic viruses. Nature reviews Cancer 2: 938–950. [DOI] [PubMed] [Google Scholar]
- 5. Russell SJ, Peng KW (2007) Viruses as anticancer drugs. Trends in pharmacological sciences 28: 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Curiel DT (2000) The development of conditionally replicative adenoviruses for cancer therapy. Clinical cancer research 6: 3395–3399. [PubMed] [Google Scholar]
- 7. Alemany R, Balague C, Curiel DT (2000) Replicative adenoviruses for cancer therapy. Nature biotechnology 18: 723–727. [DOI] [PubMed] [Google Scholar]
- 8.Shenk TE (2001) Adenoviridae: the viruses and their replication; Knipe DM, Howley PM, editors. Philadelphia: Lippincott Williams & Wilkins. 1053–1088 p. [Google Scholar]
- 9. Glasgow JN, Bauerschmitz GJ, Curiel DT, Hemminki A (2004) Transductional and transcriptional targeting of adenovirus for clinical applications. Current gene therapy 4: 1–14. [DOI] [PubMed] [Google Scholar]
- 10. Ugai H, Dobbins GC, Wang M, Le LP, Matthews DA, et al. (2012) Adenoviral protein V promotes a process of viral assembly through nucleophosmin 1. Virology 432: 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramirez PJ, Vickers SM, Ono HA, Davydova J, Takayama K, et al. (2008) Optimization of conditionally replicative adenovirus for pancreatic cancer and its evaluation in an orthotopic murine xenograft model. Am J Surg 195: 481–490. [DOI] [PubMed] [Google Scholar]
- 12. Russell SJ, Peng KW, Bell JC (2012) Oncolytic virotherapy. Nat Biotechnol 30: 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lichtenstein DL, Spencer JF, Doronin K, Patra D, Meyer JM, et al. (2009) An acute toxicology study with INGN 007, an oncolytic adenovirus vector, in mice and permissive Syrian hamsters; comparisons with wild-type Ad5 and a replication-defective adenovirus vector. Cancer gene therapy 16: 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuhn I, Harden P, Bauzon M, Chartier C, Nye J, et al. (2008) Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS One 3: e2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim KH, Dmitriev IP, Saddekni S, Kashentseva EA, Harris RD, et al. (2013) A phase I clinical trial of Ad5/3-Delta24, a novel serotype-chimeric, infectivity-enhanced, conditionally-replicative adenovirus (CRAd), in patients with recurrent ovarian cancer. Gynecol Oncol. [DOI] [PMC free article] [PubMed]
- 16. Li JL, Liu HL, Zhang XR, Xu JP, Hu WK, et al. (2009) A phase I trial of intratumoral administration of recombinant oncolytic adenovirus overexpressing HSP70 in advanced solid tumor patients. Gene Ther 16: 376–382. [DOI] [PubMed] [Google Scholar]
- 17. Nokisalmi P, Pesonen S, Escutenaire S, Sarkioja M, Raki M, et al. (2010) Oncolytic adenovirus ICOVIR-7 in patients with advanced and refractory solid tumors. Clin Cancer Res 16: 3035–3043. [DOI] [PubMed] [Google Scholar]
- 18. Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, et al. (2010) Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther 18: 1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cerullo V, Pesonen S, Diaconu I, Escutenaire S, Arstila PT, et al. (2010) Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res 70: 4297–4309. [DOI] [PubMed] [Google Scholar]
- 20. Nemunaitis J, Tong AW, Nemunaitis M, Senzer N, Phadke AP, et al. (2010) A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Molecular therapy: the journal of the American Society of Gene Therapy 18: 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Freytag SO, Khil M, Stricker H, Peabody J, Menon M, et al. (2002) Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer research 62: 4968–4976. [PubMed] [Google Scholar]
- 22. Kimball KJ, Preuss MA, Barnes MN, Wang M, Siegal GP, et al. (2010) A phase I study of a tropism-modified conditionally replicative adenovirus for recurrent malignant gynecologic diseases. Clinical cancer research 16: 5277–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramesh N, Ge Y, Ennist DL, Zhu M, Mina M, et al. (2006) CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor–armed oncolytic adenovirus for the treatment of bladder cancer. Clinical cancer research 12: 305–313. [DOI] [PubMed] [Google Scholar]
- 24. Kirn D (2001) Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: what have we learned? Gene therapy 8: 89–98. [DOI] [PubMed] [Google Scholar]
- 25. Harrison D, Sauthoff H, Heitner S, Jagirdar J, Rom WN, et al. (2001) Wild-type adenovirus decreases tumor xenograft growth, but despite viral persistence complete tumor responses are rarely achieved–deletion of the viral E1b-19-kD gene increases the viral oncolytic effect. Human Gene Therapy 12: 1323–1332. [DOI] [PubMed] [Google Scholar]
- 26. Sauthoff H, Hu J, Maca C, Goldman M, Heitner S, et al. (2003) Intratumoral spread of wild-type adenovirus is limited after local injection of human xenograft tumors: virus persists and spreads systemically at late time points. Human Gene Therapy 14: 425–433. [DOI] [PubMed] [Google Scholar]
- 27. Cascallo M, Alonso MM, Rojas JJ, Perez-Gimenez A, Fueyo J, et al. (2007) Systemic toxicity-efficacy profile of ICOVIR-5, a potent and selective oncolytic adenovirus based on the pRB pathway. Molecular therapy: the journal of the American Society of Gene Therapy 15: 1607–1615. [DOI] [PubMed] [Google Scholar]
- 28. Sauthoff H, Heitner S, Rom WN, Hay JG (2000) Deletion of the adenoviral E1b-19kD gene enhances tumor cell killing of a replicating adenoviral vector. Human Gene Therapy 11: 379–388. [DOI] [PubMed] [Google Scholar]
- 29. Adachi Y, Reynolds PN, Yamamoto M, Wang M, Takayama K, et al. (2001) A midkine promoter-based conditionally replicative adenovirus for treatment of pediatric solid tumors and bone marrow tumor purging. Cancer research 61: 7882–7888. [PubMed] [Google Scholar]
- 30. Yamamoto M, Alemany R, Adachi Y, Grizzle WE, Curiel DT (2001) Characterization of the cyclooxygenase-2 promoter in an adenoviral vector and its application for the mitigation of toxicity in suicide gene therapy of gastrointestinal cancers. Molecular therapy: the journal of the American Society of Gene Therapy 3: 385–394. [DOI] [PubMed] [Google Scholar]
- 31. DeWeese TL, van der Poel H, Li S, Mikhak B, Drew R, et al. (2001) A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer research 61: 7464–7472. [PubMed] [Google Scholar]
- 32. Glasgow JN, Everts M, Curiel DT (2006) Transductional targeting of adenovirus vectors for gene therapy. Cancer gene therapy 13: 830–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beatty MS, Curiel DT (2012) Chapter two–Adenovirus strategies for tissue-specific targeting. Advances in cancer research 115: 39–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen CY, Weaver EA, Khare R, May SM, Barry MA (2011) Mining the adenovirus virome for oncolytics against multiple solid tumor types. Cancer gene therapy 18: 744–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Subramanian T, Vijayalingam S, Chinnadurai G (2006) Genetic identification of adenovirus type 5 genes that influence viral spread. J Virol 80: 2000–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan W, Kitzes G, Dormishian F, Hawkins L, Sampson-Johannes A, et al. (2003) Developing novel oncolytic adenoviruses through bioselection. J Virol 77: 2640–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fields BN, Knipe DM, Howley PM (2007) Fields virology. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins.
- 38. Green M, Pina M, Kimes RC (1967) Biochemical studies on adenovirus multiplication. XII. Plaquing efficiencies of purified human adenoviruses. Virology 31: 562–565. [DOI] [PubMed] [Google Scholar]
- 39. Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, et al. (1997) Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275: 1320–1323. [DOI] [PubMed] [Google Scholar]
- 40. Kanegae Y, Makimura M, Saito I (1994) A simple and efficient method for purification of infectious recombinant adenovirus. Japanese journal of medical science & biology 47: 157–166. [DOI] [PubMed] [Google Scholar]
- 41. Ugai H, Suzuki E, Inabe K, Murata T, Hamada H, et al. (2003) Spontaneous mutations in the human gene for p53 in recombinant adenovirus during multiple passages in human embryonic kidney 293 cells. Biochem Biophys Res Commun 300: 448–456. [DOI] [PubMed] [Google Scholar]
- 42. Maizel JV Jr, White DO, Scharff MD (1968) The polypeptides of adenovirus. I. Evidence for multiple protein components in the virion and a comparison of types 2, 7A, and 12. Virology 36: 115–125. [DOI] [PubMed] [Google Scholar]
- 43.Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, a laboratory manual. NY: Cold Spring Harbor Laboratory Press.
- 44. Ugai H, Wang M, Le LP, Matthews DA, Yamamoto M, et al. (2010) In vitro dynamic visualization analysis of fluorescently labeled minor capsid protein IX and core protein V by simultaneous detection. J Mol Biol 395: 55–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krasnykh VN, Mikheeva GV, Douglas JT, Curiel DT (1996) Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. J Virol 70: 6839–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Saito I, Oya Y, Yamamoto K, Yuasa T, Shimojo H (1985) Construction of nondefective adenovirus type 5 bearing a 2.8-kilobase hepatitis B virus DNA near the right end of its genome. J Virol 54: 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hsu KH, Lonberg-Holm K, Alstein B, Crowell RL (1988) A monoclonal antibody specific for the cellular receptor for the group B coxsackieviruses. J Virol 62: 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim M, Sumerel LA, Belousova N, Lyons GR, Carey DE, et al. (2003) The coxsackievirus and adenovirus receptor acts as a tumour suppressor in malignant glioma cells. Br J Cancer 88: 1411–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Murakami M, Ugai H, Belousova N, Pereboev A, Dent P, et al. (2010) Chimeric adenoviral vectors incorporating a fiber of human adenovirus 3 efficiently mediate gene transfer into prostate cancer cells. The Prostate 70: 362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gros A, Martinez-Quintanilla J, Puig C, Guedan S, Mollevi DG, et al. (2008) Bioselection of a gain of function mutation that enhances adenovirus 5 release and improves its antitumoral potency. Cancer research 68: 8928–8937. [DOI] [PubMed] [Google Scholar]
- 51. Ugai H, Borovjagin AV, Le LP, Wang M, Curiel DT (2007) Thermostability/infectivity defect caused by deletion of the core protein V gene in human adenovirus type 5 is rescued by thermo-selectable mutations in the core protein X precursor. J Mol Biol 366: 1142–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Arnberg N (2009) Adenovirus receptors: implications for tropism, treatment and targeting. Reviews in medical virology 19: 165–178. [DOI] [PubMed] [Google Scholar]
- 53. Zhang Y, Bergelson JM (2005) Adenovirus receptors. J Virol 79: 12125–12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Roelvink PW, Lizonova A, Lee JG, Li Y, Bergelson JM, et al. (1998) The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J Virol. 72: 7909–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kirby I, Lord R, Davison E, Wickham TJ, Roelvink PW, et al. (2001) Adenovirus type 9 fiber knob binds to the coxsackie B virus-adenovirus receptor (CAR) with lower affinity than fiber knobs of other CAR-binding adenovirus serotypes. J Virol 75: 7210–7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Roelvink PW, Kovesdi I, Wickham TJ (1996) Comparative analysis of adenovirus fiber-cell interaction: adenovirus type 2 (Ad2) and Ad9 utilize the same cellular fiber receptor but use different binding strategies for attachment. J Virol 70: 7614–7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tang Y, Wu H, Ugai H, Matthews QL, Curiel DT (2009) Derivation of a triple mosaic adenovirus for cancer gene therapy. PLoS One 4: e8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hsu KH, Lonberg-Holm K, Alstein B, Crowell RL (1988) A monoclonal antibody specific for the cellular receptor for the group B coxsackieviruses. J Virol 62: 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Havenga MJ, Lemckert AA, Ophorst OJ, van Meijer M, Germeraad WT, et al. (2002) Exploiting the natural diversity in adenovirus tropism for therapy and prevention of disease. J Virol 76: 4612–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prage L, Hoglund S, Philipson L (1972) Structural proteins of adenoviruses. 8. Characterization of incomplete particles of adenovirus type 3. Virology 49: 745–757. [DOI] [PubMed] [Google Scholar]
- 61. Tiemessen CT, Kidd AH (1994) Adenovirus type 40 and 41 growth in vitro: host range diversity reflected by differences in patterns of DNA replication. J Virol 68: 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yamasaki S, Miura Y, Brown E, Davydova J, Yamamoto M (2010) Development of a method for effective amplification of human adenovirus 40. Arch Virol 155: 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vogels R, Zuijdgeest D, van Rijnsoever R, Hartkoorn E, Damen I, et al. (2003) Replication-deficient human adenovirus type 35 vectors for gene transfer and vaccination: efficient human cell infection and bypass of preexisting adenovirus immunity. J Virol 77: 8263–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Geisbert TW, Bailey M, Hensley L, Asiedu C, Geisbert J, et al. (2011) Recombinant adenovirus serotype 26 (Ad26) and Ad35 vaccine vectors bypass immunity to Ad5 and protect nonhuman primates against ebolavirus challenge. J Virol 85: 4222–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Abbink P, Lemckert AA, Ewald BA, Lynch DM, Denholtz M, et al. (2007) Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J Virol. 81: 4654–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kibrick S, Melendez L, Enders JF (1957) Clinical associations of enteric viruses with particular reference to agents exhibiting properties of the ECHO group. Ann N Y Acad Sci 67: 311–325. [DOI] [PubMed] [Google Scholar]
- 67. De Jong JC, Wermenbol AG, Verweij-Uijterwaal MW, Slaterus KW, Wertheim-Van Dillen P, et al. (1999) Adenoviruses from human immunodeficiency virus-infected individuals, including two strains that represent new candidate serotypes Ad50 and Ad51 of species B1 and D, respectively. J Clin Microbiol 37: 3940–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dey SK, Shimizu H, Phan TG, Hayakawa Y, Islam A, et al. (2009) Molecular epidemiology of adenovirus infection among infants and children with acute gastroenteritis in Dhaka City, Bangladesh. Infect Genet Evol 9: 518–522. [DOI] [PubMed] [Google Scholar]
- 69. Liu EB, Wadford DA, Seto J, Vu M, Hudson NR, et al. (2012) Computational and serologic analysis of novel and known viruses in species human adenovirus D in which serology and genomics do not correlate. PLoS One 7: e33212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kemp MC, Hierholzer JC, Cabradilla CP, Obijeski JF (1983) The changing etiology of epidemic keratoconjunctivitis: antigenic and restriction enzyme analyses of adenovirus types 19 and 37 isolated over a 10-year period. The Journal of infectious diseases 148: 24–33. [DOI] [PubMed] [Google Scholar]
- 71. Ford E, Nelson KE, Warren D (1987) Epidemiology of epidemic keratoconjunctivitis. Epidemiologic reviews 9: 244–261. [DOI] [PubMed] [Google Scholar]
- 72. Blusch JH, Deryckere F, Windheim M, Ruzsics Z, Arnberg N, et al. (2002) The novel early region 3 protein E3/49K is specifically expressed by adenoviruses of subgenus D: implications for epidemic keratoconjunctivitis and adenovirus evolution. Virology 296: 94–106. [DOI] [PubMed] [Google Scholar]
- 73. Hierholzer JC (1992) Adenoviruses in the immunocompromised host. Clin Microbiol Rev 5: 262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Khoo SH, Bailey AS, de Jong JC, Mandal BK (1995) Adenovirus infections in human immunodeficiency virus-positive patients: clinical features and molecular epidemiology. J Infect Dis 172: 629–637. [DOI] [PubMed] [Google Scholar]
- 75. De Jong JC, Wermenbol AG, Verweij-Uijterwaal MW, Slaterus KW, Wertheim-Van Dillen P, et al. (1999) Adenoviruses from human immunodeficiency virus-infected individuals, including two strains that represent new candidate serotypes Ad50 and Ad51 of species B1 and D, respectively. J Clin Microbiol 37: 3940–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Arnberg N, Edlund K, Kidd AH, Wadell G (2000) Adenovirus type 37 uses sialic acid as a cellular receptor. J Virol 74: 42–48. [PMC free article] [PubMed] [Google Scholar]
- 77. Arnberg N, Kidd AH, Edlund K, Olfat F, Wadell G (2000) Initial interactions of subgenus D adenoviruses with A549 cellular receptors: sialic acid versus alpha(v) integrins. J Virol 74: 7691–7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Arnberg N, Kidd AH, Edlund K, Nilsson J, Pring-Akerblom P, et al. (2002) Adenovirus type 37 binds to cell surface sialic acid through a charge-dependent interaction. Virology 302: 33–43. [DOI] [PubMed] [Google Scholar]
- 79. Gaggar A, Shayakhmetov DM, Lieber A (2003) CD46 is a cellular receptor for group B adenoviruses. Nature medicine 9: 1408–1412. [DOI] [PubMed] [Google Scholar]
- 80. Asaoka K, Tada M, Sawamura Y, Ikeda J, Abe H (2000) Dependence of efficient adenoviral gene delivery in malignant glioma cells on the expression levels of the Coxsackievirus and adenovirus receptor. Journal of neurosurgery 92: 1002–1008. [DOI] [PubMed] [Google Scholar]
- 81. Li Y, Pong RC, Bergelson JM, Hall MC, Sagalowsky AI, et al. (1999) Loss of adenoviral receptor expression in human bladder cancer cells: a potential impact on the efficacy of gene therapy. Cancer research 59: 325–330. [PubMed] [Google Scholar]
- 82. Sachs MD, Rauen KA, Ramamurthy M, Dodson JL, De Marzo AM, et al. (2002) Integrin alpha(v) and coxsackie adenovirus receptor expression in clinical bladder cancer. Urology 60: 531–536. [DOI] [PubMed] [Google Scholar]
- 83. Anders M, Vieth M, Rocken C, Ebert M, Pross M, et al. (2009) Loss of the coxsackie and adenovirus receptor contributes to gastric cancer progression. Br J Cancer 100: 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Auer D, Reimer D, Porto V, Fleischer M, Roessler J, et al. (2009) Expression of coxsackie-adenovirus receptor is related to estrogen sensitivity in breast cancer. Breast cancer research and treatment 116: 103–111. [DOI] [PubMed] [Google Scholar]
- 85. Korn WM, Macal M, Christian C, Lacher MD, McMillan A, et al. (2006) Expression of the coxsackievirus- and adenovirus receptor in gastrointestinal cancer correlates with tumor differentiation. Cancer gene therapy 13: 792–797. [DOI] [PubMed] [Google Scholar]
- 86. Matsumoto K, Shariat SF, Ayala GE, Rauen KA, Lerner SP (2005) Loss of coxsackie and adenovirus receptor expression is associated with features of aggressive bladder cancer. Urology 66: 441–446. [DOI] [PubMed] [Google Scholar]
- 87. Okegawa T, Pong RC, Li Y, Bergelson JM, Sagalowsky AI, et al. (2001) The mechanism of the growth-inhibitory effect of coxsackie and adenovirus receptor (CAR) on human bladder cancer: a functional analysis of car protein structure. Cancer research 61: 6592–6600. [PubMed] [Google Scholar]
- 88. Rauen KA, Sudilovsky D, Le JL, Chew KL, Hann B, et al. (2002) Expression of the coxsackie adenovirus receptor in normal prostate and in primary and metastatic prostate carcinoma: potential relevance to gene therapy. Cancer research 62: 3812–3818. [PubMed] [Google Scholar]
- 89. Zeimet AG, Muller-Holzner E, Schuler A, Hartung G, Berger J, et al. (2002) Determination of molecules regulating gene delivery using adenoviral vectors in ovarian carcinomas. Gene therapy 9: 1093–1100. [DOI] [PubMed] [Google Scholar]
- 90. Waddington SN, McVey JH, Bhella D, Parker AL, Barker K, et al. (2008) Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 132: 397–409. [DOI] [PubMed] [Google Scholar]
- 91. Kalyuzhniy O, Di Paolo NC, Silvestry M, Hofherr SE, Barry MA, et al. (2008) Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proceedings of the National Academy of Sciences of the United States of America 105: 5483–5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Standard curves of the HAdV-C5 and D9 genomes for qPCR. The copy numbers of purified HAdV genomes was calculated as described in the Materials and Methods section. A 100-fold serial dilution of purified HAdV genomes ranging from 102 to 108 copies per reaction was used to generate both plots; HAdV-C5 (black squares) and HAdV-D9 (white squares). Each data point represents the threshold cycle (Ct) average of samples prepared in triplicate. One copy number of the HAdV-C5 genome is equivalent to 1.04 copies of the HAdV-D9 genome.
(TIF)
Comparative analysis of plaque morphology of HAdVs on A549 cells. Plaque assay was performed as described in the Materials and Methods section. Monolayers of A549 cells in a six-well plate were infected with HAdVs, overlaid with 0.75% agar in growth medium containing 2% FBS, and stained with 0.033% neutral red at 14 days post-infection. The pictures showed microscopic view of three individual plaques formed on A549 cells infected with HAdVs.
(TIF)
Comparative analysis of plaque morphology of HAdVs on 293A cells. Monolayers of 293A cells in a six-well plate were infected with HAdVs which were propagated in 293A cells. After 1 hour post-infection, infected 293A cells were overlaid with medium containing 0.75% agar and stained with 0.033% neutral red at 14 days post-infection. The pictures showed microscopic view of three individual plaques formed on 293A cells infected with HAdVs.
(TIF)
Cell killing activity of HAdV-D9 and D51 in cancer cell lines. Nine cancer cell lines were infected with HAdV-C5 (black squares), HAdV-D9 (white squares) or HAdV-D51 (black diamonds) at indicated MOIs. Cell survival in each well was measured at 6 days post-infection using MTS assay and plotted on y-axis as the percentage of the control values obtained from uninfected cells. Data points represent mean + standard error of the mean (n = 3).
(TIF)
Genome copy numbers of HAdVs at an absorbance of 1.0 at 260 nm.
(DOC)
Classification and cellular receptors of HAdVs.
(DOC)