

Abstract
Myelin-specific, pro-inflammatory TH17 cells are widely regarded as the drivers of experimental autoimmune encephalomyelitis (EAE), an animal model for Multiple sclerosis (MS). The factors, responsible for the generation and maintenance of TH17 cells as well as their participation in the pathogenic cascade leading to the demyelinating disease, have been studied extensively. However, how these harmful autoreactive cells are controlled in vivo remains unclear. By comparing TCR transgenic mice on a disease susceptible and a disease resistant genetic background, we show here that pathogenic TH17 cells are sequestered within the intestine of spontaneous EAE resistant B10.S mice. Disease resistant B10.S mice harbored higher frequencies of TH17 cells in the intestine compared to EAE susceptible SJL/J mice. Moreover, transferred TH17 cells selectively migrated to intestinal lymphoid organs of B10.S mice. The sequestration of TH17 cells in the gut was partially dependent on the gut homing receptor α4β7-mediated adhesion to the intestine. Administration of α4β7 blocking-antibodies increased the peripheral availability of TH17 cells, resulting in increased EAE severity after immunization in B10.S mice. Together, these results support the concept that the intestine is a check-point for controlling pathogenic, organ-specific T cells.
Introduction
Evidence suggests that autoreactive T cells are commonly present in the healthy immune repertoire, but are kept in check by numerous tolerance mechanisms. Although several details of the tolerance mechanisms have yet to be elucidated, mechanisms including negative selection of autoreactive T cells in the thymus, ignorance, anergy, cytokine immune deviation, tolerogenic antigen presenting cells and induction of regulatory cells have been demonstrated to be involved in mediating self-tolerance [1], [2]. Understanding the mechanisms of self-tolerance, which limits the aberrant activation of self-reactive T cells, is crucial for the development of strategies to treat autoimmune diseases, like Multiple sclerosis (MS).
MS is an extremely complex autoimmune disease caused by various cellular and molecular mechanisms. From the animal models of MS, it became evident that distinct T helper cell subsets, such as IFN-γ-producing TH1 and IL-17-producing TH17 cells, either alone or in combination are capable of mediating a neurological disease in animals, resembling the human disease [3]–[6]. However, recently, considerable efforts focused on the role of TH17 cells in EAE as well as MS, due to their close association with several other autoimmune diseases [7]. Moreover, TH17 cells are crucial for controlling the invasion of pathogenic microorganisms and play an important role in intestinal immune homeostasis [8]. In EAE studies, IL-17-deficient mice show attenuated disease symptoms [9], while neutralization of IL-17 during EAE induction greatly delayed the development and reduced the severity of the disease [10]. In addition, transfer of polarized TH17 cells induced neurological disease, supporting the idea that this T helper cell subset plays an important role in EAE pathogenesis [3], [6]. Classical TH17 cells secrete IL-17a, IL-17f, IL-21 and IL-22 as their key effector cytokines and make use of the chemokine receptor CCR6 to enter the target tissues [11]–[13]. In vitro as well as in vivo generation of TH17 cells requires the induction of their master transcription factor retinoic acid-related orphan receptor-γt (ROR-γt) [14]. Despite the extensive knowledge about the generation and maintenance of TH17 cells, how these cells are regulated in vivo during autoimmune disease settings needs further investigation.
In this report, we compared the development of spontaneous EAE in myelin-specific TCR transgenic mice on the disease-susceptible SJL/J genetic background with animals on the disease-resistant B10.S background. We found that the pro-inflammatory, myelin-reactive TH17 cells were enriched in the intestine of B10.S mice, but failed to reach the peripheral immune organs, resulting in the absence of spontaneous EAE in B10.S mice. The release of intestine-sequestered TH17 cells by treating B10.S mice with α4β7-specific monoclonal antibodies worsened EAE. We propose that the immune tolerance against myelin-specific self-antigens could be achieved by selective sequestration of pro-inflammatory T cells in the intestine.
Results
Absence of spontaneous EAE in MOG-specific TCR transgenic B10.S mice
We recently described a transgenic mouse strain that expresses a myelin oligodendrocyte glycoprotein (MOG)-specific T cell receptor (TCR) on the SJL/J genetic background (RR mice). The TCR was derived from an encephalitogenic T cell clone generated from recombinant MOG protein (rMOG) immunized wild-type SJL/J mice [15]. To get insights into the mechanisms of precipitation of spontaneous autoimmunity and tolerance, we backcrossed RR SJL/J animals to MHC congenic B10.S mice. Similar to SJL/J mice, B10.S mice harbor the MHC class II allele I-As, but on the C57BL/10 background. Although RR SJL/J and RR B10.S mice express the same pair of TCR Vα8.3 and Vβ4 chains on their CD4+ T cells, the incidence of spontaneous EAE was strikingly different. While more than 80% of the RR SJL/J cohort presented spontaneous EAE symptoms between 6–28 weeks of age, none of the B10.S mice displayed any clinical signs of disease ( Figure 1A ). In our colony of more than 300 RR B10.S mice maintained during a period of 5 years, we noted only 3 mice with spontaneous EAE symptoms, accounting for less than 1% of disease incidence.
Figure 1. Absence of spontaneous EAE in RR B10.S mice.

A. Incidence of spontaneous EAE in a cohort of RR SJL/J (n = 14) or RR B10.S mice (n = 17). B. Frequencies of CD3+ CD4+ T cells in spleen, pooled axillary and inguinal lymph nodes (pLN), mesenteric lymph nodes (mLN), Peyer's patches (PP) and small intestinal lamina propria (siLP) were measured by flow cytometry. C. Frequencies of Foxp3+ CD3+ CD4+ regulatory T cells in the indicated organs were measured by flow cytomtery. Results are from n = 4–6 mice per group. Data were pooled from 2–3 different experiments (B–C). Error bars indicate SEM (B–C).
T cell and APC functions in B10.S mice
To determine whether resistance to spontaneous EAE in B10.S animals is due to alterations in T cells, we analyzed T cell populations in the peripheral lymphoid organs. Flow cytometric analysis of the peripheral lymphoid organs showed that the frequencies of CD4+ T cells in various lymphoid organs of B10.S mice were comparable to those of their SJL/J counterparts ( Figure 1B ). The resistance to EAE in B10.S has been attributed to an increase in the frequencies of Foxp3+ regulatory T cells (Treg cells) [16]. However, the frequencies of Treg cells were comparable in most of the lymphoid organs of RR SJL/J and B10.S mice. In the spleen of RR B10.S mice, we even observed 4-fold lower frequencies of Treg cells than in SJL/J animals ( Figure 1C ). Thus, we conclude that Treg cells do not vitally contribute to the observed EAE resistance in RR B10.S mice.
Next, we examined the functional status of the antigen presenting cells (APCs) in B10.S mice. A previous report showed that APCs from B10.S mice express lower amounts of MHC class II [17]. Flow cytometric analysis of MHC class II (I-As) on B cells from various lymphoid organs confirmed the lower MHC class II expression levels on B10.S APCs compared to APCs from SJL/J mice ( Figure 2A ). In addition, we evaluated the expression levels of several surface markers related to co-stimulation, such as CD86, PDL-1 or ICOS-L. However, there were no significant differences in the expression of these co-stimulatory/co-inhibitory molecules on splenic B cells (Figure S1). Stimulation of the APCs innate immune response by microbial products can result in the upregulation of MHC class II on the surface of B10.S APCs [17]. However, in vivo activation of APCs as well as T cells via immunization with rMOG in CFA did not result in a massive upregulation of the MHC class II-expression on APCs from B10.S mice (Figure S1). Moreover, in the gut associated lymphoid tissues (GALT), where APCs constantly encounter microbial products due to their close proximity to the commensal microbiota, B10.S B cells expressed lower amounts of MHC class II ( Figure 2A ).
Figure 2. Activation of T cells is not affected by lower MHC class II expression on APCs of B10.S mice.

A. Expression of the MHC class II molecule I-As on B cells was determined by flow cytometry in various lymphoid organs of RR SJL/J and B10.S mice. Bar graphs show the mean fluorescent intensity (MFI) + SEM of I-As on the gated B220+ population. *, p<0.05; **, p<0.01 (Mann-Whitney U test). Results are from n = 5–7 mice per group from 3 different experiments. B. Splenocytes from RR SJL/J or RR B10.S mice were isolated and their proliferative response to the indicated concentrations of rMOG or anti-CD3 tested in vitro. Data are shown as mean counts per minute (cpm) with error bars indicating SEM. Data were pooled from 3 different experiments.
We subsequently analyzed if the lower MHC class II-expression on APCs from B10.S mice would result in defective T cell activation and thus, provide an explanation for the absence of spontaneous EAE in RR B10.S mice. However, despite their lower MHC class II-expression, B10.S APCs were capable of processing and presenting the cognate antigen MOG as efficiently to T cells as APCs from SJL/J mice. Moreover, T cells from B10.S mice showed a higher proliferative response to anti-CD3 stimulation than SJL/J T cells, suggesting no intrinsic or extrinsic defect in T cell activation or functional capacity of APCs in B10.S mice ( Figure 2B ). Taken together, we conclude that the resistance to spontaneous EAE is not due to aberrant functional alterations of neither T cells nor APCs in RR B10.S mice.
Impaired accumulation of TH17 cells in the peripheral lymphoid organs of B10.S mice
Having excluded any general defect in T cell functionality, we sought to characterize the distribution of T helper cell-subsets in B10.S mice. EAE induction in mice has been attributed to distinct T helper cell subsets, which produce either IFN-γ or IL-17, TH1 or TH17 cells, respectively [3]. Recent set of seminal papers showed that at steady state, distinct components of the gut commensal microbiota are necessary to trigger TH17 cell differentiation, which subsequently contribute to the development of autoimmune diseases [18]–[20]. Moreover, we were previously able to demonstrate that spontaneous EAE development in RR SJL/J mice is critically dependent on an intact commensal microbiota, which activates and directs CD4+ T cells to differentiate into pathogenic TH17 cells [21]. To validate the TH17-inducing potential of the gut microbiota of B10.S mice, we compared the frequency of this T helper cell subset in the GALT of RR SJL/J and B10.S mice using flow cytometric analysis. The frequencies of IL-17-producing CD4+ T cells were elevated in the small intestinal lamina propria (LP) of B10.S mice compared to their SJL/J counterparts ( Figure 3A ). The increased frequency of LP TH17 cells, however, was not due to a higher abundance of TH17 cell-inducing segmented filamentous bacteria (SFB) in the intestines of B10.S mice, since colonization levels of SFB were comparable between SJL/J and B10.S animals (Figure S2).
Figure 3. B10.S TH17 cells fail to accumulate in peripheral lymphoid organs.
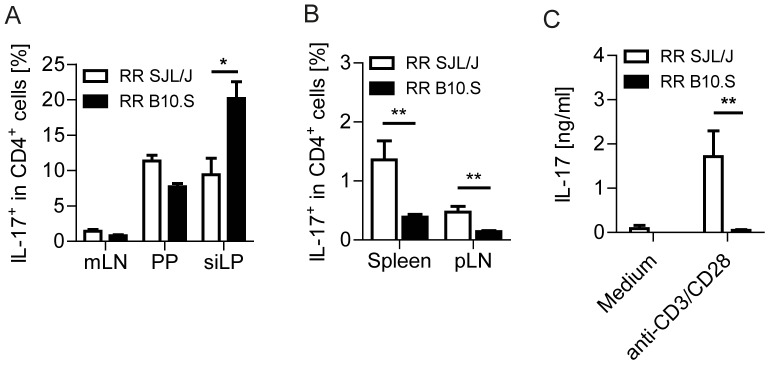
A. Flow cytometric analysis of IL-17 expression by CD4+ T cells in the gut associated lymphoid tissues. Data represent the percentage of cytokine producing cells in the gated CD4+ population. *, p<0.05; (Mann-Whitney U test). B. Flow cytometric analysis of IL-17 expression by T cells in the peripheral lymphoid organs. Data represent the percentage of cytokine producing cells in the gated CD4+ population. **, p<0.01 (Mann-Whitney U test). Results are from n = 5–6 mice per group. Data were pooled from 2–3 different experiments (A–B). Error bars indicate SEM (A–B). C. Cytokine production by splenocytes from RR SJL/J or B10.S mice. After a 72 hours culture period, production of IL-17 by RR SJL/J or B10.S spleen cells in response to medium alone or anti-CD3/anti-CD28 stimulation were measured by ELISA. Bars depict mean + SEM. **, p<0.01 (Mann-Whitney U test).
Next, we checked the fate of the pro-inflammatory TH17 cells, which are triggered in the intestine. Interestingly, in SJL/J mice, TH17 cells, which were activated in the gut, populate peripheral lymphoid organs and form a pool of pathogenic CD4+ T cells, ready to invade the central nervous system and mediate neurological disease [21]. Direct ex vivo analysis of TH17 cells in B10.S mice, however, revealed that despite the higher abundance of TH17 cells in the GALT, frequencies of TH17 cells were greatly reduced in spleen and lymph nodes ( Figure 3B ). The lower IL-17 production by splenic T cells from B10.S mice was confirmed by measuring the recall response after in vitro stimulation with anti-CD3/CD28 antibodies ( Figure 3C ).
Mechanisms of accumulation of TH17 cells in the intestine and EAE development
We next wanted to identify the mechanism(s) responsible for the accumulation of TH17 cells in the intestine and hence, for the lack of a peripheral TH17 cell-pool in B10.S mice. The specific enrichment might be the result of either impaired emigration of TH17 cells from the intestine after their induction by gut commensals or the increased migration from the periphery to the intestine [22]. We transferred in vitro polarized CFSE-labeled TH17 cells into naïve SJL/J or B10.S recipients and traced their homing to various lymphoid organs. Whereas we detected comparable frequencies of TH17 cells migrating to peripheral lymphoid organs, the intestine, i.e. the lamina propria, of B10.S mice accumulated higher frequencies of transferred TH17 cells than the intestine of SJL/J recipients ( Figure 4A ).
Figure 4. Integrin α4β7 mediated accumulation of TH17 cells in the intestine of RR B10.S mice.

A. Enhanced migration of TH17 cells to the intestine. Flow cytometric analysis of transferred CFSE-labeled TH17 cells in various lymphoid organs. Data represent the percentage of IL-17+ T cells in the gated CFSE+ CD4+ population. **, p<0.01 (Mann-Whitney U test). Results are from n = 5–6 mice per group. Data were pooled from 2–3 different experiments. B. Blockade of α4β7 increases peripheral availability of IL-17-producing T cells. RR B10.S mice were treated weekly twice with anti-α4β7 or isotype control antibodies for two consecutive weeks. Splenocytes were stimulated in vitro with anti-CD3/anti-CD28 antibodies. After a 72 hours culture period, production of IL-17 was measured by ELISA. Bars show mean + SEM. **, p<0.01 (Mann-Whitney U test). n = 11–12 mice per group. Data were from 3 independent experiments. C. Effect of blockade of α4β7 on EAE pathogenesis. RR B10.S mice were immunized with rMOG in CFA and treated weekly twice with anti-α4β7 or isotype control antibodies. Mean clinical scores are depicted. *, p<0.05. n = 9–11 mice per group. Data were from 3 independent experiments.
Gut-tropic T cells express the integrin receptor α4β7, whose ligand MAdCAM-1 is expressed on post capillary venules in the intestinal LP [23]. To test the role of the gut homing receptor α4β7 in intestinal accumulation of TH17 cells, we treated RR B10.S mice with a blocking antibody to α4β7. Interestingly, blockade of α4β7 led to an enhanced production of IL-17 in the spleen ( Figure 4B ), suggesting that the α4β7 integrin receptor at least partially contributes to the migration and retention of TH17 cells in the B10.S intestine. In addition, enhanced peripheral availability of TH17 cells in B10.S mice through blockade of α4β7 resulted in an increased EAE severity ( Figure 4C ). In summary, these data suggests that while pathogenic TH17 cells are efficiently induced in the intestine of B10.S mice, they fail to accumulate in peripheral lymphoid organs, a process partly dependent on the α4β7 integrin-mediated adhesion to the intestine.
Discussion
In this study, we describe a mechanism of tolerance to the development of spontaneous EAE. We compared transgenic mice that bear the same rearranged MOG-specific TCR in the EAE-susceptible SJL/J or the EAE-resistant B10.S genetic background. A lack of spontaneous EAE in B10.S mice in contrast to SJL/J animals led us to investigate the cellular mechanisms responsible for the resistance to the disease. We found that the pathogenic TH17 cells were contained within the intestine of B10.S mice. The retention of TH17 cells is presumably mediated by the enhanced migration as well as interaction of their gut homing integrin α4β7 with MAdCAM-1, expressed in the intestine.
The genetic background profoundly influences the purging of the autoimmune T cell repertoire. It is assumed that the presentation of low avidity self-peptide – MHC complexes in the thymus promotes the development of potentially autoreactive T cells, which may escape thymic negative selection. This is reflected by the fact that various inbred strains differ in their response to distinct myelin antigens, thus presenting varied immune and clinical responses to these autoantigens [24]. To avoid potential artifacts due to the expansion of a different T cell repertoire, we used transgenic mice expressing a MOG-specific T cell receptor, which recognizes the MOG-peptide 92–106 in the context of I-As, but on two different genetic backgrounds.
RR SJL/J mice develop high incidence of spontaneous EAE, whereas B10.S mice remained free of clinical disease symptoms. EAE resistance offers a unique opportunity to study the regulatory mechanisms that control autoimmunity. Initially, we speculated that MOG-specific T cells might be different in these two mouse strains, but flow cytometric analysis of various peripheral and intestinal lymphoid organs revealed no abnormalities in T cell development in B10.S mice with frequencies of CD4+ CD3+ cells being comparable between the two mouse strains. APCs play an important role in determining the type and degree of T cell responses by providing co-stimulatory signals and establishing the local cytokine-milieu. We confirmed an earlier report describing lower basal levels of MHC class II molecules on APCs of B10.S mice [17]. Nevertheless, B10.S APCs were still efficient processors and presenters of MOG protein to T cells.
Various reasons were suggested, explaining the resistance of B10.S mice to EAE-development after immunization with MBP or PLP peptides. Differences in the blood-brain barrier permeability between B10.S and SJL/J mice were demonstrated, with later reports unable to confirm these findings [25], [26]. Increased frequencies of regulatory T (Treg) cells have been observed in naïve B10.S mice [27]. Depletion of Treg cells partly restored EAE susceptibility with a concomitant increase in the production of IFN-γ, IL-6 and IL-17 [16]. We, however, did not observe any changes in the Treg cells in our TCR transgenic mice on B10.S background. This may be due to the fact that this particular TCR may not favor Treg cell differentiation on certain genetic backgrounds. Previous reports also indicated a defect in T helper cell responses in B10.S mice [28]. The resistance to EAE in B10.S mice was partially associated with the reduced production of the pro-inflammatory cytokine IFN-γ or the elevated production of IL-4 and IL-10 after immunization [29], [30].
While studying immunization models, the effect of the adjuvants on skewing the T helper cell responses cannot be ruled out, we therefore decided to investigate the phenotypes of the T helper cell subsets in a “natural” (unimmunized) state. Striking differences were observed in the frequencies of IL-17-producing TH17 cells. B10.S mice contained far lower frequencies of TH17 cells in all secondary peripheral lymphoid organs compared to their SJL/J counterparts. To confirm that these cells actually produce low levels of their specific pro-inflammatory cytokine, IL-17, we measured the in vitro recall response. B10.S mice selectively lacked the production of IL-17. At steady state, TH17 cells are induced in the intestine, which is essential for maintaining mucosal immune homeostasis [14]. The differentiation of TH17 cells in the mucosal tissue is controlled at least in parts by specific commensal gut microbial species, segmented filamentous bacteria (SFB), respectively [18], [31], [32]. Moreover, commensal microbiota induced TH17 cells have been shown to be critically regulating the susceptibility to autoimmune diseases [19], [33]. Recently, we showed that spontaneous EAE development in the susceptible SJL/J background requires the activation and differentiation of T cells into pro-inflammatory lineages in the gut [21]. While analyzing the GALT, we found that in contrast to the peripheral lymphoid tissues, B10.S mice harbored higher frequencies of TH17 cells in the small intestinal lamina propria than SJL/J animals. However, the TH17 cell-inducing load of the segmented filamentous bacteria was similar between SJL/J and B10.S mice. This finding suggests that B10.S mice are indeed capable of mounting a robust TH17 response, but these T cells fail to reach the peripheral lymphoid organs. To investigate whether TH17 cells may have an enhanced migratory affinity towards the intestine of B10.S mice, we transferred TH17 cells into naïve SJL/J and B10.S recipients. Interestingly, we found elevated numbers of TH17 cells in the intestine of B10.S mice compared to SJL/J recipients, suggesting that the B10.S gut harbors a milieu, which is attractive for TH17 cells.
It is well appreciated that chemokines and their receptors along with adhesion molecules control the homing pattern of lymphocytes to lymphoid and non-lymphoid tissues. Gut tropism of T cells are mediated by the integrin receptor α4β7 and the chemokine receptor CCR9 that is induced by the gut associated CD103+ dendritic cells [23], [34]. Our experiments showed that the blocking of the gut homing integrin receptor α4β7-mediated adhesion to the intestine with a monoclonal antibody increased the peripheral availability of TH17 cells, resulting in a more severe EAE. Our results suggest that multiple mechanisms are employed to target effector pathogenic TH17 cells to the intestinal mucosa, making them unavailable to participate in pro-inflammatory responses at peripheral sites.
In summary, we show here that the resistance to spontaneous EAE in B10.S mice is partly due to enhanced accumulation of activated MOG-specific TH17 cells in the intestine leading to a reduced frequency of this T helper cell subset in the peripheral repertoire. Understanding the mechanisms that promote the accumulation of pro-inflammatory TH17 cells may help in devising new therapeutic approaches to control MS.
Materials and Methods
Animals
A wild-type B10.S mice colony was established from a breeding pair obtained from the McLaughlin Research Institute (Montana, USA). TCR transgenic RR SJL/J mice [15] were backcrossed for more than 9 generations to B10.S mice. Mice were bred at the animal facility of the Max Planck Institute of Neurobiology (Martinsried, Germany). All animal procedures were approved by the Regierung von Oberbayern (Munich, Germany).
Immunization and evaluation of EAE
Mice were immunized subcutaneously with 200 µg rMOG emulsified in Freund's adjuvant supplemented with 5 mg/ml Mycobacterium tuberculosis (strain H37Ra; Difco) (CFA). 200 ng of pertussis toxin (List Biological Laboratories) were injected intraperitoneally on the day of immunization and 48 hours later. In addition, mice were injected with 250 µg of anti-α4β7 (DATK 32) or isotype control antibodies weekly twice. Clinical signs of EAE were assessed daily according to the standard 5 point scale [15].
Proliferation assay
Splenocytes from RR SJL/J or RR B10.S mice were cultured in the presence of the indicated concentration of rMOG or anti-CD3/anti-CD28 antibodies (BD Pharmingen). The proliferative response was measured by the incorporation of [3H]thymidine during the final 16 hours of a 72 hour culture period. Results are expressed as mean thymidine uptake (cpm) of triplicate cultures.
CFSE-labeled TH17 cell transfer
Polarization of splenic CD4+ T cells towards TH17 cells was performed as previously described [3]. After 6 days in culture, TH17 cells were purified by Nycoprep (Axis-Shield). Purified T cells were labelled for 10 min at 37°C with 5 mM CFSE (Life Technologies) in PBS containing 1% fetal bovine serum (FBS). Subsequently, cells were washed twice in ice-cold PBS. 5×106 CFSE-labelled TH17 cells were injected intravenously into wild-type SJL/J or B10.S mice. The frequencies of CFSE+ TH17 cells were measured by flow cytometry after 3 days.
Cell isolation and flow cytometry
Single-cell suspensions were prepared from spleen, pooled peripheral lymph nodes (axillary plus inguinal) or Peyer's patches by mechanical disruption via forcing through 40 µm cell strainers (BD Biosciences). Lamina propria lymphocytes were isolated as previously described [21]. For detection of cell surface markers, cells were stained in FACS buffer (PBS containing 1% BSA and 0.1% NaN3) with the following fluorochrome-labeled monoclonal antibodies: anti-CD4 (RM4-5), anti-CD3 (145-2C11), anti-B220 (RA3-6B2), anti-I-Aq (KH116), anti-CD86 (GL1), anti-PDL-1 (MIH5), anti-ICOS-L (HK5.3). For intracellular cytokine staining, cells were activated with 50 ng/ml PMA (Sigma) and 500 ng/ml ionomycin (Sigma) in the presence of 5 mg/ml brefeldin A (Sigma) for 4 hours at 37°C. After surface staining, cells were fixed and permeabilized using the Transcription Factor Staining Buffer Set (eBioscience) and stained intracellularly with the following antibodies: anti-IL17 (TC11-18H10) or anti-FoxP3 (FJK-16s). All antibodies were purchased from BD Pharmingen or eBioscience. Cells were acquired on a FACSCalibur or FACSVerse (BD Biosciences) and analysis was performed using FlowJo (TreeStar) software.
ELISA
Cytokine levels in cell culture supernatants were determined using matching antibody pairs for IL-17 (eBioscience) according to manufacturer's instructions.
Fecal Bacterial DNA Extraction and qPCR
Bacterial genomic DNA was extracted from fecal pellets using QIAamp DNA Stool mini kit (Qiagen). Real-time PCR targeted to 16S rDNA was performed using Absolute QPCR SYBR Green Mix (Thermo Fisher Scientific) and a 7900HT Real-time PCR System (Applied Biosystems). Values were normalized to total bacteria. The following primer sets were used [35]: total bacteria, ACTCCTACGGGAGGCAGCAGT and ATTACCGCGGCTGCTGGC; Segmented filamentous bacteria, GACGCTGAGGCATGAGAGCAT and GACGGCACGGATTGTTATTCA.
Supporting Information
Stimulation of the innate immune response does not result in enhanced MHC class II-expression on APCs of B10.S mice. Expression-levels of MHC class II (I-As) and the co-stimulatory/co-inhibitory molecules (CD86, PDL-1 and ICOS-L) on B cells were determined by flow cytometry in the spleen of naïve wild type SJL/J and B10.S mice and wild-type animals 10 days after immunization with rMOG/CFA. Bar graphs show the mean fluorescent intensity (MFI) + SEM on the gated B220+ population. *, p<0.05 (Mann-Whitney U test). Results are from n = 4–5 mice per group. Data were pooled from 2 independent experiments.
(TIF)
Fecal SFB content in B10.S and SJL/J mice. 16S rDNA PCR for the presence of SFB in the feces of RR SJL/J or B10.S mice. Values are shown as relative amount to total bacterial 16S rDNA.
(TIF)
Acknowledgments
We thank Lydia Penner and Birgit Kunkel for technical support. The DATK32 hybridoma was kindly provided by Dr. Melanie Laschinger.
Funding Statement
Laboratory work is supported by SFB 571 (Project B6), SFB TR 128 (Project A1), the German Competence Network on Multiple Sclerosis (KKNMS), Hertie foundation and by the Max Planck Society. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Walker LSK, Abbas AK (2002) The enemy within: Keeping self-reactive T cells at bay in the periphery. Nature Reviews Immunology 2: 11–19. [DOI] [PubMed]
- 2. Mueller DL (2010) Mechanisms maintaining peripheral tolerance. Nature Immunology 11: 21–27. [DOI] [PubMed] [Google Scholar]
- 3. Domingues HS, Mues M, Lassmann H, Wekerle H, Krishnamoorthy G (2010) Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. Plos One 5: e15531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM (2008) Differential regulation of central nervous system autoimmunity by TH1 and TH17 cells. Nature Medicine 14: 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Connor RA, Prendergast CT, Sabatos CA, Lau CWZ, Lech MD, et al. (2008) Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. Journal of Immunology 181: 3750–3754. [DOI] [PMC free article] [PubMed]
- 6. Jäger A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK (2009) Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. Journal of Immunology 183: 7169–7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL-17 and Th17 Cells. Annual Review of Immunology 27: 485–517. [DOI] [PubMed] [Google Scholar]
- 8. Atarashi K, Honda K (2011) Microbiota in autoimmunity and tolerance. Current Opinion in Immunology 23: 761–768. [DOI] [PubMed] [Google Scholar]
- 9. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, et al. (2006) IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis Journal of Immunology. 177: 566–573. [DOI] [PubMed] [Google Scholar]
- 10. Park H, Li ZX, Yang XXO, Chang SH, Nurieva R, et al. (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nature Immunology 6: 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, et al. (2009) C–C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nature Immunology 10: 514–523. [DOI] [PubMed] [Google Scholar]
- 12. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, et al. (2007) Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. Journal of Experimental Medicine 204: 2803–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bettelli E, Korn T, Oukka M, Kuchroo VK (2008) Induction and effector functions of TH17 cells. Nature 453: 1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, et al. (2006) The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126: 1121–1133. [DOI] [PubMed] [Google Scholar]
- 15. Pöllinger B, Krishnamoorthy G, Berer K, Lassmann H, Bösl M, et al. (2009) Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. Journal of Experimental Medicine 206: 1303–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reddy J, Waldner H, Zhang XM, Illés Z, Wucherpfennig KW, et al. (2005) CD4+CD25+ regulatory T cells contribute to gender differences in susceptibility to experimental autoimmune encephalomyelitis. Journal of Immunology 175: 5591–5595. [DOI] [PubMed] [Google Scholar]
- 17. Waldner H, Collins M, Kuchroo VK (2004) Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J Clin Invest 113: 990–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ivanov II, Atarashi K, Manel N, Brodie FL, Shima T, et al. (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu HJ, Ivanov II, Darce D, Hattori K, Shima T, et al. (2010) Gut residing filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32: 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK (2010) Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences (USA) 108: 4615–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berer K, Mues M, Koutroulos M, Al Rasbi Z, Boziki M, et al. (2011) Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479: 538–541. [DOI] [PubMed] [Google Scholar]
- 22. Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, et al. (2011) Control of TH17 cells occurs in the small intestine. Nature 475: 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berlin C, Bargatze RF, Campbell JJ, von Andrian UH, Szabo MC, et al. (1995) α4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell 80: 413–422. [DOI] [PubMed] [Google Scholar]
- 24. Krishnamoorthy G, Wekerle H (2009) EAE: An immunologist's magic eye. European Journal of Immunology 39: 2031–2035. [DOI] [PubMed] [Google Scholar]
- 25. Jemison LM, Williams SK, Lublin FD, Knobler RL, Korngold R (1993) Interferon-gamma-inducible endothelial cell class II major histocompatibility complex expression correlates with strain- and site-specific susceptibility to experimental allergic encephalomyelitis. Journal of Neuroimmunology 47: 15–22. [DOI] [PubMed] [Google Scholar]
- 26. Chen F, Shaw MK, Li JZ, Lisak RP, Tse HY (2006) Adoptive transfer of myelin basic protein-induced experimental autoimmune encephalomyelitis between SJL and B10.S mice: Correlation of priming milieus with susceptibility and resistance phenotypes. Journal of Neuroimmunology 173: 146–154. [DOI] [PubMed] [Google Scholar]
- 27. Del Rio R, Sun YF, Alard P, Tung KSK, Teuscher C (2011) H2 Control of Natural T Regulatory Cell Frequency in the Lymph Node Correlates with Susceptibility to Day 3 Thymectomy-Induced Autoimmune Disease. Journal of Immunology 186: 382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blankenhorn EP, Butterfield R, Case LK, Wall EH, Del Rio R, et al. (2011) Genetics of experimental allergic encephalomyelitis supports the role of T helper cells in multiple sclerosis pathogenesis. Annals of Neurology 70: 887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Segal BM, Shevach EM (1996) IL-12 unmasks latent autoimmune disease in resistant mice. Journal of Experimental Medicine 184: 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maron R, Hancock WW, Slavin A, Hattori M, Kuchroo VK, et al. (1999) Genetic susceptibility or resistance to autoimmune encephalomyelitis in MHC congenic mice is associated with differential production of pro- and anti-inflammatory cytokines. International Immunology 11: 1573–1580. [DOI] [PubMed] [Google Scholar]
- 31. Ivanov II, De Llanos Frutos R, Manel N, Yoshinaga K, Rifkin DB, et al. (2008) Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host & Microbe 4: 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gaboriau-Routhiau V, Rakotobe S, Lévuyer E, Mulder I, Lan A, et al. (2009) The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31: 677–689. [DOI] [PubMed] [Google Scholar]
- 33. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK (2011) Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences (USA) 108: 4615–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, et al. (2005) Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. Journal of Experimental Medicine 202: 1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salzman NH, Hung KC, Haribhai D, Chu HT, Karlsson-Sjöberg J, et al. (2010) Enteric defensins are essential regulators of intestinal microbial ecology. Nature Immunology 11: 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Stimulation of the innate immune response does not result in enhanced MHC class II-expression on APCs of B10.S mice. Expression-levels of MHC class II (I-As) and the co-stimulatory/co-inhibitory molecules (CD86, PDL-1 and ICOS-L) on B cells were determined by flow cytometry in the spleen of naïve wild type SJL/J and B10.S mice and wild-type animals 10 days after immunization with rMOG/CFA. Bar graphs show the mean fluorescent intensity (MFI) + SEM on the gated B220+ population. *, p<0.05 (Mann-Whitney U test). Results are from n = 4–5 mice per group. Data were pooled from 2 independent experiments.
(TIF)
Fecal SFB content in B10.S and SJL/J mice. 16S rDNA PCR for the presence of SFB in the feces of RR SJL/J or B10.S mice. Values are shown as relative amount to total bacterial 16S rDNA.
(TIF)