

Abstract
Background
Zellweger syndrome (ZS) is a fatal inherited disease caused by peroxisome biogenesis deficiency. Patients are characterized by multiple disturbances of lipid metabolism, profound hypotonia and neonatal seizures, and distinct craniofacial malformations. Median live expectancy of ZS patients is less than one year. While the molecular basis of peroxisome biogenesis and metabolism is known in considerable detail, it is unclear how peroxisome deficiency leads to the most severe neurological symptoms. Recent analysis of ZS mouse models has all but invalidated previous hypotheses.
Hypothesis
We suggest that a regulatory rather than a metabolic defect is responsible for the drastic impairment of brain function in ZS patients.
Testing the hypothesis
Using microarray analysis we identify diazepam binding inhibitor/acyl-CoA binding protein (DBI) as a candidate protein that might be involved in the pathogenic mechanism of ZS. DBI has a dual role as a neuropeptide antagonist of GABA(A) receptor signaling in the brain and as a regulator of lipid metabolism. Repression of DBI in ZS patients could result in an overactivation of GABAergic signaling, thus eventually leading to the characteristic hypotonia and seizures. The most important argument for a misregulation of GABA(A) in ZS is, however, provided by the striking similarity between ZS and "benzodiazepine embryofetopathy", a malformation syndrome observed after the abuse of GABA(A) agonists during pregnancy.
Implications of the hypothesis
We present a tentative mechanistic model of the effect of DBI misregulation on neuronal function that could explain some of the aspects of the pathology of Zellweger syndrome.
Background
Clinical background of Zellweger syndrome
Zellweger syndrome (ZS) is a class of inherited metabolic disorders caused by a deficiency in peroxisomal biogenesis (reviewed in [1,2]). The incidence of ZS is about 1/25,000 – 1/50,000 births. ZS patients are characterized by profound hypotonia and neonatal seizures, which – accompanied by feeding difficulties and psychomotor retardation – lead to an early death, usually in the first year of infancy. Another characteristic symptom is a distinct craniofacial malformation complex, consisting of high forehead, low/broad nasal bridge, epicanthus, high arched palate, and micrognathia. In addition, the severe disturbance of many peroxisomal metabolic pathways in ZS leads to a variety of defects in almost every organ, most prominently the liver (hepatomegaly in 78% of patients, fibrosis in 76%), the kidney (renal cysts in 98%), and the brain. In the latter, white matter abnormalities and a unique neuronal migration defect in the cerebral hemispheres, the cerebellum, and the inferior olivary complex are the most striking symptoms.
Biochemically, ZS patients are characterized by the absence of functional peroxisomes and an almost complete disruption of peroxisomal beta-oxidation, leading to the accumulation of branched and very long-chain fatty acids, abnormal bile acids, and leukotrienes. In addition, the biosynthesis of docosahexaenoic acid (DHA) and plasmalogens is impaired, resulting in a drastic decrease of these important lipid components of the central nervous system. The metabolism of pipecolic acid and the peroxisomal steps of isoprenoid biosynthesis are also impaired to some extent.
Pathogenetic mechanism of Zellweger syndrome
ZS is caused by mutations in any one of about a dozen components of the peroxisome assembly machinery [3]. The cell biology of the disease has been elucidated in considerable detail during the last decade. However, it is still completely unknown, how the deficiency in peroxisomal metabolism leads to the severe phenotype and extremely poor prognosis of ZS patients.
Several mechanisms have been suggested:
 Accumulation of branched and very long-chain fatty acids could lead to a structural disruption of cell membranes, particularly in the central nervous system (reviewed in [4]).
Accumulation of branched and very long-chain fatty acids could lead to a structural disruption of cell membranes, particularly in the central nervous system (reviewed in [4]).
The disturbance of lipid metabolism could also lead to a more subtle disturbance of membrane micro-domains (rafts, caveolae) involved in intercellular signaling.
Deficiency of major myelin constituents, such as plasmalogens, DHA, and cholesterol, might lead to impaired neuronal development (see for example [5] for a review of the beneficial effects of DHA supplementation in ZS patients).
Increased levels of leukotrienes due to beta-oxidation deficiency could precipitate an inflammatory process that might be involved in the pathogenesis of the brain defects [6,7].
Studies in a mouse model of ZS have shown that reduced levels of the ether phospholipid platelet activating factor (PAF) and consequent impairment of glutamatergic signaling might cause the neuronal migration defects by an unknown mechanism [8].
The importance of lipid metabolism for the pathogenesis of ZS is underlined by the phenotype of peroxisomal single enzyme-deficient patients. Disruption of peroxisomal beta-oxidation at the level of multifunctional protein type 2 leads to the same pathological syndrome as ZS, although the patients have a slightly better prognosis [9]. Patients with acyl-CoA:dihydroxyacetone phosphate acyltransferase deficiency have rhizomelic chondrodysplasia punctata, which shares many of the key symptoms with ZS, including hypotonia, feeding problems, growth retardation, and early neonatal death [9]. On the other hand, defects of many other peroxisomal enzymes (catalase, glutaryl-CoA oxidase, phytanoyl-CoA hydroxylase, alanine:glyoxylate aminotransferase, 2-methylacyl-CoA racemase, pipecolic acid oxidase, and mevalonate kinase) as well as deficiencies in non-peroxisomal metabolism do not lead to ZS.
The most important recent data on the pathogenesis of ZS come from the analysis of three mouse models, that are deficient in different components of the peroxisomal biogenesis machinery (Pex2, Pex5, and Pex11beta, respectively [10-12]). All three mice show an almost exact phenocopy of the human ZS, including severe hypotonia at birth, impaired neuronal migration in the same brain areas, and early neonatal death. However, some important pathologies such as hepatomegaly, renal cysts, and bone abnormalities are not recapitulated. Mice disrupted in Pex11alpha are externally indistinguishable from wild-type animals [13]. Detailed analysis of these ZS models has demonstrated that DHA and isoprenoid/cholesterol biosynthesis are unlikely to be central to the pathogenic mechanism [14-16]. A more detailed study of Pex5 knock-out mice further established that changes in very long chain fatty acid, docosahexaenoic acid, or plasmalogen levels in brain are unlikely to be responsible for the neuronal migration defects [17]. Most strikingly, however, the Pex11beta knockout mouse (which has intact peroxisomes) dies of ZS, but shows almost normal peroxisomal metabolism, including normal levels of very long-chain fatty acids and plasmalogens, thus confuting the major pathogenic hypotheses listed above [12]. This conclusively establishes that the obvious biochemical disturbances are concomitant but not causal to the most serious symptoms of ZS patients.
In addition, analysis of a mouse model of peroxisomal beta-oxidation deficiency indicates that the most severe ZS-like symptoms are independent of the characteristic neuronal migration defects [18,19]. Figure 1 summarizes the molecular conditions leading to Zellweger-like syndromes in human and mouse.
Figure 1.
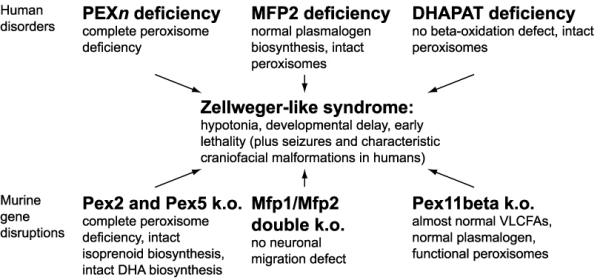
Molecular conditions leading to Zellweger-like syndromes in human and mouse. Biochemical and pathological consequences that are not common to all disorders are indicated. PEXn = any of the peroxisome biogenesis proteins that can be mutated in classical Zellweger syndrome. MFP1/2 = multifunctional proteins of peroxisomal beta-oxidation. DHAPAT = dihydroxyacetone phosphate acyl transferase. VLCFAs = very long-chain fatty acids. DHA = docosahexaenoic acid.
Identification of candidate genes by microarray analysis
We suggest that a regulatory defect is central to the severe disease phenotype of ZS patients. Even a slight disturbance of specific pathway intermediates, such as seen in the Pex11beta knockout mouse, could trigger a catastrophic regulatory response that escalates by inappropriate activation of feedback loops. Such a mechanism has recently been identified in peroxisome-deficient yeast, where a slight increase in cytosolic aminoadipate levels is suggested to lead to a full-blown activation of the lysine biosynthetic pathway via a positive feedback mechanism [20].
We decided to use microarray data to identify possible players involved in such a misregulation mechanism in human ZS. Microarrays provide an unbiased survey of gene expression levels in human tissues and cells under various experimental conditions. Many hundred data sets, examining most of the predicted genes of the human genome, are now publicly available for exploratory analysis. Based on the known clinical data of ZS patients, we defined three criteria for candidate genes:
1. A candidate should be regulated positively or negatively in concert with lipid metabolism genes, because changes in acyl-CoA derivatives are the only common factor in ZS and the ZS-like single enzyme deficiencies.
2. To be active in a regulatory pathway, a candidate should be involved in a signaling pathway or in transcriptional regulation, so that feedback regulation is possible.
3. Because the most serious symptoms of ZS affect the brain, an ideal candidate would be involved in brain or neuron function.
Using these criteria we identified only a single candidate gene, Diazepam Binding Inhibitor, DBI, in all data sets available in the Stanford Microarray Database http://genome-www5.stanford.edu/MicroArray/SMD/. Co-regulation of DBI with lipid metabolism is found consistently in experiments representing the two current microarray methodologies, Affymetrix oligonucleotide arrays [21] and spotted two-color cDNA arrays [22] (Table 1). DBI is also among the most highly upregulated genes in lovastatin-treated squamous cell carcinomas [23].
Table 1.
Ten genes that have expression profiles most similar to diazepam binding inibitor, DBI, in two large-scale microarray expression profiling studies, as quantified by Pearson correlation coefficient r (the top 10 neighbors of DBI are shown for each sample set, corresponding to r > 0.73 for the fibroblast samples). Genes that are mentioned twice refer to two independent clones on the array.
| 50 human fibroblast samples [22] | 174 human carcinoma samples [21] | ||
| FDPS | farnesyl diphosphate synthase | DKFZP564B167 | unknown function |
| HMGCS1 | 3-hydroxy-3-methylglutaryl-CoA synthase | IDI1 | isopentenyl diphosphate isomerase |
| SC4MOL | sterol-C4-methyl oxidase | INSIG1 | insulin induced gene 1 |
| INSIG1 | insulin induced gene 1 | UAP1 | UDP-N-acetylglucosamine pyrophosphorylase |
| H2AV | histone H2A variant | FASN | fatty acid synthase |
| FADS1 | fatty acid desaturase | CYP51 | lanosterol 14-alpha-demethylase |
| EBP | sterol-delta8,delta7-isomerase | C6orf34 | unknown function |
| SQLE | squalene epoxidase | SC4MOL | sterol-C4-methyl oxidase |
| INSIG1 | insulin induced gene 1 | SORD1 | sorbitol dehydrogenase |
| HMGCS1 | 3-hydroxy-3-methylglutaryl-CoA synthase | SYBL1 | synaptobrevin-like 1 |
Bold genes are involved in lipid metabolism. The expression profiles can be explored and compared at http://genome-www.stanford.edu/fibroblast/figures.shtml (fibroblast samples) and http://genome-www5.stanford.edu/cgi-bin/source/sourceSearch (carcinoma samples).
Properties of Diazepam Binding Inhibitor
DBI is a particularly promising candidate for the regulatory pathway responsible for the brain and morphological defects associated with ZS. It has a dual function as a secreted GABA(A) receptor neuroregulatory peptide and as an acyl-CoA binding protein involved in lipid metabolism (reviewed in [24,25]). DBI is expressed almost ubiquitously, including in a variety of glial and neuronal cell types in many brain regions [26-32]. In the brain DBI is processed to various neuropeptides (triakontatetraneuropeptide TTN = DBI17-50, octadecaneuropeptide ODN = DBI33-50, eikosaneuropeptide ENP = DBI51-70, and others [33]) with slightly varying activity. DBI was first identified by its ability to displace the GABA(A) agonist diazepam and hence is able to antagonistically modulate the GABA(A) receptor in the brain. GABA is the most important inhibitory neurotransmitter in the central nervous system, and the GABA(A) receptor is the main target of benzodiazepine sedatives, such as diazepam. Increases in DBI serum levels were found in epileptic patients [34]. In contrast, DBI was found to be the only identifiable protein downregulated in both schizophrenic and Alzheimer's disease patients [35].
In addition to GABA(A) receptor, DBI and its processing products bind to the ubiquitous mitochondrial benzodiazepine receptor pBR and play a crucial role in the acute stimulation of steroidogenesis [36-38]. A third type of DBI receptor mediates rapid release of intracellular calcium, but remains to be characterized in detail [39,40].
In its role as an acyl-CoA binding protein, DBI specifically binds to medium and long-chain acyl-CoA esters (but not to fatty acids, acyl carnitines, cholesterol and a number of nucleotides; [41]) and is involved in fatty acid-mediated regulation of gene expression and the formation of intracellular acyl-CoA pools (reviewed in [42]).
In vitro studies have identified functional sterol response elements (SREBP binding sites) and peroxisome proliferator gamma response elements (PPARgamma binding sites) in the DBI gene promoter [43,44].
Finally, DBI has a stimulating effect on the pancreatic cholecystokinin and insulin system [45,46], that might be related to the pancreatic islet hyperplasia seen in many ZS patients.
Benzodiazepines, GABAergic signaling, and Zellweger syndrome
Neonatal epileptic seizures and floppy infant syndrome are two important symptoms that point to an involvement of defective GABAergic signaling in ZS pathogenesis. However, the most striking connection between Zellweger syndrome and DBI-mediated misregulation of GABAergic signaling is provided by the report of severe developmental malformations in babies born to mothers that had used benzodiazepines such as diazepam during pregnancy. Laegreid et al. [47-49] have described a "benzodiazepine embryofetopathy" that involves extreme hypotonia, craniofacial abnormalities (epicanthus, low nasal bridge, abnormal ears, high arched palate), feeding difficulties, and delayed neuromotor development (table 2). The similarity to ZS was so striking that one of the first patients described by Laegreid et al. [47] was later recognized to be a genuine ZS case [50,51]. However, two others of their patients were tested biochemically and did not show accumulation of very long and branched-chain fatty acids, a biochemical hallmark of ZS. One of these two died at 11 weeks of age, and upon autopsy was found to have slight cortical dysplasia and single-cell neuronal heterotopias in white matter [48].
Table 2.
Comparison of selected symptoms in "fetal benzodiazepine syndrome", Zellweger syndrome and peroxisomal beta-oxidation (multifunctional protein-2) deficiency.
| "benzodiazepine embryofetopathy" [48] | Zellweger syndrome [2] | MFP2 deficiency [9] | |
| hypotonia/floppy-baby syndrome | 100% (8/8) | 99% (94/95) | 98% (41/42) |
| epicanthic folds | 100% (8/8) | 92% (33/36) | present1 |
| feeding difficulties | 88% (7/8) | 96% (74/77) | 91% (10/11) |
| mental retardation | 86% (6/7) | 100% (45/45) | yes |
| short upturned nose with low nasal bridge | 75% (6/8) | 100% (23/23) | present |
| micrognathia | 63% (5/8) | 100% (18/18) | present |
| Highly arched palate | 50% (4/8) | 95% (35/37) | present |
| abnormal ears | 50% (4/8) | 98% (39/40) | present |
| high forehead | present | 97% (58/60) | present |
| neonatal seizures | no | 92% (56/61) | 95% (36/38) |
| neuronal migration defects | yes, in single case examined | yes, characteristic | 88% (15/17) |
1 79% (30/38) MFP2-deficient patients have a combination of dysmorphic features [9]. Numbers of affected and examined cases are indicated in parentheses.
The work by Laegreid et al. was heavily criticized for its epidemiological validity (e.g. [52]) and later studies did not find a significant correlation between benzodiazepine use during pregnancy and birth defects (reviewed in [53]). However, some reports have linked diazepam to oral clefts, distal limb defects, microcephaly, and cardiovascular anomalies (reviewed in [53]). In addition, it is well established that high doses of diazepam lead to craniofacial malformations in rodents [54-57] and mice carrying targeted disruptions of the GABAergic pathway consistently develop a cleft palate, a rather common feature of Zellweger patients [58,59].
We are not aware of any other drug-induced teratogenic syndrome that mimics ZS. It is quite unlikely that all of Laegreid's patients just had a mild form of ZS, as they did neither show the characteristic biochemical abnormalities nor the disease progression with age. Also, consistent with the fact that benzodiazepine exposure is largely terminated at birth, they did not show the neonatal seizures that affect ZS patients.
Presentation of the hypothesis
We suggest the following tentative mechanistic model for the pathogenesis of DBI misregulation in ZS (figure 2):
Figure 2.

Possible regulatory network leading to disturbed GABAergic signaling in peroxisome-deficient patients. Arrows indicate the predicted direction of change in the patients. See text for details.
Peroxisome deficiency leads to an accumulation of peroxisomal metabolic intermediates (metabolite X), most likely acyl-CoA derivatives that are also disturbed by the single enzyme defects that cause ZS-like syndromes. Directly or indirectly, metabolite X causes an inactivation and down-regulation of DBI and a decrease in DBI release, possibly by replacing its physiological acyl-CoA ligands. Decreased activity of DBI affects two intertwined feedback loops. In the first loop, decrease of DBI releases the inhibition of GABA(A) receptor [60]. Overactivation of GABA(A) receptor inhibits glutamate release [61] and consequently causes a decrease in NMDA receptor-mediated glutamatergic signaling [62]. This in turn causes a lack of calcium release in the target cells and a deficiency of DBI secretion and expression [63]. The second loop starts by a decrease in DBI action on its non-GABA(A) receptor and a subsequent lack of DBI-stimulated calcium release [39,40,64]. Both pathways converge on the decrease in intracellular calcium, that results in an inhibition of DBI release [65] and closes the loop. A hypothetical ancillary loop could involve positive crosstalk between the over-activated GABA(A) receptor and its counterpart GABA(B), which is known to inhibit DBI release [66]. The two loops are not necessarily present in the same cells, and each of them may involve several adjacent cells. Also, different processing products of DBI act on intracellular calcium using different pathways [67], and may lead to additional differentiation of the DBI misregulation effect. The main feature is that both loops tend to enhance the down-regulation of DBI, thus amplifying and maintaining a small initial disturbance caused by the peroxisomal defect. Both loops involve a deficiency of NMDA receptor-mediated calcium release, in agreement with observations in ZS mouse models [8]. Gressens et al. (2000) found that administration of GABA at a concentration of 0.25 mg/kg twice a day had no effect on neuronal migration in peroxisome-deficient and control mice. This finding might indicate that the neuronal migration defect is mediated by the direct effect of DBI-derived peptides on intracellular calcium through its non-GABA(A) receptors.
Misregulation of DBI and the consequent disinhibition of GABAergic signaling should lead to diverse defects in various brain areas. A region of special interest is the inferior olive, which is malformed or absent in classical ZS. Olivary axon collaterals do not only innervate non-GABAergic neurons in the cerebellar nuclei, but also GABAergic nucleo-olivary cells, thus establishing a direct feedback loop to the inferior olive [68]. Purkinje cells, which show characteristic heterotopias in the cerebellum of ZS patients are also GABAergic [69,70].
Furthermore, it has been demonstrated recently that excessive GABA(A) receptor activation by neurosteroids or benzodiazepines initiates a slow form of neuronal death in cultured hippocampal neurons [71]. This could be the basis of the increased neuronal apoptosis observed in ZS. It should also be noted that the suggested mechanism for ZS brain disturbances bears conceptual similarity to that for fetal alcohol syndrome, where a combination of NMDA blockade and GABA(A) over-activation leads to widespread apoptosis in the developing brain [72].
The observation of cleft palate in GABA signaling-deficient mouse models indicates that disregulation of the GABA(A) pathway might also be responsible for the malformation syndromes observed in ZS patients and in benzodiazepine embryofetopathy [58,59].
DBI is an especially suitable candidate for ZS pathogenesis because it is shown to integrate signals from a variety of lipid-related pathways (fatty acids, sterols, steroids). Its restricted binding properties narrow down the search for a specific metabolite X that is responsible for the initial repression of DBI, and the feedback loops explain how even a slight disturbance that is common to all ZS-like syndromes can tip the balance towards a disinhibition of the GABAergic system, resulting in the severe disorders that are found in all patients.
Testing the hypothesis
The recent availability of several peroxisome-deficient mouse models provides an excellent opportunity to directly test the GABA/DBI model of Zellweger pathogenesis. This would involve a detailed analysis of DBI isoform levels in wild type and mutant brains, as well as a study of GABA agonist and antagonist effects, both in vivo and in neuronal explants. Additional in vitro studies could be used to determine the molecular details of DBI regulation and processing, to test the existence of the feedback loops postulated by the model. After the general mechanism has been established, identification of "metabolite X" would be crucial and could be achieved, e.g., by examining the effect of fractionated brain extracts on a DBI reporter cell line.
Implications of the hypothesis
Currently, no successful therapy for peroxisome deficient patients is available. Identification of a GABA component to the pathogenesis of Zellweger syndrome has obvious implications for the treatment of affected patients. Straightforward pharmacological intervention with well-known GABA antagonists could be used to counteract the misregulation and to interrupt the pathological feedback loop. In addition, even if the molecular details of the proposed mechanism turn out to be incorrect, consideration of an amplification of minor metabolic changes by positive feedback changes could influence the search for "metabolite X", which by implication of the present model would be a low-concentration intermediate that binds DBI, and hence is probably a Co-enzyme A thioester.
Conclusions
We suggest that peroxisomal deficiency leads to an unidentified disturbance of lipid metabolism that causes misregulation of the diazepam binding inhibitor, DBI. This misregulation affects two independent systems and is amplified by specific feedback loops: The DBI/GABA(A) system and the DBI/calcium system. The independence of the two systems agrees with the observation in peroxisomal beta-oxidation-deficient mice, where hypotonia and early death occur without neuronal migration defects. The activation and subsequent escalation of feedback loops would explain why several defects with a widely varying biochemical background lead to the same complex of symptoms.
Our rather simplistic model necessarily ignores many details that complicate the picture, e.g. we do not consider the regional and developmental heterogeneity in the GABAergic system (e.g. excitatory GABA action in early development and the enormous plasticity of the central nervous system that will result in important compensatory regulation phenomena. Also, relatively little is known about the functional distribution of DBI in the central nervous system and its autocrine vs. paracrine effects.
In fact, the DBI hypothesis is not able to explain all observations in peroxisome deficiencies. However, it seems that the DBI hypothesis is able to explain a larger part of the observations than previous ideas, e.g. the Pex11beta k.o. phenotype seems to follow naturally from the suggestion that slight initiating changes in a metabolite might lead to an escalating response via feed-back mechanisms. It is very well possible that the DBI hypothesis is incomplete and/or over-simplified (for example, explaining some differences between Zellweger and Zellweger-like syndromes might require postulating different mechanisms of action for DBI in these conditions) but we hope that even a partially correct hypothesis may be able to point research into a useful direction, e.g. concerning the importance of small perturbations amplified by misactivated feedback mechanisms.
The existence of several mouse models of ZS should allow for rapid testing and refinement of our idea that misregulation of DBI is involved in the pathogenesis of Zellweger syndrome.
Competing interests
None declared.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
I thank Dr. D. Höller and Dr. S. K. Krisans for helpful discussions. This work was supported in part by National Institutes of Health grants DK58238 and DK58040.
References
- Baumgartner Matthias R., Saudubray Jean Marie. Peroxisomal disorders. Seminars in Neonatology. 2002;7:85–94. doi: 10.1053/siny.2001.0089. [DOI] [PubMed] [Google Scholar]
- Gould SJ, Raymond GV, Valle D. The peroxisome biogenesis disorders. In: Scriver C R, Beaudet A L, Valle D and Sly W S, editor. The metabolic & molecular bases of inherited disease. 8th. Vol. 3. New York, McGraw-Hill; 2001. p. 3181–3217. [Google Scholar]
- Sacksteder KA, Gould SJ. The genetics of peroxisome biogenesis. Annu Rev Genet. 2000;34:623–652. doi: 10.1146/annurev.genet.34.1.623. [DOI] [PubMed] [Google Scholar]
- Powers JM, Moser HW. Peroxisomal disorders: genotype, phenotype, major neuropathologic lesions, and pathogenesis. Brain Pathol. 1998;8:101–120. doi: 10.1111/j.1750-3639.1998.tb00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez M, Vazquez E, Garcia-Silva MT, Manzanares J, Bertran JM, Castello F, Mougan I. Therapeutic effects of docosahexaenoic acid ethyl ester in patients with generalized peroxisomal disorders. Am J Clin Nutr. 2000;71:376S–85S. doi: 10.1093/ajcn/71.1.376s. [DOI] [PubMed] [Google Scholar]
- Mayatepek E, Lehmann WD, Fauler J, Tsikas D, Frolich JC, Schutgens RB, Wanders RJ, Keppler D. Impaired degradation of leukotrienes in patients with peroxisome deficiency disorders. J Clin Invest. 1993;91:881–888. doi: 10.1172/JCI116309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedlitschky G, Mayatepek E, Keppler D. Peroxisomal leukotriene degradation: biochemical and clinical implications. Adv Enzyme Regul. 1993;33:181–194. doi: 10.1016/0065-2571(93)90017-8. [DOI] [PubMed] [Google Scholar]
- Gressens P, Baes M, Leroux P, Lombet A, Van Veldhoven P, Janssen A, Vamecq J, Marret S, Evrard P. Neuronal migration disorder in Zellweger mice is secondary to glutamate receptor dysfunction. Ann Neurol. 2000;48:336–343. doi: 10.1002/1531-8249(200009)48:3<336::AID-ANA8>3.3.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Wanders RJA, Barth PG, Heymanns HS. Single peroxisomal enzyme deficiencies. In: Scriver C R, Beaudet A L, Valle D and Sly W S, editor. The metabolic & molecular bases of inherited disease. Vol. 3. New York, McGraw-Hill; 2001. p. 3219–3256. [Google Scholar]
- Faust PL, Hatten ME. Targeted deletion of the PEX2 peroxisome assembly gene in mice provides a model for Zellweger syndrome, a human neuronal migration disorder. J Cell Biol. 1997;139:1293–1305. doi: 10.1083/jcb.139.5.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baes M, Gressens P, Baumgart E, Carmeliet P, Casteels M, Fransen M, Evrard P, Fahimi D, Declercq PE, Collen D, van Veldhoven PP, Mannaerts GP. A mouse model for Zellweger syndrome. Nat Genet. 1997;17:49–57. doi: 10.1038/ng0997-49. [DOI] [PubMed] [Google Scholar]
- Li X, Baumgart E, Morrell JC, Jimenez-Sanchez G, Valle D, Gould SJ. PEX11 beta deficiency is lethal and impairs neuronal migration but does not abrogate peroxisome function. Mol Cell Biol. 2002;22:4358–4365. doi: 10.1128/MCB.22.12.4358-4365.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Baumgart E, Dong GX, Morrell JC, Jimenez-Sanchez G, Valle D, Smith KD, Gould SJ. PEX11alpha is required for peroxisome proliferation in response to 4-phenylbutyrate but is dispensable for peroxisome proliferator-activated receptor alpha-mediated peroxisome proliferation. Mol Cell Biol. 2002;22:8226–8240. doi: 10.1128/MCB.22.23.8226-8240.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante JP, Huszagh VA. Zellweger syndrome knockout mouse models challenge putative peroxisomal beta-oxidation involvement in docosahexaenoic acid (22:6n-3) biosynthesis. Mol Genet Metab. 2001;72:1–7. doi: 10.1006/mgme.2000.3101. [DOI] [PubMed] [Google Scholar]
- Janssen A, Baes M, Gressens P, Mannaerts GP, Declercq P, Van Veldhoven PP. Docosahexaenoic acid deficit is not a major pathogenic factor in peroxisome-deficient mice. Lab Invest. 2000;80:31–35. doi: 10.1038/labinvest.3780005. [DOI] [PubMed] [Google Scholar]
- Vanhorebeek I, Baes M, Declercq PE. Isoprenoid biosynthesis is not compromised in a Zellweger syndrome mouse model. Biochim Biophys Acta. 2001;1532:28–36. doi: 10.1016/S1388-1981(01)00108-1. [DOI] [PubMed] [Google Scholar]
- Janssen A, Gressens P, Grabenbauer M, Baumgart E, Schad A, Vanhorebeek I, Brouwers A, Declercq PE, Fahimi D, Evrard P, Schoonjans L, Collen D, Carmeliet P, Mannaerts G, Van Veldhoven P, Baes M. Neuronal migration depends on intact peroxisomal function in brain and in extraneuronal tissues. J Neurosci. 2003;23:9732–9741. doi: 10.1523/JNEUROSCI.23-30-09732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baes M, Gressens P, Huyghe S, De NK, Qi C, Jia Y, Mannaerts GP, Evrard P, Van VP, Declercq PE, Reddy JK. The neuronal migration defect in mice with Zellweger syndrome (Pex5 knockout) is not caused by the inactivity of peroxisomal beta-oxidation. J Neuropathol Exp Neurol. 2002;61:368–374. doi: 10.1093/jnen/61.4.368. [DOI] [PubMed] [Google Scholar]
- Baes M, Huyghe S, Carmeliet P, Declercq PE, Collen D, Mannaerts GP, Van Veldhoven PP. Inactivation of the peroxisomal multifunctional protein-2 in mice impedes the degradation of not only 2-methyl-branched fatty acids and bile acid intermediates but also of very long chain fatty acids. J Biol Chem. 2000;275:16329–16336. doi: 10.1074/jbc.M001994200. [DOI] [PubMed] [Google Scholar]
- Breitling R, Sharif O, Hartman ML, Krisans SK. Loss of compartmentalization causes misregulation of lysine biosynthesis in peroxisome-deficient yeast cells. Eukaryot Cell. 2002;1:978–986. doi: 10.1128/EC.1.6.978-986.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Welsh JB, Sapinoso LM, Kern SG, Dimitrov P, Lapp H, Schultz PG, Powell SM, Moskaluk CA, Frierson H. F., Jr., Hampton GM. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 2001;61:7388–7393. [PubMed] [Google Scholar]
- Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, Brown PO. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99:12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitroulakos J, Marhin WH, Tokunaga J, Irish J, Gullane P, Penn LZ, Kamel-Reid S. Microarray and biochemical analysis of lovastatin-induced apoptosis of squamous cell carcinomas. Neoplasia. 2002;4:337–346. doi: 10.1038/sj.neo.7900247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa E, Guidotti A. Diazepam binding inhibitor (DBI): a peptide with multiple biological actions. Life Sci. 1991;49:325–344. doi: 10.1016/0024-3205(91)90440-M. [DOI] [PubMed] [Google Scholar]
- Knudsen J, Mandrup S, Rasmussen JT, Andreasen PH, Poulsen F, Kristiansen K. The function of acyl-CoA-binding protein (ACBP)/diazepam binding inhibitor (DBI) Mol Cell Biochem. 1993;123:129–138. doi: 10.1007/BF01076484. [DOI] [PubMed] [Google Scholar]
- Bürgi B, Lichtensteiger W, Lauber ME, Schlumpf M. Ontogeny of diazepam binding inhibitor/acyl-CoA binding protein mRNA and peripheral benzodiazepine receptor mRNA expression in the rat. J Neuroendocrinol. 1999;11:85–100. doi: 10.1046/j.1365-2826.1999.00292.x. [DOI] [PubMed] [Google Scholar]
- Bovolin P, Schlichting J, Miyata M, Ferrarese C, Guidotti A, Alho H. Distribution and characterization of diazepam binding inhibitor (DBI) in peripheral tissues of rat. Regul Pept. 1990;29:267–281. doi: 10.1016/0167-0115(90)90089-F. [DOI] [PubMed] [Google Scholar]
- Alho H, Harjuntausta T, Schultz R, Pelto-Huikko M, Bovolin P. Immunohistochemistry of diazepam binding inhibitor (DBI) in the central nervous system and peripheral organs: its possible role as an endogenous regulator of different types of benzodiazepine receptors. Neuropharmacology. 1991;30:1381–1386. doi: 10.1016/s0028-3908(11)80005-5. [DOI] [PubMed] [Google Scholar]
- Alho H, Fremeau R. T., Jr., Tiedge H, Wilcox J, Bovolin P, Brosius J, Roberts JL, Costa E. Diazepam binding inhibitor gene expression: location in brain and peripheral tissues of rat. Proc Natl Acad Sci U S A. 1988;85:7018–7022. doi: 10.1073/pnas.85.18.7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmer M, Rovio A, Alho H. The characterization of two diazepam binding inhibitor (DBI) transcripts in humans. Biochem J. 1995;306 ( Pt 2):327–330. doi: 10.1042/bj3060327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocchetti I, Santi MR. Diazepam binding inhibitor peptide: cloning and gene expression. Neuropharmacology. 1991;30:1365–1371. doi: 10.1016/s0028-3908(11)80003-1. [DOI] [PubMed] [Google Scholar]
- Owens GP, Sinha AK, Sikela JM, Hahn WE. Sequence and expression of the murine diazepam binding inhibitor. Brain Res Mol Brain Res. 1989;6:101–108. doi: 10.1016/0169-328X(89)90043-0. [DOI] [PubMed] [Google Scholar]
- Guarneri P, Berkovich A, Guidotti A, Costa E. A study of diazepam binding inhibitor (DBI) processing products in human cerebrospinal fluid and in postmortem human brain. Neuropharmacology. 1990;29:419–428. doi: 10.1016/0028-3908(90)90162-K. [DOI] [PubMed] [Google Scholar]
- Ferrarese C, Cogliati T, Tortorella R, Zucca C, Bogliun G, Beghi E, Passoni D, Zoia C, Begni B, Airoldi L, Alho H, Frattola L. Diazepam binding inhibitor (DBI) in the plasma of pediatric and adult epileptic patients. Epilepsy Res. 1998;29:129–134. doi: 10.1016/S0920-1211(97)00074-0. [DOI] [PubMed] [Google Scholar]
- Edgar PF, Schonberger SJ, Dean B, Faull RL, Kydd R, Cooper GJ. A comparative proteome analysis of hippocampal tissue from schizophrenic and Alzheimer's disease individuals. Mol Psychiatry. 1999;4:173–178. doi: 10.1038/sj.mp.4000463. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Berkovich A, Krueger KE, Costa E, Guidotti A. Diazepam binding inhibitor and its processing products stimulate mitochondrial steroid biosynthesis via an interaction with mitochondrial benzodiazepine receptors. Endocrinology. 1991;129:1481–1488. doi: 10.1210/endo-129-3-1481. [DOI] [PubMed] [Google Scholar]
- Cavallaro S, Korneyev A, Guidotti A, Costa E. Diazepam-binding inhibitor (DBI)-processing products, acting at the mitochondrial DBI receptor, mediate adrenocorticotropic hormone-induced steroidogenesis in rat adrenal gland. Proc Natl Acad Sci U S A. 1992;89:10598–10602. doi: 10.1073/pnas.89.22.10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boujrad N, Hudson J. R., Jr., Papadopoulos V. Inhibition of hormone-stimulated steroidogenesis in cultured Leydig tumor cells by a cholesterol-linked phosphorothioate oligodeoxynucleotide antisense to diazepam-binding inhibitor. Proc Natl Acad Sci U S A. 1993;90:5728–5731. doi: 10.1073/pnas.90.12.5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino M, Marino F, Cattaneo S, Di Grazia L, Francioli C, Fietta AM, Lecchini S, Frigo G. Diazepam-binding inhibitor-derived peptides induce intracellular calcium changes and modulate human neutrophil function. J Leukoc Biol. 2000;67:637–643. doi: 10.1002/jlb.67.5.637. [DOI] [PubMed] [Google Scholar]
- Gandolfo P, Patte C, Leprince J, Thoumas JL, Vaudry H, Tonon MC. The stimulatory effect of the octadecaneuropeptide (ODN) on cytosolic Ca2+ in rat astrocytes is not mediated through classical benzodiazepine receptors. Eur J Pharmacol. 1997;322:275–281. doi: 10.1016/S0014-2999(97)00012-5. [DOI] [PubMed] [Google Scholar]
- Rosendal J, Ertbjerg P, Knudsen J. Characterization of ligand binding to acyl-CoA-binding protein. Biochem J. 1993;290 ( Pt 2):321–326. doi: 10.1042/bj2900321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen J, Neergaard TB, Gaigg B, Jensen MV, Hansen JK. Role of acyl-CoA binding protein in acyl-CoA metabolism and acyl-CoA-mediated cell signaling. J Nutr. 2000;130:294S–298S. doi: 10.1093/jn/130.2.294S. [DOI] [PubMed] [Google Scholar]
- Helledie T, Grontved L, Jensen SS, Kiilerich P, Rietveld L, Albrektsen T, Boysen MS, Nohr J, Larsen LK, Fleckner J, Stunnenberg HG, Kristiansen K, Mandrup S. The gene encoding the Acyl-CoA-binding protein is activated by peroxisome proliferator-activated receptor gamma through an intronic response element functionally conserved between humans and rodents. J Biol Chem. 2002;277:26821–26830. doi: 10.1074/jbc.M111295200. [DOI] [PubMed] [Google Scholar]
- Swinnen JV, Alen P, Heyns W, Verhoeven G. Identification of diazepam-binding Inhibitor/Acyl-CoA-binding protein as a sterol regulatory element-binding protein-responsive gene. J Biol Chem. 1998;273:19938–19944. doi: 10.1074/jbc.273.32.19938. [DOI] [PubMed] [Google Scholar]
- Borboni P, Condorelli L, De Stefanis P, Sesti G, Lauro R. Modulation of insulin secretion by diazepam binding inhibitor and its processing products. Neuropharmacology. 1991;30:1399–1403. doi: 10.1016/s0028-3908(11)80008-0. [DOI] [PubMed] [Google Scholar]
- Ostenson CG, Ahren B, Karlsson S, Sandberg E, Efendic S. Effects of porcine diazepam-binding inhibitor on insulin and glucagon secretion in vitro from the rat endocrine pancreas. Regul Pept. 1990;29:143–151. doi: 10.1016/0167-0115(90)90077-A. [DOI] [PubMed] [Google Scholar]
- Laegreid L, Olegard R, Wahlstrom J, Conradi N. Abnormalities in children exposed to benzodiazepines in utero. Lancet. 1987;1:108–109. doi: 10.1016/S0140-6736(87)91951-9. [DOI] [PubMed] [Google Scholar]
- Laegreid L, Olegard R, Walstrom J, Conradi N. Teratogenic effects of benzodiazepine use during pregnancy. J Pediatr. 1989;114:126–131. doi: 10.1016/s0022-3476(89)80619-5. [DOI] [PubMed] [Google Scholar]
- Laegreid L, Olegard R, Conradi N, Hagberg G, Wahlstrom J, Abrahamsson L. Congenital malformations and maternal consumption of benzodiazepines: a case-control study. Dev Med Child Neurol. 1990;32:432–441. doi: 10.1111/j.1469-8749.1990.tb16962.x. [DOI] [PubMed] [Google Scholar]
- Laegreid L, Olegard R, Wahlstrom J, Conradi N, Sisfontes L. Benzodiazepine overconsumption in pregnancy. Lancet. 1987;2:1405–1406. doi: 10.1016/S0140-6736(87)91303-1. [DOI] [PubMed] [Google Scholar]
- Winter RM. In utero Exposure to Benzodiazepines. Lancet. 1987;1:627–627. doi: 10.1016/S0140-6736(87)90265-0. [DOI] [PubMed] [Google Scholar]
- Iqbal MM, Sobhan T, Ryals T. Effects of commonly used benzodiazepines on the fetus, the neonate, and the nursing infant. Psychiatr Serv. 2002;53:39–49. doi: 10.1176/appi.ps.53.1.39. [DOI] [PubMed] [Google Scholar]
- Katz RA. Effect of diazepam on the embryonic development of the palate in the rat. J Craniofac Genet Dev Biol. 1988;8:155–166. [PubMed] [Google Scholar]
- Tocco DR, Renskers K, Zimmerman EF. Diazepam-induced cleft palate in the mouse and lack of correlation with the H-2 locus. Teratology. 1987;35:439–445. doi: 10.1002/tera.1420350316. [DOI] [PubMed] [Google Scholar]
- Zimmerman EF. Role of neurotransmitters in palate development and teratologic implications. Prog Clin Biol Res. 1985;171:283–294. [PubMed] [Google Scholar]
- Tucker JC. Benzodiazepines and the developing rat: a critical review. Neurosci Biobehav Rev. 1985;9:101–111. doi: 10.1016/0149-7634(85)90036-3. [DOI] [PubMed] [Google Scholar]
- Condie BG, Bain G, Gottlieb DI, Capecchi MR. Cleft palate in mice with a targeted mutation in the gamma-aminobutyric acid-producing enzyme glutamic acid decarboxylase 67. Proc Natl Acad Sci U S A. 1997;94:11451–11455. doi: 10.1073/pnas.94.21.11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culiat CT, Stubbs LJ, Woychik RP, Russell LB, Johnson DK, Rinchik EM. Deficiency of the beta 3 subunit of the type A gamma-aminobutyric acid receptor causes cleft palate in mice. Nat Genet. 1995;11:344–346. doi: 10.1038/ng1195-344. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Forchetti CM, Corda MG, Konkel D, Bennett CD, Costa E. Isolation, characterization, and purification to homogeneity of an endogenous polypeptide with agonistic action on benzodiazepine receptors. Proc Natl Acad Sci U S A. 1983;80:3531–3535. doi: 10.1073/pnas.80.11.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schousboe A. Pharmacologic and therapeutic aspects of the developmentally regulated expression of GABA(A) and GABA(B) receptors: cerebellar granule cells as a model system. Neurochem Int. 1999;34:373–377. doi: 10.1016/S0197-0186(99)00044-3. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Iribe Y, Tepper JM. GABAA receptor stimulation blocks NMDA-induced bursting of dopaminergic neurons in vitro by decreasing input resistance. Brain Res. 1999;832:145–151. doi: 10.1016/S0006-8993(99)01484-5. [DOI] [PubMed] [Google Scholar]
- Katsura M, Takesue M, Shuto K, Mohri Y, Tarumi C, Tsujimura A, Shirotani K, Ohkuma S. NMDA receptor activation enhances diazepam binding inhibitor and its mRNA expressions in mouse cerebral cortical neurons. Brain Res Mol Brain Res. 2001;88:161–165. doi: 10.1016/S0169-328X(01)00030-4. [DOI] [PubMed] [Google Scholar]
- Lamacz M, Tonon MC, Smih-Rouet F, Patte C, Gasque P, Fontaine M, Vaudry H. The endogenous benzodiazepine receptor ligand ODN increases cytosolic calcium in cultured rat astrocytes. Brain Res Mol Brain Res. 1996;37:290–296. doi: 10.1016/0169-328X(95)00330-U. [DOI] [PubMed] [Google Scholar]
- Ferrarese C, Vaccarino F, Alho H, Mellstrom B, Costa E, Guidotti A. Subcellular location and neuronal release of diazepam binding inhibitor. J Neurochem. 1987;48:1093–1102. doi: 10.1111/j.1471-4159.1987.tb05632.x. [DOI] [PubMed] [Google Scholar]
- Patte C, Gandolfo P, Leprince J, Thoumas JL, Fontaine M, Vaudry H, Tonon MC. GABA inhibits endozepine release from cultured rat astrocytes. Glia. 1999;25:404–411. doi: 10.1002/(SICI)1098-1136(19990215)25:4<404::AID-GLIA9>3.3.CO;2-H. [DOI] [PubMed] [Google Scholar]
- De Stefanis P, Impagnatiello F, Berkovich A, Guidotti A. Inhibitory effect of ODN, a naturally occurring processing product of diazepam binding inhibitor, on secretagogues-induced insulin secretion. Regul Pept. 1995;56:153–165. doi: 10.1016/0167-0115(95)00002-S. [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Van Alphen AM, Hawkins RK, Ruigrok TJ. Climbing fibre collaterals contact neurons in the cerebellar nuclei that provide a GABAergic feedback to the inferior olive. Neuroscience. 1997;80:981–986. doi: 10.1016/S0306-4522(97)00249-2. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Somogyi J, Stewart MG, Hamori J. GABA-immunoreactive neurons in the rat cerebellum: a light and electron microscope study. J Comp Neurol. 1986;251:474–490. doi: 10.1002/cne.902510404. [DOI] [PubMed] [Google Scholar]
- Aoki E, Semba R, Kashiwamata S. When does GABA-like immunoreactivity appear in the rat cerebellar GABAergic neurons? Brain Res. 1989;502:245–251. doi: 10.1016/0006-8993(89)90619-7. [DOI] [PubMed] [Google Scholar]
- Xu W, Cormier R, Fu T, Covey DF, Isenberg KE, Zorumski CF, Mennerick S. Slow death of postnatal hippocampal neurons by GABA(A) receptor overactivation. J Neurosci. 2000;20:3147–3156. doi: 10.1523/JNEUROSCI.20-09-03147.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Cherubini E, Gaiarsa JL, Ben-Ari Y. GABA: an excitatory transmitter in early postnatal life. Trends Neurosci. 1991;14:515–519. doi: 10.1016/0166-2236(91)90003-D. [DOI] [PubMed] [Google Scholar]
- Leinekugel X, Khalilov I, McLean H, Caillard O, Gaiarsa JL, Ben-Ari Y, Khazipov R. GABA is the principal fast-acting excitatory transmitter in the neonatal brain. Adv Neurol. 1999;79:189–201. [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/S0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]