

Abstract
Local side-chain interactions in lyophilized protein formulations were mapped using solid-state photolytic labeling-mass spectrometry (ssPL-MS). Photoactive amino acid analogs (PAAs) were used as probes and either added to the lyophilized matrix or incorporated within the amino acid sequence of a peptide. In the first approach, apomyoglobin was lyophilized with sucrose and varying concentrations of photo-leucine (L-2-amino-4, 4′-azipentanoic acid; pLeu). The lyophilized solid was irradiated at 365 nm to initiate photolabeling. The rate and extent of labeling were measured using ESI-HPLC-MS, with labeling reaching a plateau at ∼ 30 min, forming up to 6 labeled populations. Bottom-up MS/MS analysis was able to provide peptidelevel resolution of the location of pLeu. ssPL-MS was also able to detect differences in side-chain environment between sucrose and guanidine hydrochloride formulations. In the second approach, peptide GCG (1-8)* containing p-benzoyl-L-phenylalanine (pBpA) in the amino acid sequence was lyophilized with various excipients and irradiated. Peptide-peptide and peptide-excipient adducts were detected using MS. Top-down MS/MS on the peptide dimer provided amino acidlevel resolution regarding interactions and the cross-linking partner for pBpA in the solid state. The results show that ssPL-MS can provide high-resolution information about protein interactions in the lyophilized environment.
Keywords: Photolytic labeling, mass spectrometry, lyophilized formulations, protein-protein interactions, apomyoglobin, photo-leucine, p-benzoyl-L-phenylalanine
Introduction
Protein drugs are an increasingly important part of the global pharmaceuticals market. In 2009, the ten best-selling protein drugs had a combined sales value of close to $50 billion1. The number of approved protein drugs is expected to increase in the next few years, particularly given the expiration of patents and the growth of biosimilars. According to a report by Global Industry Analysts, Inc., biosimilars are expected to be valued at $17.9 million by 20172. However, the inherent instability of proteins and their tendency to aggregate is an obstacle to the development of these life-saving medicines. In an attempt to maintain stability and provide adequate shelf-life, many proteins are lyophilized. In addition to those products marketed as lyophilized powders, the protein itself may be lyophilized for storage prior to final formulation in either solution or solid forms. Although lyophilized formulations usually confer greater stability when compared to solution, degradation may still occur in the solid state and during the freeze-drying process3-8. Retention of native protein structure in the lyophilized solid has generally been associated with improved stability during shelf-storage and a decreased propensity for aggregate formation9-11. Ensuring the retention of native conformation would benefit from analytical methods that could identify subtle protein structural perturbations in lyophilized solids with high resolution. Such information could be used to design formulations rationally and to screen candidate formulations efficiently.
Most of the current analytical techniques used to characterize proteins in the solid state lack sufficient resolution to serve as design tools, however. Methods such as Fourier transforminfrared spectroscopy (FTIR), Raman spectroscopy and differential scanning calorimetry (DSC) have been used to study structural changes in lyophilized proteins12-16. These methods are semiquantitative at best, suffer from low sensitivity and can provide only low-resolution information on protein structure. Solid-state nuclear magnetic resonance spectroscopy (ssNMR) can provide site-specific information about conformational changes17, 18, but requires extensive sample preparation and isotopic labeling, and is less sensitive in amorphous samples than in those that are crystalline. Thus, ssNMR is not always useful for lyophilized protein formulations, which are usually amorphous. Recently, our group has developed solid-state hydrogen-deuterium exchange with mass spectrometric analysis (ssHDX-MS) to allow the protein environment in amorphous solids to be probed with higher resolution. ssHDX-MS provides structural information with peptide level resolution, and has been used previously by our group to characterize protein conformations in lyophilized solids containing various excipients19, 20.
In the work reported here, we have developed a complementary analytical technique, solid-state photolytic labeling with mass spectrometric analysis (ssPL-MS), to probe protein structure and matrix interactions in lyophilized formulations. In solution, PL-MS with photoreactive amino acid analogs (PAAs) has been used to study protein/peptide conformation and protein-protein interactions (PPIs)21-23. The approach has also been used in living cells to map the interactome24-26. In solution PL-MS, a solution containing protein and PAA is irradiated with UV light (350-365 nm), activating the PAA photoreactive functional group, which then forms a covalent bond between the PAA and protein in its immediate vicinity. The labeled protein is analyzed by MS at the intact protein level and by MS/MS fragmentation after enzymatic digestion (bottom-up) or direct fragmentation (top-down). The location of the label identifies sites on the protein that are accessible to the photoreactive probe, providing information about the side-chain environment. This makes the method complementary to HDX-MS, which probes backbone environment and secondary structure. Moreover, the covalently attached label is permanent and does not undergo back-exchange, a limitation of HDX. Solution state PL-MS has also been carried out by incorporating the PAA within a protein or peptide sequence22, 27. Exposure to UV light generates photoadducts of the PAA-containing protein/peptide with interacting molecules (e.g. ligand) in the microenvironment. These photoadducts can then be digested enzymatically and analyzed to identify the reactive sites at the interface of the complex.
In the current work, we have adapted PL-MS for proteins in lyophilized solids. PAAs were used to probe the side chain protein environment in two different ways: (i) by incorporating a PAA into the lyophilized solid as an excipient and (ii) by incorporating a PAA into the sequence of a model peptide. In studies using a PAA probe as an excipient (i), apomyoglobin (ApoMb) was selected as a model protein and L-photo-leucine (L-2-amino-4, 4′-azipentanoic acid; pLeu) was used as an excipient. In studies with the PAA incorporated into the protein sequence (ii), an octapeptide derived from the N-terminus of human glucagon (1-HSQGTFTS-8) with the phenylalanine residue (F6) replaced by the PAA p-benzoyl-L-phenylalanine (pBpA) was used. The two PAAs have different reaction mechanisms upon exposure to UV light. The diazirine functional group of pLeu forms a reactive carbene intermediate that inserts non-specifically into any C-C or X-H bond (X= C, N, O, S), or adds to a C=C bond in its immediate molecular cage. The benzophenone group in pBpA forms a reactive ketyl radical that reacts preferentially with C-H bonds and forms new C-C covalent linkages28, 29. The results demonstrate that photolytic labeling occurs in lyophilized solids when the label is either incorporated into the matrix (i) or into a model peptide (ii). The results also show that the extent of labeling varies with position in the protein sequence and with solid composition. To our knowledge, this is the first reported use of photolytic labeling to map the protein environment in lyophilized solids. The findings support further development of the method to probe the amorphous solid state and in formulation development.
Materials And Methods
Materials
Apomyoglobin (apoMb) from equine skeletal muscle, monobasic and dibasic potassium hydrogen phosphate, L-methionine (Met), L-leucine (Leu), sucrose, trehalose, urea and guanidine hydrochloride were purchased from Sigma Aldrich (St. Louis, MO). Two commercially available PAAs, L-photo-leucine (L-2-amino-4, 4′-azipentanoic acid; pLeu) and p-benzoyl-L-phenylalanine (pBpA) were used for photolytic studies. pLeu was obtained from Thermo Scientific (Rockford, IL). An octapeptide derived from the N-terminus of glucagon (HSQGT-pBpA-TS; henceforth referred to as GCG (1-8)*) containing the photoreactive amino acid pBpA within its sequence was synthesized by American Peptide Company (Sunnyvale, CA) and received as a lyophilized powder. Trypsin and chymotrypsin were purchased from Promega (Madison, WI) and mass spectrometry-grade water, acetonitrile and formic acid from Fisher Scientific (Fair Lawn, NJ).
Sample Preparation
ApoMb was dissolved in potassium phosphate buffer (20 mM, pH 7.4) to produce a stock solution of 200 μM protein and the solution was dialyzed using Biotech Cellulose Ester dialysis tubing (MWCO 8,000-10,000 Da, Spectrum Laboratories, Rancho Dominguez, CA) for 24 h into the same buffer. After dialysis, the solution was filtered through a 0.2 μm syringe filter (Gelman Nylon Acrodisc 13) and used for further experiments. Sucrose was selected as a cryoprotectant to preserve native protein structure during lyophilization30, 31. Sucrose stock solution (33.9 mg/mL) was prepared by dissolving sucrose in potassium phosphate buffer and filtering through a 0.2 μm syringe filter. The resulting solution was stored at 4 °C until use. A stock solution of pLeu (30 mM) was prepared similarly. Lyophilization was carried out with different ratios of protein to pLeu using a VirTis Plus AdVantage freeze dryer (SP Industries Inc., Gardiner, NY). ApoMb, sucrose and pLeu stock solutions were mixed such that the final protein concentration was 100 μM, the protein to sucrose ratio was 1:2 w/w and the protein to pLeu molar ratio was 1:20, 1:50 or 1:100. Control samples contained apoMb and sucrose without pLeu. The final volume for lyophilization was 80 μL. In order to produce a pharmaceutically relevant formulation, ∼ 50 % or more of the solid matrix consisted of sucrose, and buffer was less than 10 % of the formulation by weight (Table 1).
Table 1. Composition of lyophilized formulations containing apomyoglobin (apoMb)a and photo-leucine (pLeu)b.
| apoMb:pLeu molar ratio | Composition (%w/w) | |||
|---|---|---|---|---|
| ApoMb | Sucrose | pLeu | Buffer | |
| No pLeu | 31.0 | 61.9 | 0.0 | 7.1 |
| 1:20 | 29.4 | 58.5 | 5.0 | 6.8 |
| 1:50 | 27.4 | 54.8 | 11.6 | 6.3 |
| 1:100 | 24.5 | 49.1 | 20.7 | 5.6 |
Equine apomyoglobin
Photo-leucine (pLeu), L-2-amino-4,4-azipentanoic acid
All samples were lyophilized in vials made of borosilicate clear glass using an established conservative freeze-drying cycle. During the lyophilization cycle, the shelves were precooled to -2 °C. Freezing was carried out at -40 °C for 50 min, followed by drying under vacuum (70 mTorr) over 5 steps (-35 °C for 10 h, -20 °C for 8 h, -5 °C for 6 h, 10 °C for 6 h and 25 °C for 6 h). The lyophilized samples were stored at -20 °C until use. Solution controls were prepared at each composition.
Photolytic Labeling and MS Analysis of Intact Protein
Photolytic labeling was carried out using a UV Stratalinker 2400 equipped with five 365 nm UV lamps (Stratagene Corp., La Jolla, CA). The lamps were allowed to warm up for 5 min. Vials containing lyophilized samples and solution controls were uncapped and placed inside the UV chamber. The distance between the lamps and the cake at the bottom of the vial was approximately 15 cm. All samples were irradiated with UV light for 40 min. After irradiation, the solid was reconstituted with 800 μL of Solution A (A= 0.1 % formic acid in MS water) to give a final protein concentration of 10 μM. The solution formulation was diluted similarly. The samples were diluted further with Solution A and 20 pmol of protein was injected into the HPLC-MS system. Intact labeled protein was analyzed using HPLC-MS equipped with an ESI source (1200 series LC, 6520 qTOF; Agilent Technologies, Santa Clara, CA). Mass spectra were processed and deconvoluted using MassHunter software (Agilent Technologies). Percentages of protein populations with 1 through 6 labels were calculated using peak heights from extracted ion chromatograms:
| (1) |
where i denotes the number of labels (1-6), PHi denotes the peak height for labeled protein Li and PHu denotes the peak height of the unlabeled protein as observed by mass spectrometry. Hereinafter, the term ‘labeled’ will refer to a protein/peptide that has been exposed to pLeu and irradiation, and was covalently modified with one or more pLeu molecules. The term ‘unlabeled’ will refer to a protein/peptide that has been exposed to pLeu and irradiation, but was not labeled, while the term ‘native’ will refer to a protein/peptide that has not been exposed to pLeu and irradiation.
To test for structural perturbations upon labeling, samples were analyzed using solid-state FTIR (ssFTIR) and solution-state far UV CD spectroscopy. Formulations containing apoMb, sucrose and 100× molar excess of pLeu were lyophilized and irradiated for 40 min in the solid state. ssFTIR was performed using a Tensor 37 spectrometer (Bruker Optics, Billerica, MA) as described by Sophocleous et al 19. For CD spectroscopy, the irradiated samples were reconstituted in DDW to a final concentration of 4 μM and placed in a quartz cuvette with 1 cm path length. Spectra were acquired using a JASCO J-815 spectrometer (JASCO Analytical Instruments, Easton, MD) with 3 acquisitions at a scan rate of 50 nm/min. Non-irradiated control formulations were analyzed similarly.
Effect of Irradiation Time and pLeu Concentration on Labeling Efficiency
ApoMb lyophilized with sucrose and pLeu (1:100 molar ratio protein: pLeu, which is equivalent to 20.7 % w/w pLeu) was used to study the kinetics of photolytic labeling. Lyophilized samples were subjected to photolysis for different periods of time (0, 0.5, 2, 4, 10, 20, 30, 40 and 60 min). The samples were reconstituted and analyzed as described above. In a separate study, apoMb was lyophilized with sucrose and varying pLeu concentrations (0, 0.3, 1.3, 1.5, 2.5, 11.6 and 20.7 % w/w). The solid was irradiated at 365 nm for 40 min, reconstituted and analyzed for labeled protein. The fraction of labeled protein (FL) was calculated using peak heights from extracted ion chromatograms:
| (2) |
where PHL denotes the peak height for labeled protein and PHu denotes the peak height of the unlabeled protein as observed by mass spectrometry. FL represents the sum of populations of apoMb with 1-6 labels.
MS- and MS/MS- Analysis of Labeled apoMb Peptides
To identify the sites of photolytic labeling, apoMb was lyophilized with sucrose and pLeu as described above using 0, 0.3, 1.3, 1.5, 2.5, 11.6 and 20.7 % w/w pLeu. The solid was irradiated at 365 nm for 40 min and then reconstituted in ammonium bicarbonate buffer (100 μM, pH 8.0) to give a protein concentration of 10 μM. Enzymatic digestion of labeled apoMb was performed for 24 h at 60 °C using a combination of trypsin and chymotrypsin (1:1 molar ratio) at a total enzyme to protein molar ratio of 1:10. The reaction was then quenched with solution A and 20 pmol was injected into the LC-MS system. The proteolytic fragments were separated on a ZORBAX 300SB-C18 column (Agilent Technologies; 1.0×50 mm, particle size 3.5 μm). The column was equilibrated with 5% Mobile Phase B (B= 0.1 % formic acid in acetonitrile) and peptides were eluted at 50 μL/min using a gradient that increased from 5 to 45% B over 22 min and then from 45 to 95% B over 0.5 min. Mass spectra were processed using MassHunter and a theoretical digest map (with known sites of enzymatic cleavage, and allowing for up to 8 missed cleavages) was used to create a mass list for peptides carrying 0 through 7 labels. This theoretical list was matched against mass values obtained experimentally.
Based on this analysis, up to 15 labeled peptides were detected that carried one, two or four labels. One of these peptides, L32-K42 (LFTGHPETLEK) with one label was selected for MS/MS analysis. This precursor peptide had m/z = 462.9133 (z = +3) and was subjected to fragmentation using low-energy CID (Agilent Technologies), which predominantly produces b- and y-ions. Product ions were identified using MassHunter software.
Formulation Effects
In order to study the effect of excipients on side-chain environment, apoMb was lyophilized with 100× molar excess of pLeu in two formulations: the first with sucrose (1:2 w/w ratio of protein to sucrose) and the second with guanidine hydrochloride (Gdn HCl; 1.5 M final concentration). The final protein concentration was 100 μM and the concentration of pLeu was 20.7 % w/w (sucrose formulation) or ∼ 1 % w/w (Gdn HCl formulation). The lyophilized formulations were subjected to photolysis at 365 nm for 40 min. After reconstitution with ammonium bicarbonate buffer, the samples were digested with trypsin and chymotrypsin and peptide-level MS analysis was carried out as described above.
Photolytic Labeling with p-Benzoyl-L-Phenylalanine (pBpA)
GCG (1-8)* was dissolved in water to give a final concentration of 1 mM. The peptide was lyophilized alone or with one of the following excipients: sucrose, trehalose, urea, L-methionine and L-leucine (1:2 w/w ratio of peptide to excipient). After lyophilization, the formulations were irradiated with UV light (365 nm) for 30 min. The irradiated samples were then reconstituted in 200 μL of MS water containing 0.1 % formic acid. Solution controls prepared with or without excipients were lyophilized and reconstituted before irradiation. The samples were further diluted to 20 pmol of peptide for injection into the LC-MS system. MassHunter software was used to detect peptide-peptide and peptide-excipient adducts.
Photolytic labeling with GCG (1-8)* was also carried out with pLeu in the matrix. Two formulations were prepared. Formulation A contained GCG (1-8)* and pLeu at a 1:1 molar ratio, while Formulation B contained GCG (1-8)* and pLeu at a 1:1 molar ratio, together with sucrose (1:2 w/w ratio of GCG (1-8)* to sucrose). Both formulations were lyophilized as described above, irradiated with UV light for 30 min, reconstituted and analyzed by ESI-LC-MS. Solution controls were prepared and analyzed as described above.
MS/MS Analysis of GCG (1-8)* Dimer
Both lyophilized and solution formulations showed the presence of GCG (1-8)* dimer. LC-MS/MS was carried out on GCG (1-8)* monomer (m/z 968.41, z= +1) and GCG (1-8)* dimer (m/z 646.28, z= +3) in the peptide-Leu formulation. CID fragmentation was performed at 10 V and the resultant b- and y-ions were monitored. The dimer from solution controls (unlyophilized solution and lyophilized-rehydrated solution) was also analyzed by MS/MS.
Results
Labeling of Intact apoMb
The mechanisms of photolytic labeling with pLeu in solution are well understood21, 28. Briefly, photolysis of pLeu at 365 nm results in the loss of N2 with the generation of a reactive carbene. The carbene labels any C-C, C=C or X-H group (X=C, O, N, S) in its proximity without bias towards a particular amino acid or functional group. Photolabeling of myoglobin with pLeu has been carried out in solution, with successful labeling at a 1:10,000 molar ratio of protein to pLeu21. Here, we investigated the covalent labeling of apoMb with pLeu in lyophilized solids. Intact protein was co-lyophilized with sucrose and pLeu as excipients in weight fractions that are pharmaceutically relevant.
Mass spectrometric analysis of lyophilized solids containing apoMb and pLeu showed that carbene labeling also occurs in the solid state. Peaks corresponding to labeled protein were observed, with masses differing by multiples of ∼115 amu (Fig. 1). The extent of labeling depended on the amount of pLeu in the matrix. Peaks corresponding to singly- and doublylabeled apoMb populations were observed when apoMb was lyophilized with a 20-fold molar excess of pLeu (Fig. 1). Similarly, peaks corresponding to up to 4 and 6 labels per protein molecule were observed for the 1:50 and 1:100 formulations, respectively. ApoMb lyophilized without pLeu showed no adduct formation after irradiation (data not shown), confirming that UV light did not cause protein cross-linking. Also, protein lyophilized with pLeu showed no labeling in the absence of UV light (data not shown). Moreover, solution controls showed no labeling of apoMb with 20-, 50-, 100- or 1000-fold molar excess of pLeu (data not shown), suggesting that the reactive carbene species was consumed by reaction with water rather than reacting with protein. The effect of labeling on apoMb structure was studied using solid-state FTIR and solution CD spectroscopy. The second-derivative FTIR spectra and the far UV CD spectra showed minor changes with no loss of secondary structure for apoMb in lyophilized solids and reconstituted solutions (Supporting Information, Fig. S1).
Figure 1.
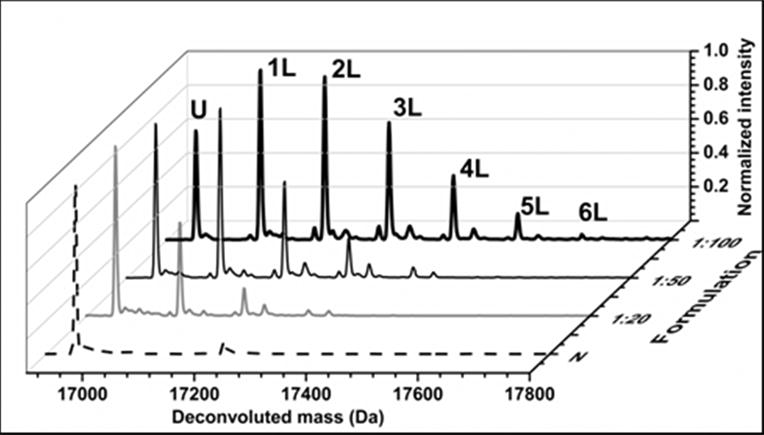
Deconvoluted mass spectra of native ApoMb (N) and ApoMb co-lyophilized with sucrose (1:2 w/w ratio of protein to sucrose) and pLeu in molar ratios 1:20, 1:50 and 1:100. Mass spectra show peaks for unlabeled apoMb (U) and labeled apoMb (nL) (n=1-6). The peaks differ by ∼ 115 amu, corresponding to the mass of one pLeu label.
Labeling Kinetics for Intact apoMb
During exposure of solid samples to UV irradiation, the fraction of labeled protein increased with time (Fig. 2A). The rate of formation of labeled protein was rapid initially and plateaued at ∼30 min with ∼20% of the protein remaining unlabeled (Fig. 2A). Labeling followed monoexponential kinetics as a function of irradiation time. To determine the effect of pLeu concentration on the plateau value, the extent of labeling was measured at different initial concentrations of pLeu with 40 min of irradiation (Fig. 2B). At 0 % w/w pLeu, no labeling occurred. As pLeu concentration was increased, the fraction of labeled protein increased until at 20.7 % w/w pLeu, ∼35 % unlabeled protein remained after 40 min of irradiation. The dependence of the extent of modification on pLeu concentration also followed monoexponential behavior.
Figure 2.
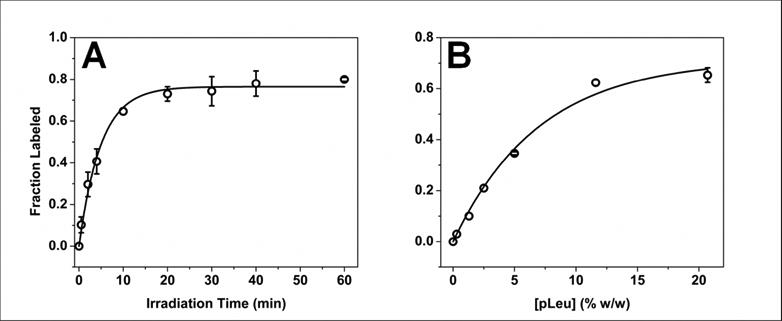
(A) Kinetics of photolytic labeling of apoMb in lyophilized solids containing 20.7 % w/w pLeu in the matrix, 365 nm irradiation. The solid line is fit to Eqn. 3. n = 3 ± SD. (B) Dependence of ApoMb photolytic labeling on the concentration of pLeu after 40 min irradiation at 365 nm. The solid line is fit to Eqn. 3. n = 3 ± SD.
An exponential model was used to simultaneously fit the rate and extent of labeling:
| (3) |
where FL(C, t) is the fraction of labeled protein as a function of pLeu concentration (C) and irradiation time (t), k1 and k2 are apparent first-order rate constants for the rate and extent of labeling, respectively, and A is the fraction of protein labeled at plateau. Nonlinear regression (Origin Pro v.8.6, OriginLab, Northampton, MA; n = 48) returned values of the regression parameters of A = 0.82 (±0.03), k1= 0.22 min-1 (±0.02) and k2= 0.12 mM-1 (±0.01).
Peptide-Level Labeling of apoMb
In order to investigate the specificity of labeling, sites of labeling were probed using bottom-up mass spectrometry. Digestion of native apoMb with trypsin/chymotrypsin produced 36 peptides, of which 13 were selected to provide 100 % sequence coverage (Supporting Information, Fig. S2). Labeled apoMb showed ∼ 96 % sequence coverage and a maximum of fifteen peptide fragments (obtained with 20.7 % w/w pLeu) with one, two or four labels (Fig. 3). As expected, the signal intensity of labeled peptides was less than that of unlabeled peptides, supporting the incomplete labeling observed at the intact protein level. Proteolytic digestion was influenced by the presence of pLeu labels: both the labeled and unlabeled peptides obtained after digestion of labeled apoMb differed from those in the native protein (Fig. 3). This suggests that the label interferes with digestion by obstructing access by the enzyme.
Figure 3.
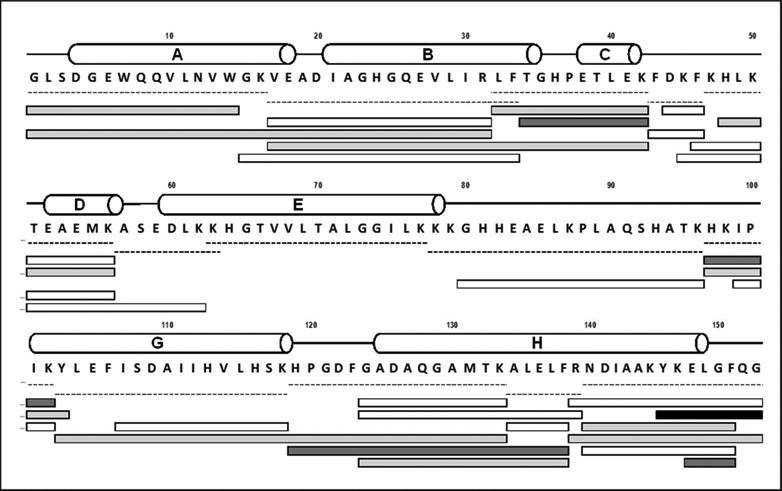
Digest map of apoMb labeled with 10 mM pLeu. Labeled apoMb was digested with a combination of trypsin and chymotrypsin. White bars represent unlabeled peptides, while labeled peptides are shown in gray (light gray bars carry one label; dark gray bars carry two labels and the black bar carries four labels). Dashed lines represent native peptides. Helical secondary structure is represented by cylinders labeled A-E, G and H. Helix F of holomyoglobin (H82-H97) is disordered in native apoMb at neutral pH (see ref. 27).
As the concentration of pLeu increased, labeling was detected in different regions of the protein (Fig. 4). Labeling at the peptide level was obtained using MS analysis of digested labeled apoMb at various pLeu concentrations (Fig. 4(b-h)). At 0 % w/w pLeu, no labeling was observed (Fig. 4b). At 0.3 % w/w pLeu, peptides L32-K42 and T34-K42 were labeled (Fig. 4c). This region forms helix C and part of helix B. At 1.3 % w/w pLeu, an additional peptide HKIPIKY (H97-Y103; located on a loop and part of helix G) was labeled (Fig. 4d). As pLeu concentration was increased to 2.5 % w/w, labeling was detected in peptide H119-F138 (helix H) in addition to L32-K42, T34-K42 and H97-Y103 (Fig. 4e). At 5 % w/w and 11.6 % w/w pLeu, Y103-K133 was labeled as well (Fig. 4f, g). At 20.7 % w/w pLeu, label was detected in G1-W14, G1-R31, V17-K42 (helices A, B and C), H48-K56 (helix D), H97-K102 (helix G), G124-F138, N140-F151, Y146-G153, E148-F151 and R139-G153 (helix H), in addition to the previously mentioned sequences (Fig. 4h). Increased label uptake at the C terminus is consistent with our previous solid-state hydrogen-deuterium exchange (ssHDX-MS) results for myoglobin, which showed greater deuterium uptake in this region even in the solid state19. Overall, labeling was observed across helices A, B, C, D, G and H. No labeling was observed on amino acids A57-K96, which form helices E and F. These two helices are involved in heme binding in holomyoglobin (holoMb), but are considerably disordered in apoMb32, 33. The absence of label suggests that this region is protected from matrix exposure at the tertiary structure level in the solid state.
Figure 4.
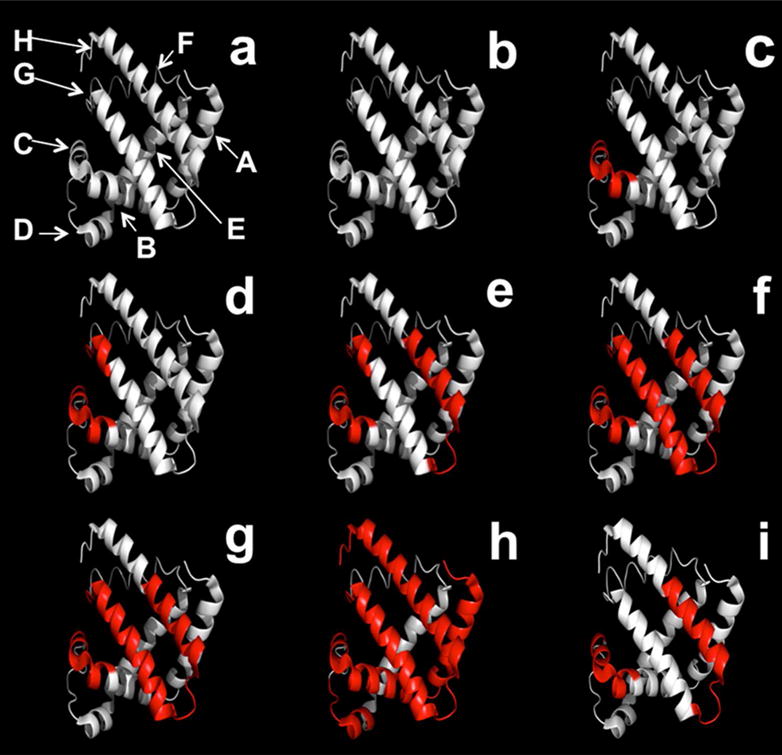
(a) Ribbon diagram of apoMb showing helices A-E, G and H. (b-h) Ribbon diagram of apoMb showing covalent labeling with increasing amounts of pLeu in the matrix in the presence of sucrose. (b) 0 (c) 0.3 (d) 1.3 (e) 2.5 (f) 5.0 (g) 11.6 (h) 20.7 %w/w pLeu. (i) Ribbon diagram of apoMb showing covalent labeling with 20.7 % w/w pLeu in the presence of Gdn HCl (1.5 M). The ribbon diagrams were generated using PyMOL (PyMOL Molecular Graphics System, Version 1.4.1, Schrödinger, LLC) and the crystal structure of myoglobin (PDB ID 1WLA; www.rcsb.org). Helix F (H82-H97) in the myoglobin structure was modified to an unstructured region, which is observed for native apoMb at neutral pH (see ref 27).
MS/MS Analysis of Peptide L32-K42 (LFTGHPETLEK)
To obtain additional information on the sites of photolytic labeling, tandem MS analysis was carried out on the singly-labeled peptide L32-K42, both in the labeled and native form. After fragmenting the native peptide, almost all b- and y-ions were observed (Supporting Information, Table S1, I and II). However, no b-ions were observed in the labeled peptide product ion mass spectrum. Six y-ions (y6, y7, y8, y9, y10 and y11) with z = +2 and three y-ions (y4, y5 and y6) with z = +1 were identified by fragmenting the labeled peptide at 13 V (Fig. 5; Supporting Information Table S1, III and IV). Unlabeled y-ions (y1-y10) were also observed upon fragmentation of the labeled peptide (Supporting Information Table S1, III and IV; dotted arrows in Fig. 5). Assuming that the ionization and fragmentation efficiencies of the labeled and native peptides are similar, and that the instrument is sensitive toward all possible labeled and unlabeled ions, the results suggest two possible reasons for the differences in fragmentation patterns: (1) Labeling is sitespecific at Thr (peptide TLEK), since unlabeled y1-y3 and labeled y4-y11 were observed. The presence of unlabeled y4-y10 could indicate loss of label from Thr during fragmentation. (2) Labeling is heterogeneous, with multiple sites of modification ranging from Leu to Thr (peptide LFTGHPET), since labeled y4-y11 and unlabeled y1-y10 were observed. The presence of unlabeled y1-y10 may be due to neutral loss of label from any of the labeled amino acids in peptide LFTGHPET. The absence of b-ions in the product ion spectrum of the labeled peptide makes it difficult to establish the cause of the differences in labeling pattern.
Figure 5.
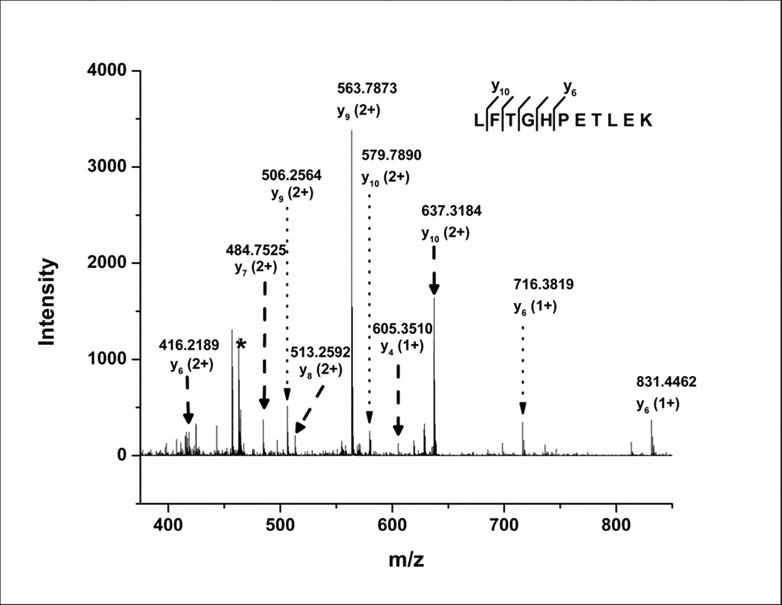
MS/MS spectrum of labeled peptide L32-K42 showing y-ion products obtained by CID fragmentation. The asterisk indicates the precursor peptide peak (m/z = 462.91). Dashed arrows represent y-ion peaks produced from the labeled peptide and dotted arrows represent y-ion peaks produced by possible loss of the label from the corresponding labeled y-ions. Labeled y-ions y11 (z=+2) and y5 (z=+1) are not shown due to low abundance.
Formulation Effects on apoMb Labeling
ApoMb lyophilized with Gdn HCl and 100× molar excess of pLeu (∼ 1% w/w pLeu) was analyzed for label uptake at the peptide level. MS analysis after enzymatic digestion showed that labeling occurred at peptides L32-K42, T34-K42 and H119-F138 (Fig. 4(i)). This is similar to the labeling observed with sucrose at 2.5 % w/w pLeu, but with no labeling on the G helix (at the BG contact interface). Gdn HCl is expected to have a chaotropic effect on protein structure and to cause increased label uptake due to protein unfolding and higher solvent exposure. In contrast, sucrose is expected to preserve the native structure of the protein through preferential exclusion and show lower labeling. These differences between expected and observed labeling patterns may be attributed to changes in protein side-chain environment caused by Gdn HCl.
Photolytic Labeling with p-benzoyl-L-phenylalanine (pBpA)
To monitor molecular interactions at the amino acid level, a known residue in peptide sequence 1-HSQGTFTS-8 derived from human glucagon was substituted with PAA. In this peptide, GCG (1-8)*, the F6 residue is substituted with pBpA. GCG (1-8)* lyophilized with and without excipients and irradiated in the solid state showed the formation of peptide-peptide adducts. GCG (1-8)* dimers and trimers were observed by ESI-LC-MS (Table 2); these adducts were also present in solution controls (Table 2). Peptide-excipient adducts were also observed for formulations with Met or Leu as excipients in solution and in the solid state, while formulations with sucrose, trehalose and urea as excipients showed no peptide-excipient adducts in either solid or solution state (Table 2).
Table 2. Cross-linked products formed after irradiation of GCG (1-8)* in various lyophilized formulations.
| Cross-linked Products | |||||
|---|---|---|---|---|---|
| Formulation | State | GCG (1-8)* Monomer | GCG (1-8)* Dimer | GCG (1-8)* Trimer | GCG (1-8)* + Excipient Adduct |
| GCG (1-8)* alone | Solid | + | + | + | N/A |
| Solution | + | + | + | N/A | |
| GCG (1-8)* + Sucrosea | Solid | + | + | + | - |
| Solution | + | + | + | - | |
| GCG (1-8)* + Trehalosea | Solid | + | + | + | - |
| Solution | + | + | + | - | |
| GCG (1-8)* + L-methioninea | Solid | + | + | + | + |
| Solution | + | + | + | + | |
| GCG (1-8)* + L-leucinea | Solid | + | + | + | + |
| Solution | + | + | + | + | |
| GCG (1-8)* + Ureaa | Solid | + | + | + | - |
| Solution | + | + | + | - | |
| Formulation Ab | Solid | + | + | + | + d,e |
| Solution | + | + | + | + d | |
| Formulation Bc | Solid | + | + | + | + d,e,f,g |
| Solution | + | + | + | + d | |
Excipients were added in a 2:1 w/w ratio with GCG (1-8)*.
Formulation A = GCG (1-8)* and pLeu in a 1:1 molar ratio.
Formulation B = GCG (1-8)*, pLeu and sucrose, with a 1:1 molar ratio of GCG (1-8)* and pLeu and 1:2 w/w ratio of GCG (1-8)* and sucrose.
GCG (1-8)* + pLeu adduct with loss of N2.
GCG (1-8)* + pLeu adduct without loss of N2.
pLeu + sucrose adduct with loss of N2.
GCG (1-8)* + pLeu + sucrose adduct with loss of N2.
Formulation A containing GCG (1-8)* and pLeu in lyophilized solids showed that cross-linking occurred between GCG (1-8)* molecules to form dimers and trimers. Cross-linking was also observed between GCG (1-8)* and pLeu both with and without the loss of N2. Similarly, Formulation B containing GCG (1-8)*, pLeu and sucrose produced GCG (1-8)* dimers, trimers and GCG (1-8)*-pLeu adducts with and without the loss of N2. pLeu-sucrose adducts with the loss of N2 and GCG (1-8)*-pLeu-sucrose adducts with the loss of N2 were also observed (Table 2). The solution controls for both Formulation A and B showed GCG (1-8)* dimer, trimer and GCG (1-8)*-pLeu adducts, but only with the loss of N2. No adducts with sucrose were observed in solution controls (Table 2).
MS/MS Analysis of GCG (1-8)* Monomer and Dimer
To identify the site of GCG (1-8)* dimerization, tandem mass spectrometry analysis was selectively carried out for GCG (1-8)* monomer and dimer. All b-ions (b1-b8; z=+2) and several y-ions were detected after CID fragmentation of GCG (1-8)* monomer (data not shown). Fragmentation of the dimer from lyophilized formulations produced cross-linked product ions in addition to internal fragment (non-cross-linked) b- and y-ions (Fig. 6; Supporting Information Table S2, I and II). In order to assign product ions to cross-linked sequences, the nomenclature proposed by Schilling and coworkers was used34. GCG (1-8)* monomer was designated as α, while b- and y-ions (from the second monomer unit in the dimer) cross-linked with α were designated as b∼α- and α∼y-ions. The following cross-linked ions were detected: b4∼α, b5∼α, b6∼α, b7∼α, α∼y5 and α∼y6. Internal fragment product ions b1, b2, b3, b5, y1, y2, y3 and y4 were also detected. The evidence suggests that, for lyophilized GCG (1-8)*, peptide-peptide cross-linking occurs preferentially between pBpA and Gly residues from two different GCG (1-8)* molecules.
Figure 6.
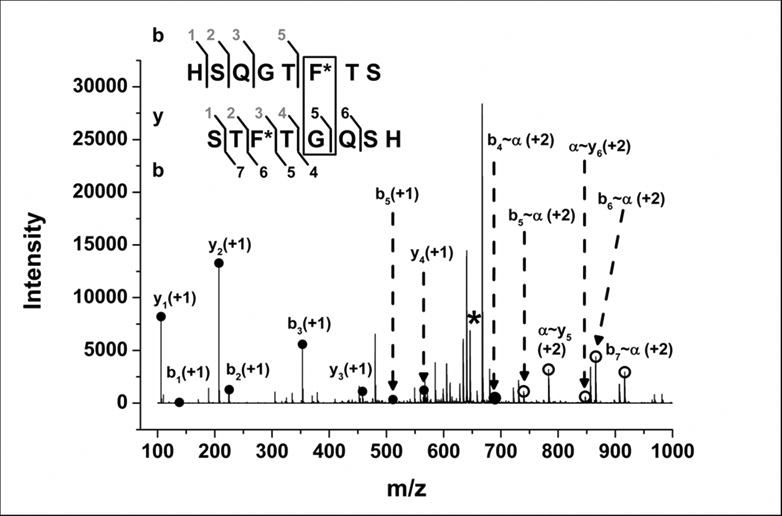
MS/MS spectrum of GCG (1-8)* dimer in the lyophilized formulation with L-Leucine showing b- and y-ion products obtained by CID fragmentation. The asterisk denotes the precursor dimer peak (m/z = 646.28). Closed circles represent simple (non-cross-linked) b- and y-ions. Open circles represent cross-linked b- and y-ions, labeled as b∼α and α∼y. Inset shows b- and y-ion sequences for internal fragment ions (numbered in grey) and cross-linked ions (numbered in black) detected.
In solution controls, fragmentation of the GCG (1-8)* dimer also produced internal fragment ions and cross-linked product ions (data not shown). An unambiguous assignment of the site of crosslinking could not be made, however, suggesting multiple sites of cross-linking in solution.
Discussion
Photolytic Labeling occurs in Lyophilized Solids
The studies presented here demonstrate successful photolytic labeling with pLeu and pBpA in lyophilized powders. To our knowledge, this is the first reported use of PAAs to study proteinprotein and protein-matrix interactions in amorphous solids, though previous studies have employed PL-MS in solutions in liquid and frozen states. For example, PL-MS using pLeu has been reported in solution for myoglobin and calmodulin21 using a 1:10,000 molar ratio of protein to pLeu and a pulsed laser for irradiation. Calmodulin was detected carrying up to 4 labels, while myoglobin showed up to 2 labels. Our studies with apoMb were unable to detect covalent labeling in solution at a 1000× molar excess of pLeu. This may be due to differences in irradiation energy in the two studies. However, solid state labeling with 100× molar excess of pLeu showed up to 6 labeled populations in our studies, suggesting that labeling with pLeu is more efficient in the solid state than in solution, perhaps due to greater proximity of protein and pLeu, low water content and/or reduced mobility in the solid state. It should be noted that nearly 20 % of the total apoMb population in solid state remains unlabeled even after irradiation for 60 min. If labeling is uniform throughout the lyophilized matrix, this is not a concern. However, non-uniform labeling could represent loss of information, particularly given the spatial and conformational heterogeneity of amorphous solids.
PL-MS with pLeu has also been used previously to study the effect of carbene diffusion and solvent accessibility in frozen calmodulin solutions35. In frozen solutions, Jumper et al observed labeling at multiple sites, with higher labeling yields at Glu and Asp and no correlation with solvent accessibility. They proposed that pre-concentration of pLeu at the protein surface prior to freezing (driven by electrostatic interaction) and carbene diffusion (driven by temperature) dictated preferential labeling at carboxylate groups. In our studies in lyophilized solids, sitespecific labeling such as this was not detected, though we were able to localize the label to the peptide level. Jumper et al used high-energy collisional dissociation (HCD) and electron-transfer dissociation (ETD), rather than the CID fragmentation used here. It is possible that CID fragmentation may have caused some loss of label, as has been reported previously35, 36. Alternatively, there could be multiple sites of labeling in the lyophilized samples, as expected given the non-specific nature of carbene reactivity. Kolbel and coworkers observed multiple cross-links between pLeu and Gly, Leu and Tyr when pLeu was incorporated within a peptide sequence and irradiated in solution22. Their results indicated preferential labeling based on secondary structural constraints, rather than chemical reactivity.
Structure Dependent Labeling of apoMb in Lyophilized Solids
Though our results do not support preferential labeling of specific functional groups, preferential labeling was observed at the peptide level in lyophilized samples, which varied with the pLeu content of the solid (Fig. 4 (b-h)). Interestingly, the peptides labeled preferentially (i.e., labeled at the lowest pLeu concentrations and at higher concentrations) correspond to those in the molten globule of apoMb in solution. In solution, apoMb has molten globule characteristics at neutral pH, with helices A, G and H forming its core37. The most commonly accepted folding pathway is AGH → ABGH → ABCDEGH38. Interactions between the BG helix pair are critical in maintaining the stability of the AGH core and promoting favorable interactions between the GH helix pair39, 40. BG and GH interactions cause the largest decrease in solvent accessible surface area upon folding41. These interactions are also thought to destabilize helices E and F, which are less stable than helices A, B, G and H40, 42. Our ssPL-MS data showed that helices B, G and H are among the first to be labeled at lower pLeu concentrations, while helices E and F show no label uptake even at pLeu higher concentrations. This suggests that the molten globule is intact in lyophilized solids and is preferentially labeled, perhaps because amino acid side chains are exposed to pLeu in the matrix when the helices are intact. Interaction of pLeu with these regions prior to lyophilization cannot be ruled out, however.
ssPL-MS was also used to examine formulation effects on the side-chain environment, with peptide level resolution. In the presence of sucrose and 100× molar excess of pLeu (20.7 % w/w pLeu), apoMb showed labeling on all helices except E and F. When Gdn HCl was included as an excipient, CD spectroscopy of the solution prior to lyophilization showed loss of signal at 222 nm and 208 nm (data not shown), confirming that the protein had lost helicity. We expected that the Gdn HCl unfolded protein would remain unfolded after lyophilization and would be labeled to a greater extent than folded protein (e.g., in sucrose, as in our previous ssHDX studies20, 43). Instead, photolytic labeling was less in solids containing Gdn HCl than in those containing sucrose. This may be due in part to the high mass fraction of Gdn HCl in the lyophilized solid (∼0.97), limiting interaction between the protein and pLeu by simple dilution. The high Gdn HCl fraction in the solid is the result of the high molar concentration used to unfold apoMb in solution, and is greater than the mass fraction of sucrose (∼0.50) in the sucrose formulation. Preferential interaction of guanidinium ions with apoMb may also contribute, blocking protein-pLeu interactions and thereby inhibiting pLeu labeling44. Similarly, the high ionic strength of the Gdn HCl solutions prior to lyophilization may inhibit ionic interactions between pLeu and apoMb, so that labeling is reduced.
Interactions of GCG (1-8)* in the Solid State
To complement studies with pLeu incorporated into the matrix as an excipient, studies were also performed with a PAA incorporated into the peptide sequence. This approach has been used to map the interactome in cells and to study PPIs in vitro24, 45, 46. The studies used an octapeptide derived from the N-terminal sequence of glucagon (GCG (1-8)*), with pBpA at the F6 position. Glucagon is a 29 amino-acid peptide used to treat insulin-induced hypoglycemia. The monomeric peptide is relatively unstructured in solution, but forms fibrils in acidic and alkaline pH47-49. Previous experimental and computational reports have assigned higher aggregation tendency to glucagon's N- and C-termini49-51. For example, Pedersen et al used experimental Ala mutation to study glucagon aggregation in solution and observed that mutations at residues F6, Y10, V23 and M27 decreased the rate of fibrillation at acidic pH49. Their results indicated that regions 6-10 and 23-27 are involved in fibrillation. Solution-state HDX-NMR studies have also indicated involvement of the N-terminus in aggregation50.
We used GCG (1-8)* to study peptide-peptide and peptide-matrix interactions of the N-terminal sequence in solution and in the solid state, in the presence of various excipients. In solid samples, adducts of pBpA with L-Met and L-Leu excipients were observed. Adducts were not detected in lyophilized solids containing sucrose, trehalose or urea. The formation of adducts with L-Met and L-Leu may be attributable to their free, electron-rich C-H groups, which are known to react with the ketyl radical of pBpA29, 52. Preferential exclusion of sucrose and trehalose from the vicinity of the peptide in the pre-lyophilized solution may contribute to the lack of adduct formation with these excipients53, 54. Urea was selected as a negative control, since it has no C-H groups and hence is not expected to form adducts with pBpA, as was observed. In addition to protein-matrix interactions, studies with GCG (1-8)* were able to capture PPIs at the interface of dimers in the solid state, with amino acid-level resolution. The studies showed that the pBpA label interacts preferentially with G6 in forming the dimer (Fig. 6). In contrast, there appear to be multiple cross-linking sites in solution, perhaps due to greater mobility of the peptide in solution and/or multiple alignments of two monomer units.
Formulations containing both GCG (1-8)* and pLeu, with and without sucrose, were used to examine interactions in the solid state and in solution. In these studies, photolytic labels are present in both the peptide sequence and in the matrix. Following photoirradiation, peptide dimers and trimers, binary adducts of GCG (1-8)* with pLeu, binary adducts of pLeu with sucrose, and ternary adducts containing GCG (1-8)*, pLeu and sucrose were detected (Table 2). The two PAAs (i.e., pLeu, pBpA) are activated at the same wavelength, but have different mechanisms of labeling. The formation of a peptide-pLeu complex with the loss of N2 is consistent with the mechanism of carbene labeling through pLeu activation, while adducts formed without the loss of N2 are consistent with labeling through pBpA activation. In solids containing binary mixtures of GCG (1-8)* and pLeu, both types of adducts were detected, indicating activation and labeling via both pLeu and pBpA (Formulation A, Table 2). In solution, products were detected only with loss of N2 indicating adduct formation via pLeu and not via pBpA, perhaps due to reaction of activated pBpA with water. In solids containing GCG (1-8)*, pLeu and sucrose (Formulation B, Table 2), ternary adducts were detected with the loss of N2, indicating participation of both PAAs in the formation of the adduct. It is unlikely that GCG (1-8)* interacts with sucrose directly in these ternary adducts, since it did not form adducts with sucrose in the binary formulation. Together, these studies with samples containing both GCG (1-8)* and pLeu show that peptide-peptide and peptide-matrix interactions can be detected, and that reactivity of the two PAAs differs in solution and in the solid state.
Applications of Solid State PL-MS
The irreversible nature of photolytic labeling and complementarity to ssHDX-MS makes ssPL-MS a useful tool to study the protein environment in lyophilized powders. The primary advantage of using a PAA in the excipient matrix is the ease of labeling; the PAA simply needs to be added in an appropriate concentration to the pre-lyophilized solution. Moreover, since the PAA is only activated at a certain wavelength range and has a very short lifetime (nanosecond scale for singlet state carbene in solution, 80-120 μs for ketyl radicals in the triplet state in solution29, 55), the photolabeling reaction can be better controlled than with other labeling reagents such as sulfo-N-hydroxysuccinimide acetate (NHSA) that require quenching and removal of excess unreacted reagent. Another benefit of the non-specific diazirine chemistry is that the entire protein structure can be probed, as opposed to reagents such as NHSA and 2,3- butanedione that target only lysine, N-terminal amino acids and arginine. However, non-specific labeling with diazirine-based probes poses analytical challenges. Our results showed that ssPL-MS with apoMb and pLeu could identify the location of the label at the peptide level, but MS/MS using CID failed to provide amino acid-level resolution. Labeling with PAAs incorporated in the protein sequence overcomes this hurdle by localizing the site of labeling to particular amino acid(s), with the attendant disadvantage that the PAA-labeled peptide/protein must first be synthesized. Incorporating the label in the protein sequence provided residue-level information about the sites of interaction, as shown with GCG (1-8)*.
The results have implications for formulation design and stability testing in the biopharmaceutical industry. The high resolution of ssPL-MS can facilitate rational design of formulations by allowing excipients to be selected and created based on their interactions with the protein sidechain. The information can also be used to improve protein drugs themselves through protein engineering. The hydration of protein side-chains, although not studied here, can be probed by tracking PAA-water adducts formed in ssPL-MS. The effects of relative humidity (% RH) on lyophilized protein secondary structure have been studied using ssHDX-MS in our lab43. Similar studies using ssPL-MS may be performed with increasing % RH or during reconstitution. Ongoing work in our laboratory is developing alternate approaches to incorporate photolytic label into the protein sequence, including the use of auxotrophic cell lines and site-directed mutagenesis 56, 57. The use of heterobifunctional cross-linkers such as succinimidyl 4,4′-azipentanoate, which contains a primary amine-specific NHS functional group and a non-specific diazirine functional group, is also being explored as an alternative approach to label incorporation.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Andreas M. Sophocleous (GlaxoSmithKline) for helpful suggestions and scientific discussion. Financial support was provided through NIH RO1 GM085293 (PI: E. M. Topp) and from the College of Pharmacy at Purdue University.
Footnotes
Supporting Information: Additional tables and figures are available in Supporting Information, as mentioned in text. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28(9):917–24. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- 2.Biosimilars- A global strategic business report. 2012 [Google Scholar]
- 3.Kasper JC, Friess W. The freezing step in lyophilization: physico-chemical fundamentals, freezing methods and consequences on process performance and quality attributes of biopharmaceuticals. Eur J Pharm Biopharm. 2011;78(2):248–63. doi: 10.1016/j.ejpb.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 4.Luthra S, Obert JP, Kalonia DS, Pikal MJ. Investigation of drying stresses on proteins during lyophilization: differentiation between primary and secondary-drying stresses on lactate dehydrogenase using a humidity controlled mini freeze-dryer. J Pharm Sci. 2007;96(1):61–70. doi: 10.1002/jps.20758. [DOI] [PubMed] [Google Scholar]
- 5.Chang LL, Pikal MJ. Mechanisms of protein stabilization in the solid state. J Pharm Sci. 2009;98(9):2886–908. doi: 10.1002/jps.21825. [DOI] [PubMed] [Google Scholar]
- 6.Lai MC, Topp EM. Solid-state chemical stability of proteins and peptides. J Pharm Sci. 1999;88(5):489–500. doi: 10.1021/js980374e. [DOI] [PubMed] [Google Scholar]
- 7.Oliyai C, Patel JP, Carr L, Borchardt RT. Solid state chemical instability of an asparaginyl residue in a model hexapeptide. J Pharm Sci Technol. 1994;48(3):167–23. [PubMed] [Google Scholar]
- 8.Li S, Patapoff TW, Overcashier D, Hsu C, Nguyen TH, Borchardt RT. Effects of reducing sugars on the chemical stability of human relaxin in the lyophilized state. J Pharm Sci. 1996;85(8):873–7. doi: 10.1021/js950456s. [DOI] [PubMed] [Google Scholar]
- 9.Carpenter JF, Pikal MJ, Chang BS, Randolph TW. Rational design of stable lyophilized protein formulations: some practical advice. Pharm Res. 1997;14(8):969–75. doi: 10.1023/a:1012180707283. [DOI] [PubMed] [Google Scholar]
- 10.Tang X, Pikal MJ. Design of freeze-drying processes for pharmaceuticals: practical advice. Pharm Res. 2004;21(2):191–200. doi: 10.1023/b:pham.0000016234.73023.75. [DOI] [PubMed] [Google Scholar]
- 11.Costantino HR, Langer R, Klibanov AM. Aggregation of a lyophilized pharmaceutical protein, recombinant human albumin: effect of moisture and stabilization by excipients. Biotechnology (N Y) 1995;13(5):493–6. doi: 10.1038/nbt0595-493. [DOI] [PubMed] [Google Scholar]
- 12.Carpenter JF, Prestrelski SJ, Dong A. Application of infrared spectroscopy to development of stable lyophilized protein formulations. Eur J Pharm Biopharm. 1998;45(3):231–8. doi: 10.1016/s0939-6411(98)00005-8. [DOI] [PubMed] [Google Scholar]
- 13.Costantino HR, Carrasquillo KG, Cordero RA, Mumenthaler M, Hsu CC, Griebenow K. Effect of excipients on the stability and structure of lyophilized recombinant human growth hormone. J Pharm Sci. 1998;87(11):1412–20. doi: 10.1021/js980069t. [DOI] [PubMed] [Google Scholar]
- 14.Sane SU, Wong R, Hsu CC. Raman spectroscopic characterization of drying-induced structural changes in a therapeutic antibody: correlating structural changes with long-term stability. J Pharm Sci. 2004;93(4):1005–18. doi: 10.1002/jps.20014. [DOI] [PubMed] [Google Scholar]
- 15.Elkordy AA, Forbes RT, Barry BW. Study of protein conformational stability and integrity using calorimetry and FT-Raman spectroscopy correlated with enzymatic activity. Eur J Pharm Sci. 2008;33(2):177–90. doi: 10.1016/j.ejps.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Valdivia AA, Barth A, Batista YR, Kumar S. Characterization of recombinant antibodies for cancer therapy by infrared spectroscopy. Biologicals. 2013;41(2):104–10. doi: 10.1016/j.biologicals.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Hu KN, Tycko R. What can solid state NMR contribute to our understanding of protein folding? Biophys Chem. 2010;151(1-2):10–21. doi: 10.1016/j.bpc.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshioka S, Miyazaki T, Aso Y. Beta-relaxation of insulin molecule in lyophilized formulations containing trehalose or dextran as a determinant of chemical reactivity. Pharm Res. 2006;23(5):961–6. doi: 10.1007/s11095-006-9907-3. [DOI] [PubMed] [Google Scholar]
- 19.Sophocleous AM, Zhang J, Topp EM. Localized hydration in lyophilized myoglobin by hydrogen-deuterium exchange mass spectrometry. 1. Exchange mapping. Mol Pharm. 2012;9(4):718–26. doi: 10.1021/mp3000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinha S, Li Y, Williams TD, Topp EM. Protein conformation in amorphous solids by FTIR and by hydrogen/deuterium exchange with mass spectrometry. Biophys J. 2008;95(12):5951–61. doi: 10.1529/biophysj.108.139899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jumper CC, Schriemer DC. Mass spectrometry of laser-initiated carbene reactions for protein topographic analysis. Anal Chem. 2011;83(8):2913–20. doi: 10.1021/ac102655f. [DOI] [PubMed] [Google Scholar]
- 22.Kolbel K, Ihling CH, Sinz A. Analysis of peptide secondary structures by photoactivatable amino acid analogues. Angew Chem Int Ed Engl. 2012;51(50):12602–5. doi: 10.1002/anie.201205308. [DOI] [PubMed] [Google Scholar]
- 23.Kauer JC, Erickson-Viitanen S, Wolfe HR, Jr, DeGrado WF. p-Benzoyl-L-phenylalanine, a new photoreactive amino acid. Photolabeling of calmodulin with a synthetic calmodulin-binding peptide. J Biol Chem. 1986;261(23):10695–700. [PubMed] [Google Scholar]
- 24.Suchanek M, Radzikowska A, Thiele C. Photo-leucine and photomethionine allow identification of protein-protein interactions in living cells. Nat Methods. 2005;2(4):261–7. doi: 10.1038/nmeth752. [DOI] [PubMed] [Google Scholar]
- 25.Hino N, Okazaki Y, Kobayashi T, Hayashi A, Sakamoto K, Yokoyama S. Protein photo-cross-linking in mammalian cells by site-specific incorporation of a photoreactive amino acid. Nat Methods. 2005;2(3):201–6. doi: 10.1038/nmeth739. [DOI] [PubMed] [Google Scholar]
- 26.Soutourina J, Wydau S, Ambroise Y, Boschiero C, Werner M. Direct interaction of RNA polymerase II and mediator required for transcription in vivo. Science. 2011;331(6023):1451–4. doi: 10.1126/science.1200188. [DOI] [PubMed] [Google Scholar]
- 27.Janz JM, Ren Y, Looby R, Kazmi MA, Sachdev P, Grunbeck A, Haggis L, Chinnapen D, Lin AY, Seibert C, McMurry T, Carlson KE, Muir TW, Hunt S, 3rd, Sakmar TP. Direct interaction between an allosteric agonist pepducin and the chemokine receptor CXCR4. J Am Chem Soc. 2011;133(40):15878–81. doi: 10.1021/ja206661w. [DOI] [PubMed] [Google Scholar]
- 28.Dubinsky L, Krom BP, Meijler MM. Diazirine based photoaffinity labeling. Bioorg Med Chem. 2012;20(2):554–70. doi: 10.1016/j.bmc.2011.06.066. [DOI] [PubMed] [Google Scholar]
- 29.Dorman G, Prestwich GD. Benzophenone photophores in biochemistry. Biochemistry. 1994;33(19):5661–73. doi: 10.1021/bi00185a001. [DOI] [PubMed] [Google Scholar]
- 30.Arakawa T, Prestrelski SJ, Kenney WC, Carpenter JF. Factors affecting short-term and long-term stabilities of proteins. Adv Drug Deliv Rev. 2001;46(1-3):307–326. doi: 10.1016/s0169-409x(00)00144-7. [DOI] [PubMed] [Google Scholar]
- 31.Wang W. Lyophilization and development of solid protein pharmaceuticals. Int J Pharm. 2000;203(1-2):1–60. doi: 10.1016/s0378-5173(00)00423-3. [DOI] [PubMed] [Google Scholar]
- 32.Fontana A, Zambonin M, Polverino de Laureto P, De Filippis V, Clementi A, Scaramella E. Probing the conformational state of apomyoglobin by limited proteolysis. J Mol Biol. 1997;266(2):223–30. doi: 10.1006/jmbi.1996.0787. [DOI] [PubMed] [Google Scholar]
- 33.Eliezer D, Yao J, Dyson HJ, Wright PE. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat Struct Biol. 1998;5(2):148–55. doi: 10.1038/nsb0298-148. [DOI] [PubMed] [Google Scholar]
- 34.Schilling B, Row RH, Gibson BW, Guo X, Young MM. MS2Assign, automated assignment and nomenclature of tandem mass spectra of chemically crosslinked peptides. J Am Soc Mass Spectrom. 2003;14(8):834–50. doi: 10.1016/S1044-0305(03)00327-1. [DOI] [PubMed] [Google Scholar]
- 35.Jumper CC, Bomgarden R, Rogers J, Etienne C, Schriemer DC. Highresolution mapping of carbene-based protein footprints. Anal Chem. 2012;84(10):4411–8. doi: 10.1021/ac300120z. [DOI] [PubMed] [Google Scholar]
- 36.Mendoza VL, Antwi K, Baron-Rodriguez MA, Blanco C, Vachet RW. Structure of the preamyloid dimer of beta-2-microglobulin from covalent labeling and mass spectrometry. Biochemistry. 2010;49(7):1522–32. doi: 10.1021/bi901748h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin L, Pinker RJ, Forde K, Rose GD, Kallenbach NR. Molten globular characteristics of the native state of apomyoglobin. Nat Struct Biol. 1994;1(7):447–52. doi: 10.1038/nsb0794-447. [DOI] [PubMed] [Google Scholar]
- 38.Jennings PA, Wright PE. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science. 1993;262(5135):892–6. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 39.Hughson FM, Baldwin RL. Use of site-directed mutagenesis to destabilize native apomyoglobin relative to folding intermediates. Biochemistry. 1989;28(10):4415–22. doi: 10.1021/bi00436a044. [DOI] [PubMed] [Google Scholar]
- 40.Nishimura C, Dyson HJ, Wright PE. Energetic frustration of apomyoglobin folding: role of the B helix. J Mol Biol. 2010;396(5):1319–28. doi: 10.1016/j.jmb.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weaver DL. Hydrophobic interaction between globin helices. Biopolymers. 1992;32(5):477–90. doi: 10.1002/bip.360320504. [DOI] [PubMed] [Google Scholar]
- 42.Pappu RV, Weaver DL. The early folding kinetics of apomyoglobin. Protein Sci. 1998;7(2):480–90. doi: 10.1002/pro.5560070229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sophocleous AM, Topp EM. Localized hydration in lyophilized myoglobin by hydrogen-deuterium exchange mass spectrometry. 2. Exchange kinetics. Mol Pharm. 2012;9(4):727–33. doi: 10.1021/mp2004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monera OD, Kay CM, Hodges RS. Protein denaturation with guanidine hydrochloride or urea provides a different estimate of stability depending on the contributions of electrostatic interactions. Protein Sci. 1994;3(11):1984–91. doi: 10.1002/pro.5560031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MacKinnon AL, Garrison JL, Hegde RS, Taunton J. Photo-leucine incorporation reveals the target of a cyclodepsipeptide inhibitor of cotranslational translocation. J Am Chem Soc. 2007;129(47):14560–1. doi: 10.1021/ja076250y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pettelkau J, Schroder T, Ihling CH, Olausson BE, Kolbel K, Lange C, Sinz A. Structural Insights into retinal guanylylcyclase-GCAP-2 interaction determined by cross-linking and mass spectrometry. Biochemistry. 2012;51(24):4932–49. doi: 10.1021/bi300064v. [DOI] [PubMed] [Google Scholar]
- 47.Panijpan B, Gratzer WB. Conformational nature of monomeric glucagon. Eur J Biochem. 1974;45(2):547–53. doi: 10.1111/j.1432-1033.1974.tb03580.x. [DOI] [PubMed] [Google Scholar]
- 48.Onoue S, Ohshima K, Debari K, Koh K, Shioda S, Iwasa S, Kashimoto K, Yajima T. Mishandling of the therapeutic peptide glucagon generates cytotoxic amyloidogenic fibrils. Pharm Res. 2004;21(7):1274–83. doi: 10.1023/b:pham.0000033016.36825.2c. [DOI] [PubMed] [Google Scholar]
- 49.Pedersen JS, Dikov D, Otzen DE. N- and C-terminal hydrophobic patches are involved in fibrillation of glucagon. Biochemistry. 2006;45(48):14503–12. doi: 10.1021/bi061228n. [DOI] [PubMed] [Google Scholar]
- 50.Svane AS, Jahn K, Deva T, Malmendal A, Otzen DE, Dittmer J, Nielsen NC. Early stages of amyloid fibril formation studied by liquid-state NMR: the peptide hormone glucagon. Biophys J. 2008;95(1):366–77. doi: 10.1529/biophysj.107.122895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pande VS. A universal TANGO? Nat Biotechnol. 2004;22(10):1240–1. doi: 10.1038/nbt1004-1240. [DOI] [PubMed] [Google Scholar]
- 52.Wittelsberger A, Thomas BE, Mierke DF, Rosenblatt M. Methionine acts as a "magnet" in photoaffinity crosslinking experiments. FEBS Lett. 2006;580(7):1872–6. doi: 10.1016/j.febslet.2006.02.050. [DOI] [PubMed] [Google Scholar]
- 53.Lee JC, Timasheff SN. The stabilization of proteins by sucrose. J Biol Chem. 1981;256(14):7193–201. [PubMed] [Google Scholar]
- 54.Xie G, Timasheff SN. The thermodynamic mechanism of protein stabilization by trehalose. Biophys Chem. 1997;64(1-3):25–43. doi: 10.1016/s0301-4622(96)02222-3. [DOI] [PubMed] [Google Scholar]
- 55.Ford F, Yuzawa T, Platz MS, Matzinger S, Fülscher M. Rearrangement of Dimethylcarbene to Propene: Study by Laser Flash Photolysis and ab Initio Molecular Orbital Theory. J Am Chem Soc. 1998;120(18):4430–4438. [Google Scholar]
- 56.Kiick KL, Weberskirch R, Tirrell DA. Identification of an expanded set of translationally active methionine analogues in Escherichia coli. FEBS Lett. 2001;502(1-2):25–30. doi: 10.1016/s0014-5793(01)02657-6. [DOI] [PubMed] [Google Scholar]
- 57.Chin JW, Martin AB, King DS, Wang L, Schultz PG. Addition of a photocrosslinking amino acid to the genetic code of Escherichiacoli. Proc Natl Acad Sci USA. 2002;99(17):11020–4. doi: 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.